
7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells

 ,
,  , ,
, , 
Abstract
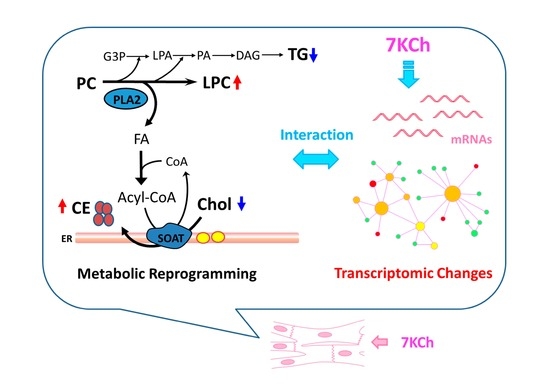
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture and Growth Curve Determination
2.3. Global Metabolite Analysis by Ultrahigh-Performance Liquid Chromatography Time-of-Flight Mass Spectrometry (UPLC-TOF-MS)
2.4. Quantification of Coenzymes and Sterols by Liquid Chromatography Coupled with Tandem Mass Spectrometry (LC-MS/MS)
2.5. Quantification of Dolichols in HL-1 Cell by UPLC-TOF-MS
2.6. Quantification of Metabolites in MVA Pathway by LC-MS/MS
2.7. Quantification of Cholesteryl Esters (CE) and Triglycerides by LC-MS/MS
2.8. mRNA-seq Profiling and Data Analysis
2.9. Statistical Analyses
3. Results
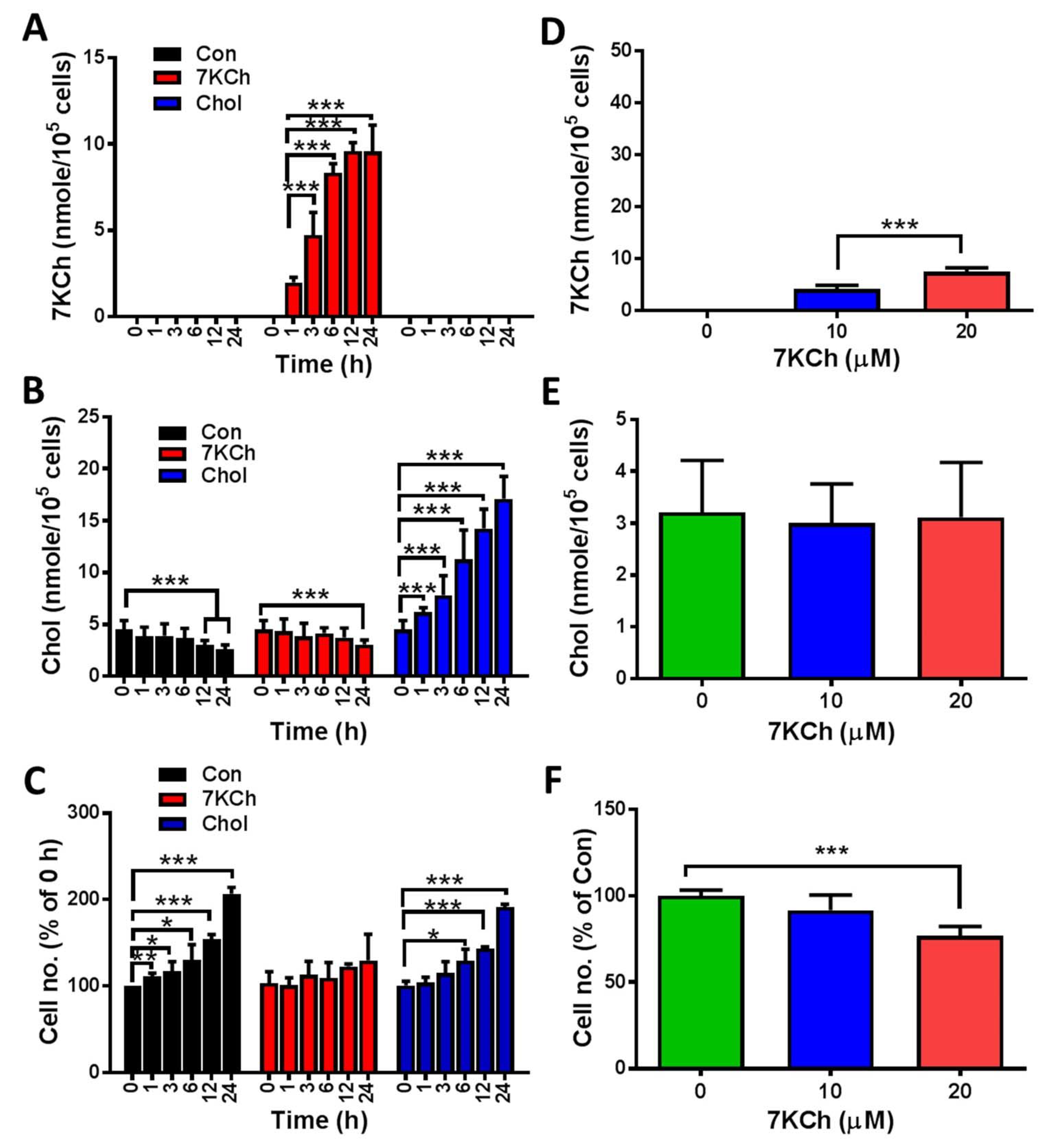
3.1. Intracellular 7KCh Accumulation Is Associated with Cell Growth Inhibition
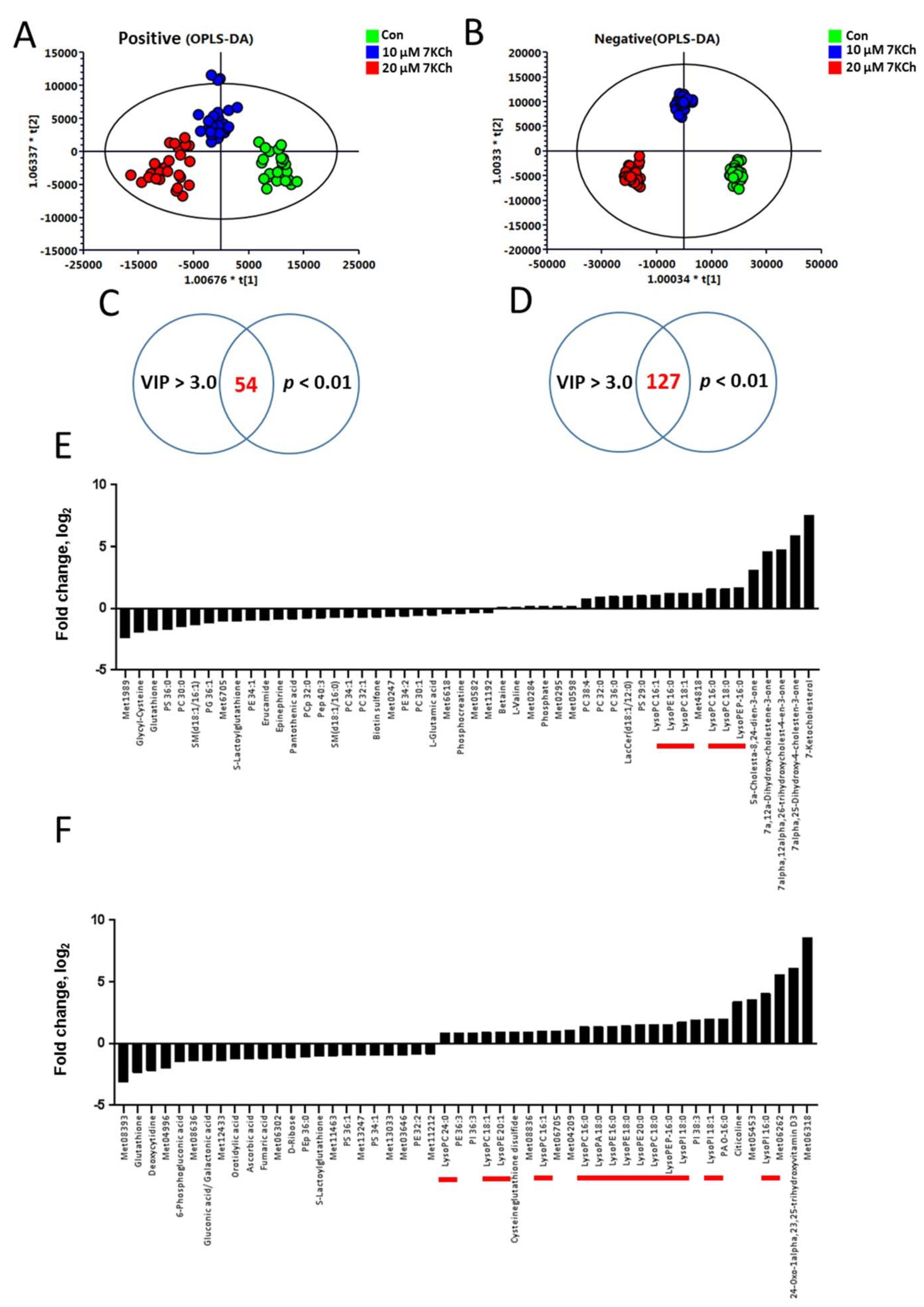
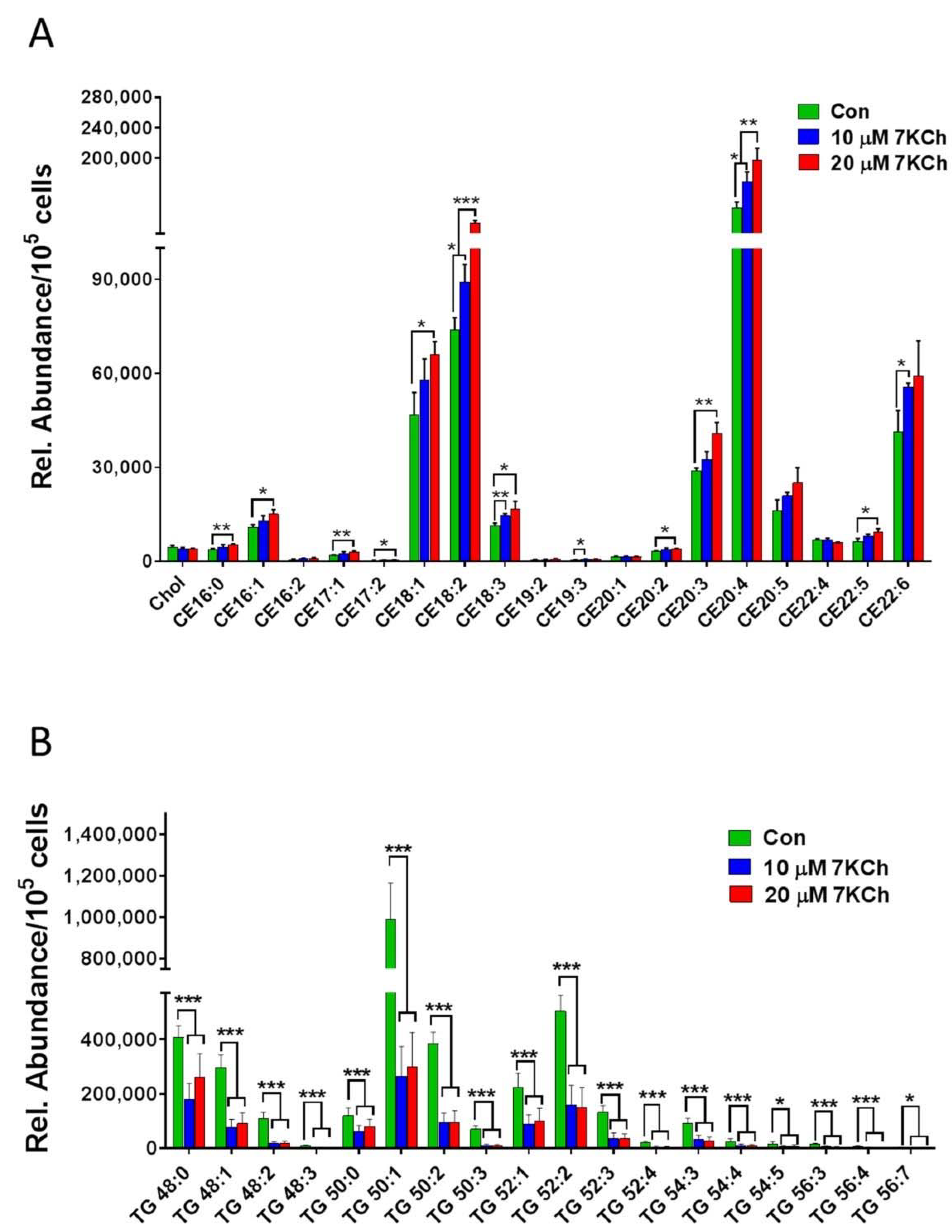
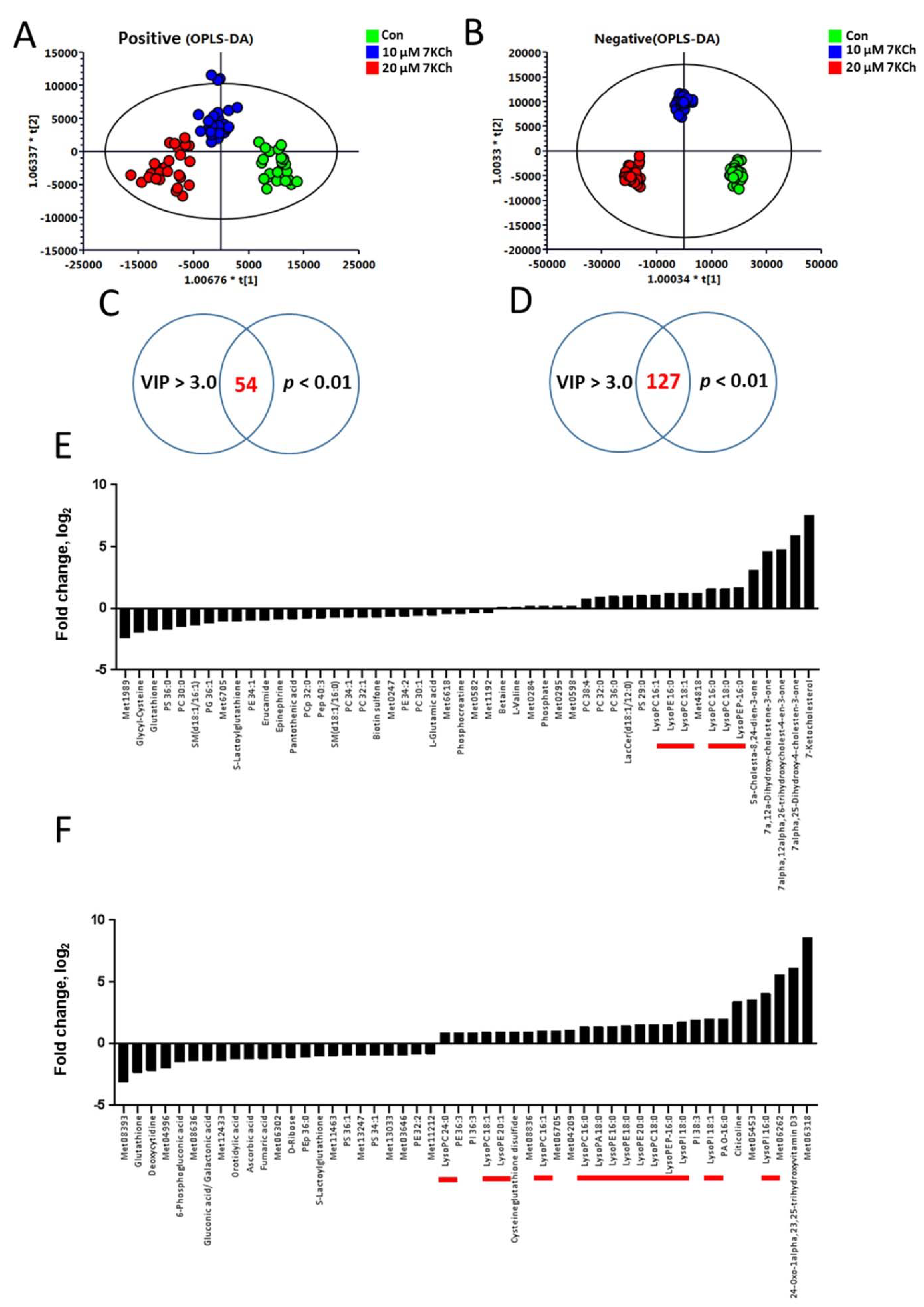
3.2. The Metabolic Profiles of 7KCh-Treated HL-1 Cells
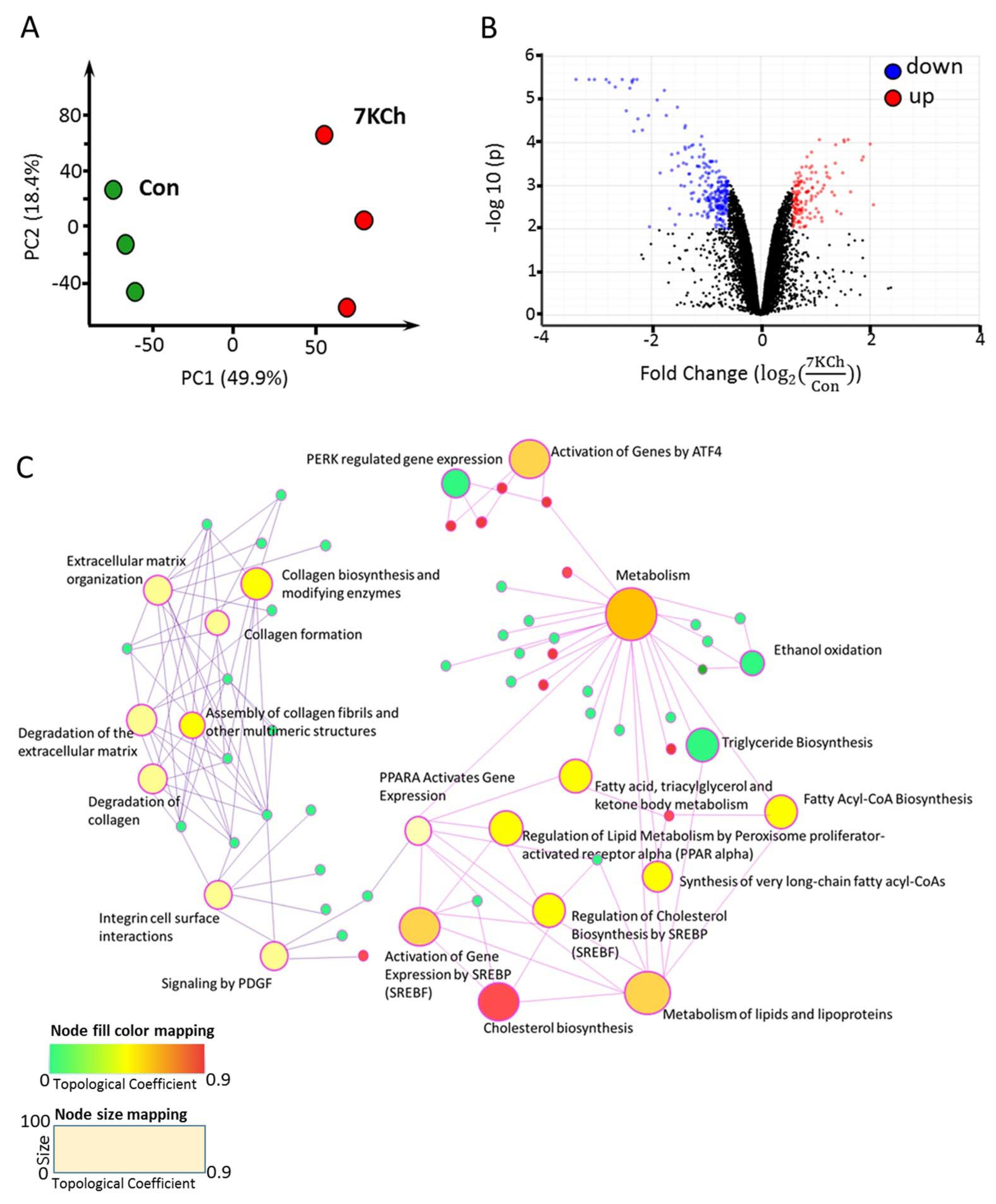
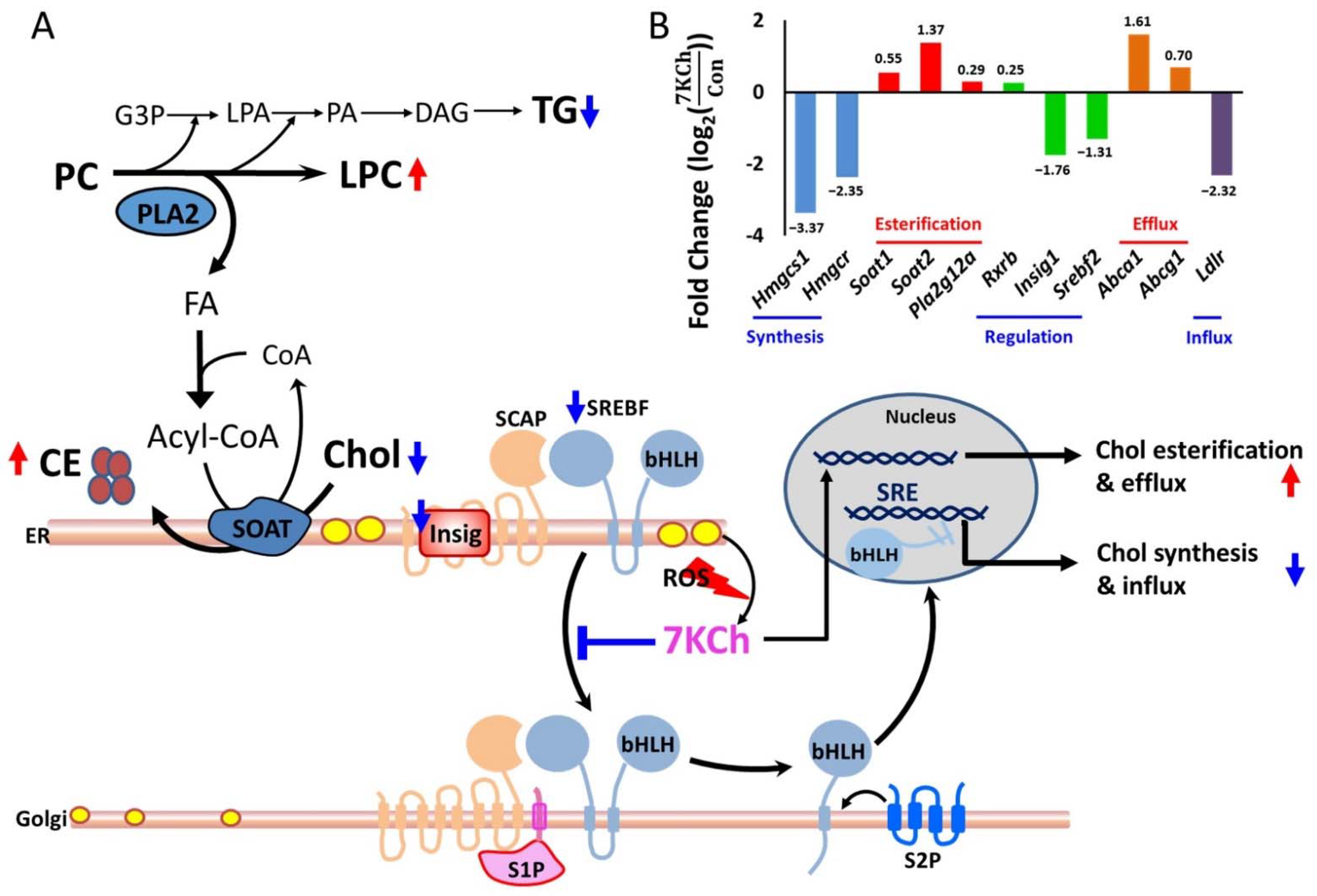
3.3. 7KCh Affects Transcription of Lipid Metabolism-Related Genes
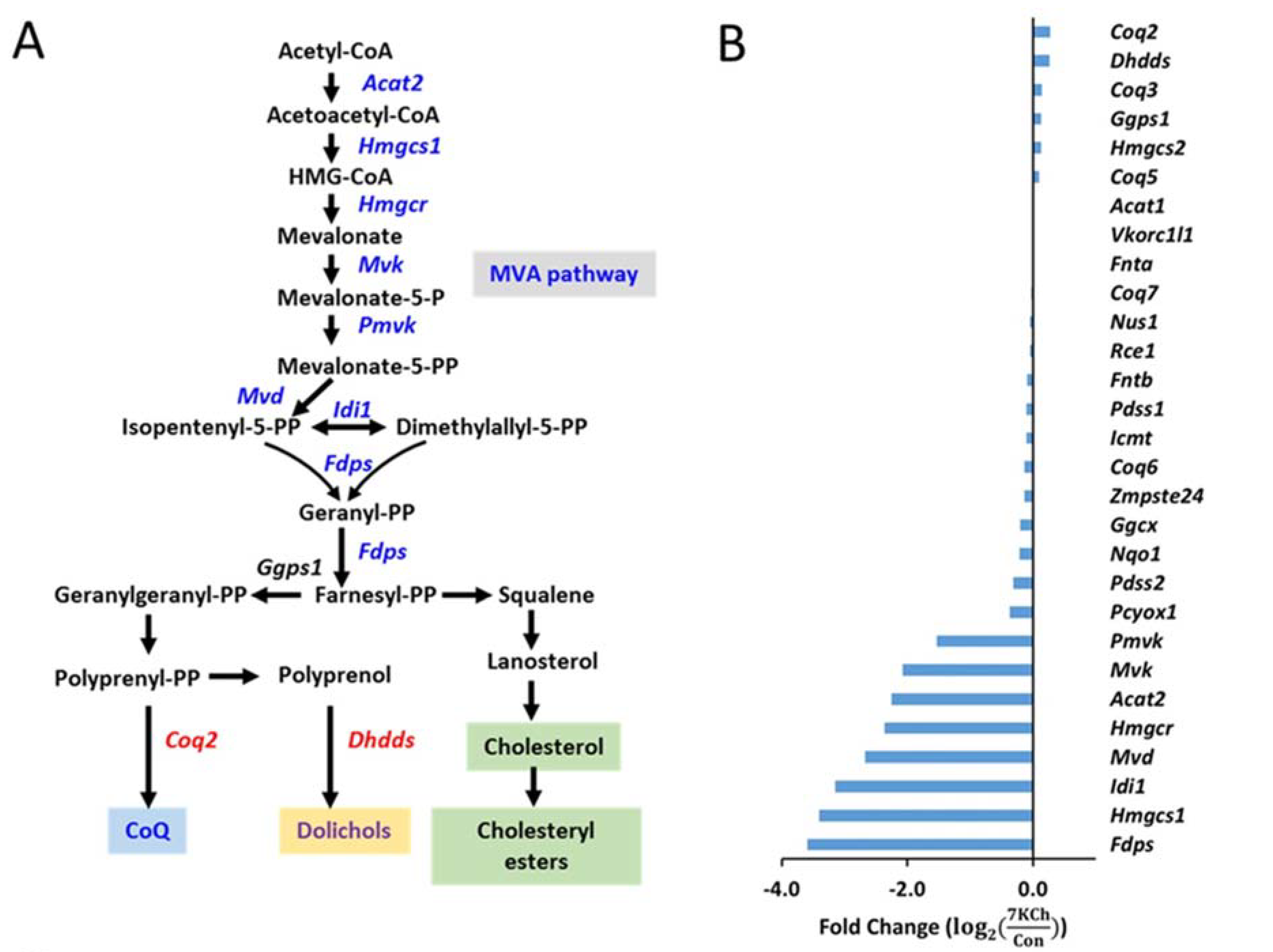
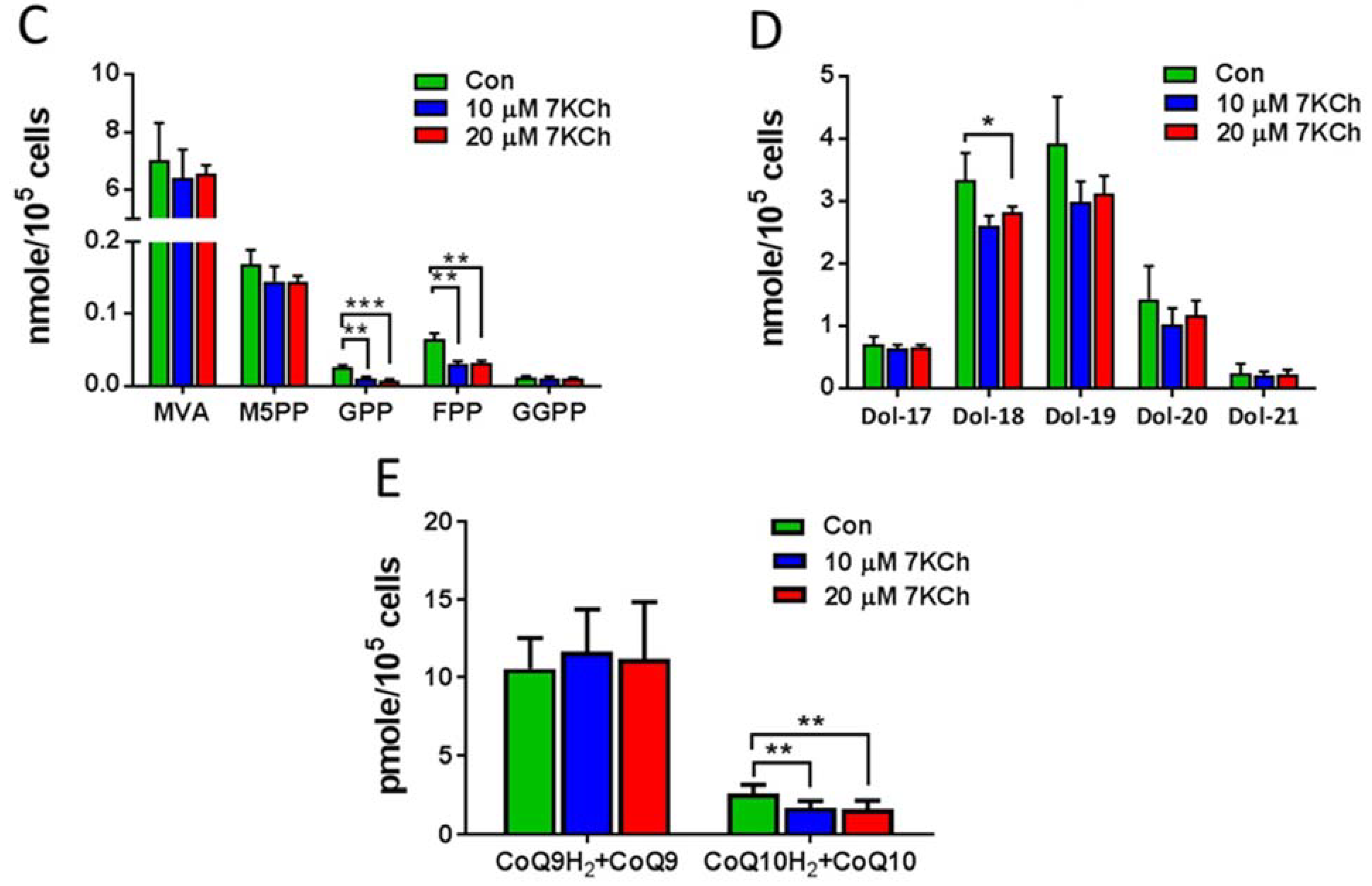
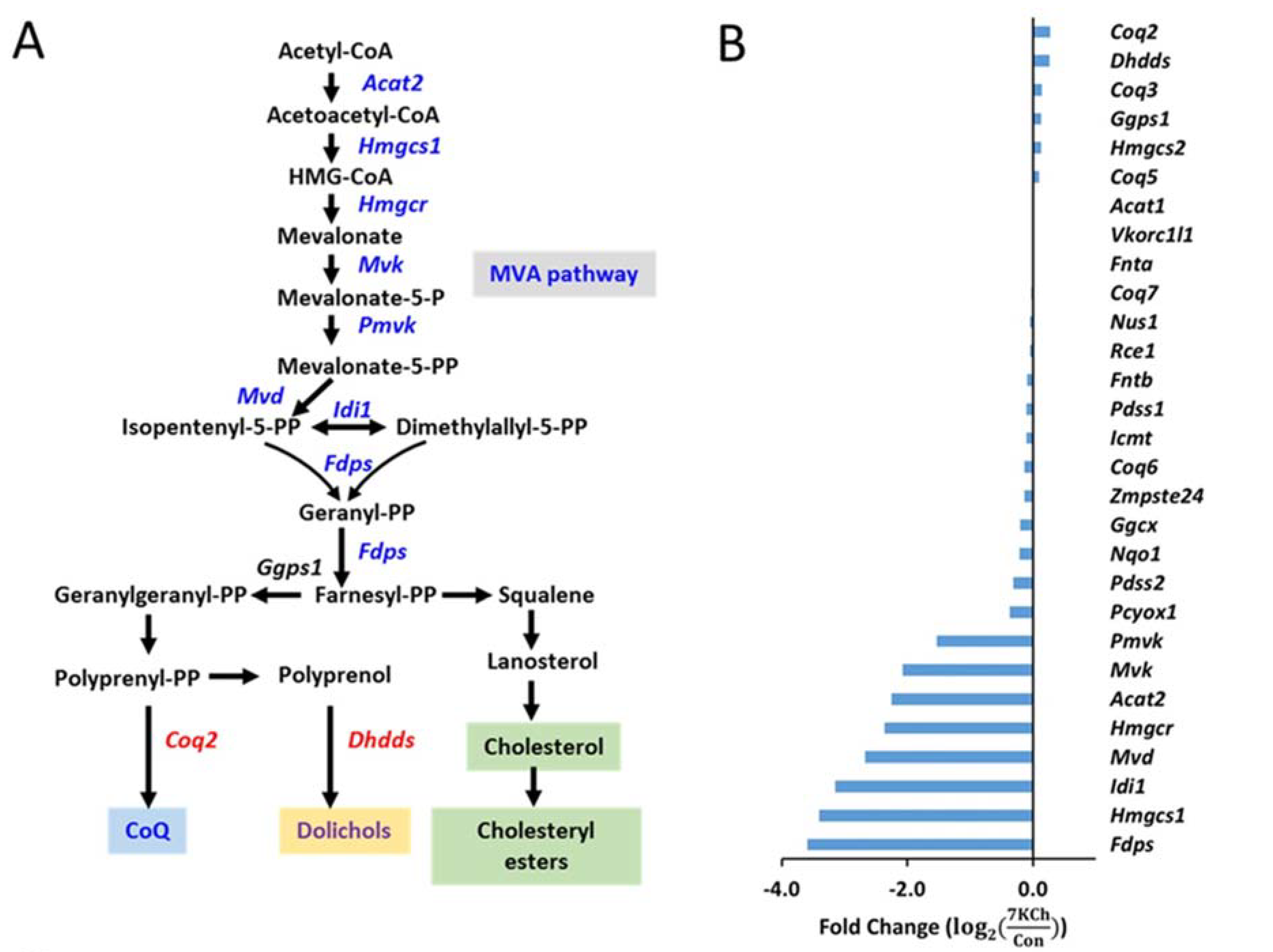
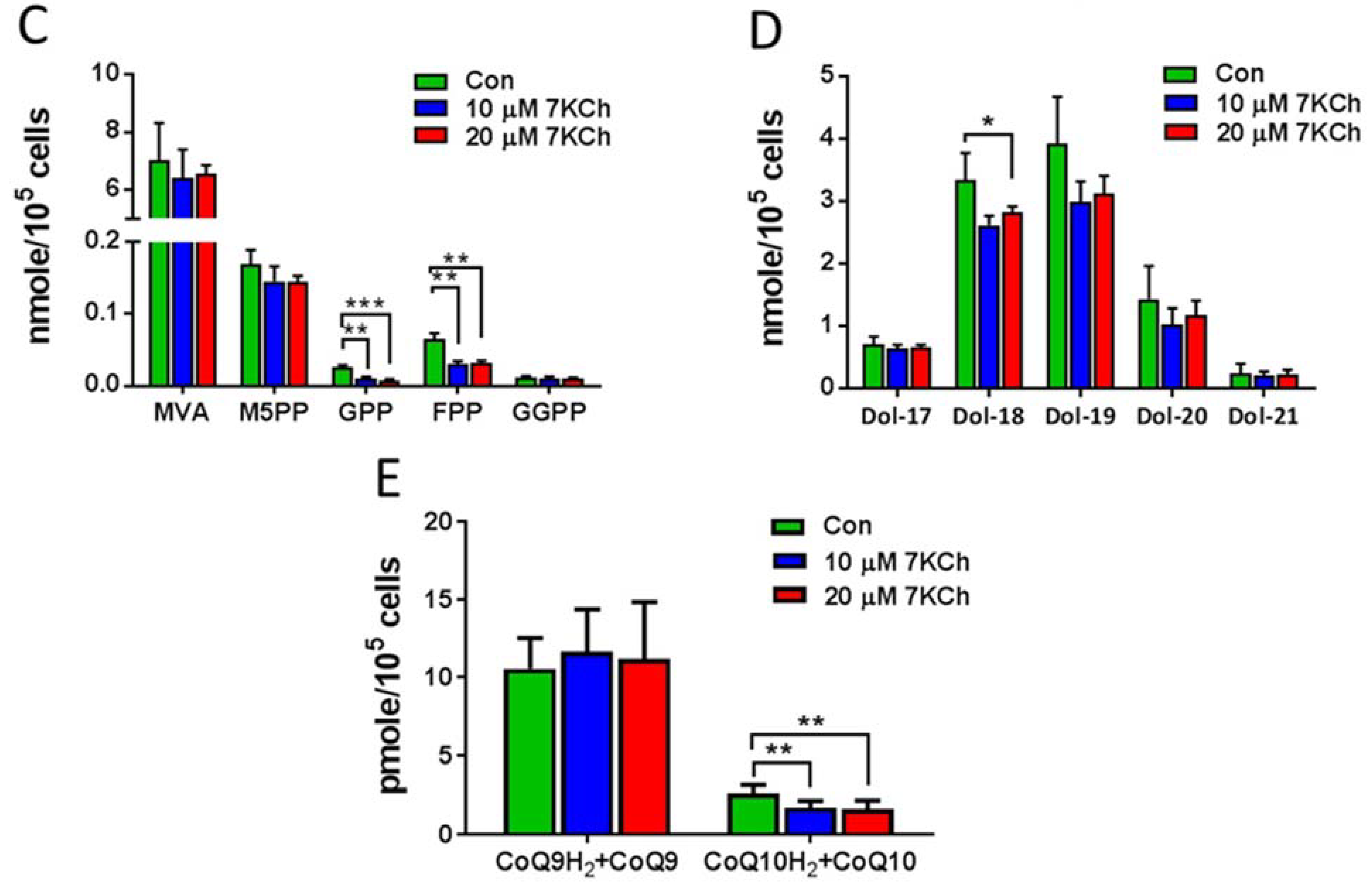
3.4. Alteration in Gene Expression and Metabolites Involved in MVA Pathway in 7KCh-Treated Cardiac Cells
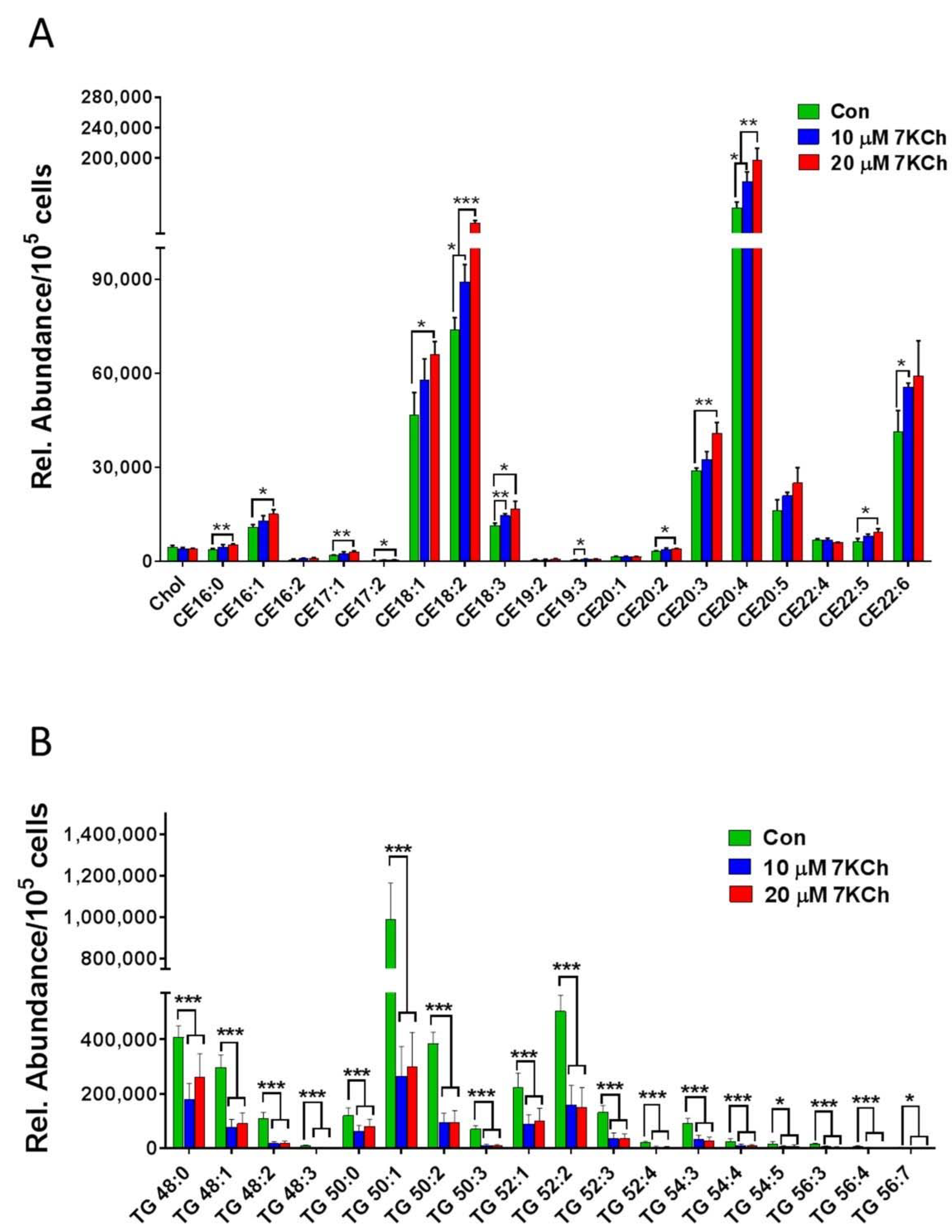
3.5. 7KCh Reduces Triacylglyceride Synthesis in Cardiac Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. Low Density Lipoprotein Oxidation and Its Pathobiological Significance. J. Biol. Chem. 1997, 272, 20963–20966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, M.A.; Brown, A.J. 7-Ketocholesterol. Int. J. Biochem. Cell Biol. 1999, 31, 369–375. [Google Scholar] [CrossRef]
- Wang, M.; Long, W.; Li, D.; Wang, D.; Zhong, Y.; Mu, D.; Song, J.; Xia, M. Plasma 7-ketocholesterol levels and the risk of incident cardiovascular events. Heart 2017, 103, 1788–1794. [Google Scholar] [CrossRef]
- Song, J.; Wang, D.; Chen, H.; Huang, X.; Zhong, Y.; Jiang, N.; Chen, C.; Xia, M. Association of Plasma 7-Ketocholesterol With Cardiovascular Outcomes and Total Mortality in Patients With Coronary Artery Disease. Circ. Res. 2017, 120, 1622–1631. [Google Scholar] [CrossRef]
- Schweizer, R.A.; Zurcher, M.; Balazs, Z.; Dick, B.; Odermatt, A. Rapid hepatic metabolism of 7-ketocholesterol by 11beta-hydroxysteroid dehydrogenase type 1: Species-specific differences between the rat, human, and hamster enzyme. J. Biol. Chem. 2004, 279, 18415–18424. [Google Scholar] [CrossRef] [Green Version]
- Lyons, M.A.; Samman, S.; Gatto, L.; Brown, A.J. Rapid hepatic metabolism of 7-ketocholesterol in vivo: Implications for dietary oxysterols. J. Lipid Res. 1999, 40, 1846–1857. [Google Scholar] [CrossRef]
- Lee, J.W.; Huang, J.-D.; Rodriguez, I.R. Extra-hepatic metabolism of 7-ketocholesterol occurs by esterification to fatty acids via cPLA2α and SOAT1 followed by selective efflux to HDL. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2015, 1851, 605–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.-Y.; Wang, C.-H.; Ho, H.-Y.; Wu, P.-T.; Hung, C.-L.; Huang, C.-Y.; Yeh, Y.-H.; Cheng, M.-L. Lipidomics reveals accumulation of the oxidized cholesterol in erythrocytes of heart failure patients. Redox Biol. 2017, 14, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Suppression of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase Activity and Inhibition of Growth of Human Fibroblasts by 7-Ketocholesterol. J. Biol. Chem. 1974, 249, 7306–7314. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Familial Hypercholesterolemia: Identification of a Defect in the Regulation of 3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Activity Associated with Overproduction of Cholesterol. Proc. Natl. Acad. Sci. USA 1973, 70, 2804–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [Green Version]
- Thurnher, M.; Gruenbacher, G. T lymphocyte regulation by mevalonate metabolism. Sci. Signal. 2015, 8, re4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidani, Y.; Bensinger, S.J. Modulating Cholesterol Homeostasis to Build a Better T Cell. Cell Metab. 2016, 23, 963–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Lu, X.; Rong, X.; Jiang, W.; Lai, D.; Ma, Y.; Zhou, K.; Fu, G.; Xu, S. Inhibition of the mevalonate pathway ameliorates anoxia-induced down-regulation of FKBP12.6 and intracellular calcium handling dysfunction in H9c2 cells. J. Mol. Cell. Cardiol. 2015, 80, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rong, X.; Lv, X.; Jiang, W.; Yang, Y.; Lai, D.; Xu, S.; Fu, G. Inhibition of mevalonate pathway prevents ischemia-induced cardiac dysfunction in rats via RhoA-independent signaling pathway. Cardiovasc. Ther. 2017, 35, 5. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Teppo, J.; Pöhö, P.; Movahedi, P.; Vaikkinen, A.; Karhu, S.T.; Trošt, K.; Suvitaival, T.; Heikkonen, J.; Pahikkala, T.; et al. Molecular Atlas of Postnatal Mouse Heart Development. J. Am. Hear. Assoc. 2018, 7, e010378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, R.J.; Parker, B.L.; Quaife-Ryan, G.; Voges, H.K.; Needham, E.; Bornot, A.; Ding, M.; Andersson, H.; Polla, M.; Elliott, D.A.; et al. Drug Screening in Human PSC-Cardiac Organoids Identifies Pro-proliferative Compounds Acting via the Mevalonate Pathway. Cell Stem Cell 2019, 24, 895–907.e6. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.-Y.; Lin, Y.-T.; Lin, G.; Wu, P.-R.; Cheng, M.-L. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol. 2017, 12, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.-Y.; Cheng, M.-L.; Shiao, M.-S.; Chiu, D.T.-Y. Characterization of global metabolic responses of glucose-6-phosphate dehydrogenase-deficient hepatoma cells to diamide-induced oxidative stress. Free. Radic. Biol. Med. 2013, 54, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-L.; Shiao, M.-S.; Chiu, D.T.-Y.; Weng, S.-F.; Tang, H.-Y.; Ho, H.-Y. Biochemical disorders associated with antiproliferative effect of dehydroepiandrosterone in hepatoma cells as revealed by LC-based metabolomics. Biochem. Pharmacol. 2011, 82, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D8907. [Google Scholar] [CrossRef] [PubMed]
- Schou-Pedersen, A.M.V.; Schemeth, D.; Lykkesfeldt, J. Determination of Reduced and Oxidized Coenzyme Q10 in Canine Plasma and Heart Tissue by HPLC-ECD: Comparison with LC-MS/MS Quantification. Antioxidants 2019, 8, 253. [Google Scholar] [CrossRef] [Green Version]
- Lin, N.; Zhang, H.; Qiu, W.; Ye, J.; Han, L.; Wang, Y.; Gu, X. Determination of 7-ketocholesterol in plasma by LC-MS for rapid diagnosis of acid SMase-deficient Niemann-Pick disease. J. Lipid Res. 2014, 55, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.A.; Guan, Z.; Raetz, C.R. Analysis of ubiquinones, dolichols, and dolichol diphosphate-oligosaccharides by liquid chromatography-electrospray ionization-mass spectrometry. Methods Enzymol. 2007, 432, 117–143. [Google Scholar] [PubMed]
- Sugimoto, H.; Iguchi, M.; Jinno, F. Bioanalysis of farnesyl pyrophosphate in human plasma by high-performance liquid chromatography coupled to triple quadrupole tandem mass spectrometry and hybrid quadrupole Orbitrap high-resolution mass spectrometry. Anal. Bioanal. Chem. 2017, 343, 425–3560. [Google Scholar] [CrossRef]
- Pataj, Z.; Liebisch, G.; Schmitz, G.; Matysik, S. Quantification of oxysterols in human plasma and red blood cells by liquid chromatography high-resolution tandem mass spectrometry. J. Chromatogr. A 2015, 1439, 82–88. [Google Scholar] [CrossRef]
- Akiba, S.; Yoneda, Y.; Ohno, S.; Nemoto, M.; Sato, T. Oxidized LDL activates phospholipase A2 to supply fatty acids required for cholesterol esterification. J. Lipid Res. 2003, 44, 1676–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, N.E.; Rusinol, A.E.; Linton, M.; Hachey, D.L.; Fazio, S.; Sinensky, M.S.; Thewke, D. Acyl-coenzyme A:cholesterol acyltransferase promotes oxidized LDL/oxysterol-induced apoptosis in macrophages. J. Lipid Res. 2005, 46, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Perrey, S.; Legendre, C.; Matsuura, A.; Guffroy, C.; Binet, J.; Ohbayashi, S.; Tanaka, T.; Ortuno, J.C.; Matsukura, T.; Laugel, T.; et al. Preferential pharmacological inhibition of macrophage ACAT increases plaque formation in mouse and rabbit models of atherogenesis. Atherosclerosis 2001, 155, 359–370. [Google Scholar] [CrossRef]
- Fazio, S.; Major, A.S.; Swift, L.L.; Gleaves, L.A.; Accad, M.; Linton, M.F.; Farese, R.V. Increased atherosclerosis in LDL receptor–null mice lacking ACAT1 in macrophages. J. Clin. Investig. 2001, 107, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.P.; Cheng, M.L.; Wang, C.H.; Shiao, M.S.; Chen, J.K.; Hung, L.M. High-fructose and high-fat feeding correspondingly lead to the development of lysoPC-associated apoptotic cardiomyopathy and adrenergic signaling-related cardiac hypertrophy. Int. J. Cardiol. 2016, 215, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Hissa, B.; Oakes, P.W.; Pontes, B.; Juan, G.R.-S.; Gardel, M.L. Cholesterol depletion impairs contractile machinery in neonatal rat cardiomyocytes. Sci. Rep. 2017, 7, 43764. [Google Scholar] [CrossRef] [Green Version]
- Chojnacki, T.; Dallner, G. The biological role of dolichol. Biochem. J. 1988, 251, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Breitling, J.; Aebi, M. N-Linked Protein Glycosylation in the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013359. [Google Scholar] [CrossRef] [Green Version]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. The protonmotive Q cycle: A general formulation. FEBS Lett. 1975, 59, 137–139. [Google Scholar] [CrossRef] [Green Version]
- Heier, C.; Haemmerle, G. Fat in the heart: The enzymatic machinery regulating cardiac triacylglycerol metabolism. Biochim. Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2016, 1861, 1500–1512. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of Glycerolipides: A Comparison of Lecithin and Triglyceride Synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Total | Expected | Hits | p Value | FDR |
|---|---|---|---|---|---|
| Upregulated genes in 7KCh-treated cardiac cell | |||||
| Activation of Genes by ATF4 | 7 | 0.047 | 4 | 6.12 × 10−8 | 8.02 × 10−5 |
| PERK regulated gene expression | 10 | 0.0671 | 4 | 3.62 × 10−7 | 2.37 × 10−4 |
| Unfolded Protein Response | 66 | 0.443 | 6 | 4.38 × 10−6 | 1.91 × 10−3 |
| Circadian Clock | 39 | 0.262 | 4 | 0.000123 | 3.22 × 10−2 |
| PPARA Activates Gene Expression | 78 | 0.523 | 5 | 0.00016 | 3.22 × 10−2 |
| Amino acid synthesis and interconversion (transamination) | 21 | 0.141 | 3 | 0.000346 | 3.48 × 10−2 |
| Downregulated genes in 7KCh-treated cardiac cell | |||||
| Cholesterol biosynthesis | 28 | 0.449 | 18 | 1.35 × 10−26 | 1.77 × 10−23 |
| Activation of Gene Expression by SREBP (SREBF) | 32 | 0.513 | 15 | 2.04 × 10−19 | 1.33 × 10−16 |
| Regulation of Cholesterol Biosynthesis by SREBP (SREBF) | 49 | 0.786 | 16 | 1.36 × 10−17 | 5.94 × 10−15 |
| Metabolism of lipids and lipoproteins | 553 | 8.87 | 37 | 1.08 × 10−14 | 3.54 × 10−12 |
| Metabolism | 1600 | 25.6 | 55 | 7.37 × 10−10 | 1.93 × 10−7 |
| Fatty Acyl-CoA Biosynthesis | 18 | 0.289 | 6 | 2.36 × 10−7 | 5.16 × 10−5 |
| Collagen biosynthesis and modifying enzymes | 52 | 0.834 | 8 | 1.42 × 10−6 | 2.56 × 10−4 |
| Triglyceride Biosynthesis | 37 | 0.594 | 7 | 1.57 × 10−6 | 2.56 × 10−4 |
| Extracellular matrix organization | 159 | 2.55 | 12 | 7.90 × 10−6 | 1.00 × 10−3 |
| Assembly of collagen fibrils and other multimeric structures | 47 | 0.754 | 7 | 8.38 × 10−6 | 1.00 × 10−3 |
| Fatty acid, triacylglycerol, and ketone body metabolism | 160 | 2.57 | 12 | 8.43 × 10−6 | 1.00 × 10−3 |
| Collagen formation | 73 | 1.17 | 8 | 1.93 × 10−5 | 2.10 × 10−3 |
| Degradation of collagen | 54 | 0.867 | 7 | 2.15 × 10−5 | 2.17 × 10−3 |
| Integrin cell surface interactions | 86 | 1.38 | 8 | 6.39 × 10−5 | 5.98 × 10−3 |
| Degradation of the extracellular matrix | 87 | 1.4 | 8 | 6.95 × 10−5 | 6.07 × 10−3 |
| Ethanol oxidation | 8 | 0.128 | 3 | 0.000212 | 1.74 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.-L.; Tang, H.-Y.; Wu, P.-T.; Yang, C.-H.; Lo, C.-J.; Lin, J.-F.; Ho, H.-Y. 7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells. Cells 2021, 10, 3597. https://doi.org/10.3390/cells10123597
Cheng M-L, Tang H-Y, Wu P-T, Yang C-H, Lo C-J, Lin J-F, Ho H-Y. 7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells. Cells. 2021; 10(12):3597. https://doi.org/10.3390/cells10123597
Chicago/Turabian StyleCheng, Mei-Ling, Hsiang-Yu Tang, Pei-Ting Wu, Cheng-Hung Yang, Chi-Jen Lo, Jui-Fen Lin, and Hung-Yao Ho. 2021. "7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells" Cells 10, no. 12: 3597. https://doi.org/10.3390/cells10123597
APA StyleCheng, M.-L., Tang, H.-Y., Wu, P.-T., Yang, C.-H., Lo, C.-J., Lin, J.-F., & Ho, H.-Y. (2021). 7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells. Cells, 10(12), 3597. https://doi.org/10.3390/cells10123597