
Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen


 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice and Intravenous Injections
2.2. Murine Cell Isolation
2.3. Human BM Cell Isolation
2.4. Flow Cytometry, Cell Sorting and ImageStream
2.5. RNA Sequencing
2.6. Confocal Microscopy
2.7. CD8 T-Cell Stimulation
2.8. DC Co-Cultures with Zymosan and Aspergillus
2.9. LSK Cell Culture Assays
2.10. Statistics
3. Results
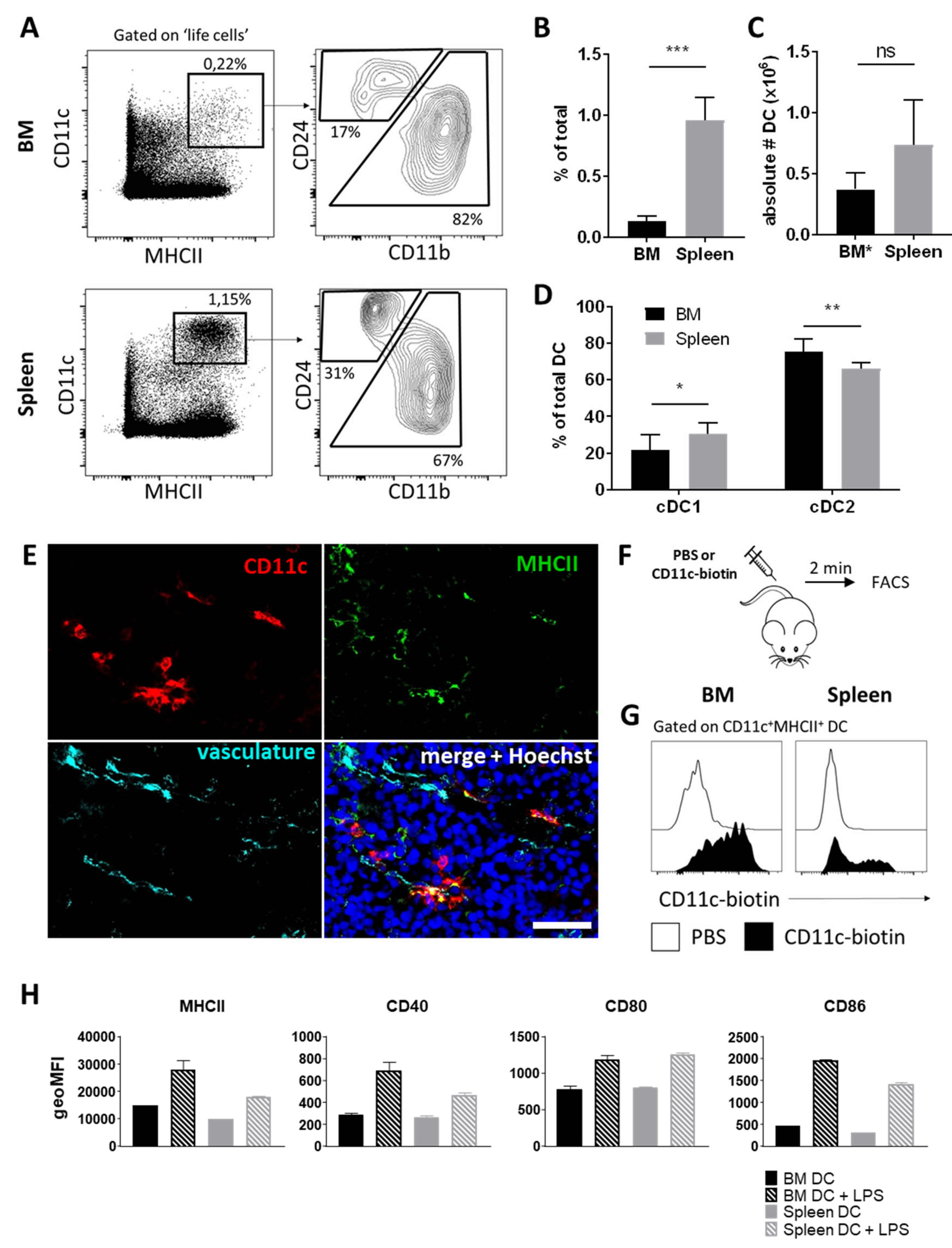
3.1. BM Harbors a Substantial Population of Dendritic Cells
3.2. Perivascular BM DCs Are Accessible to Blood-Borne Molecules and Respond to Systemic Infection
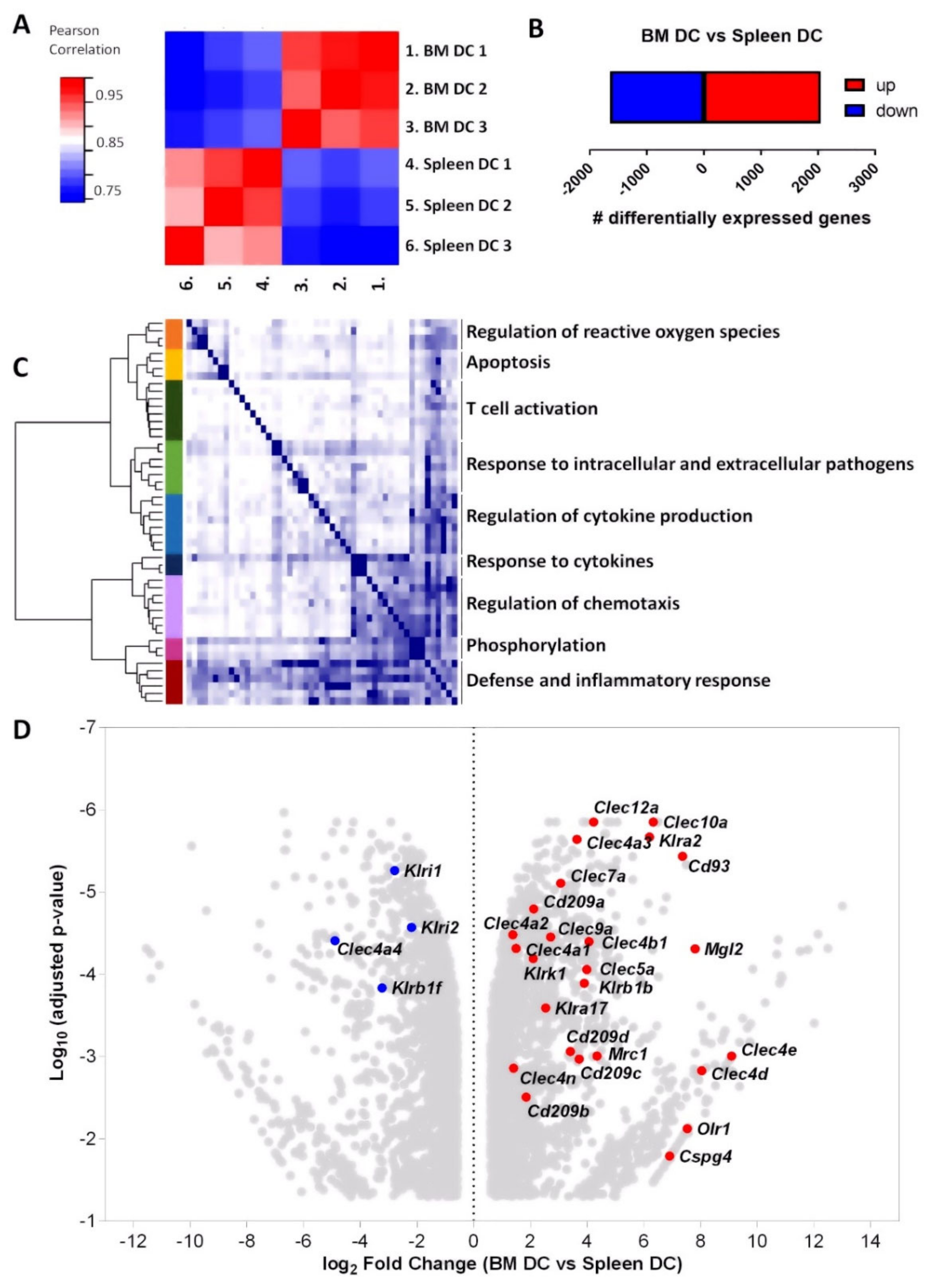
3.3. mRNA Profiling Reveals High Expression of C-Type Lectin Family Receptors by BM DCs
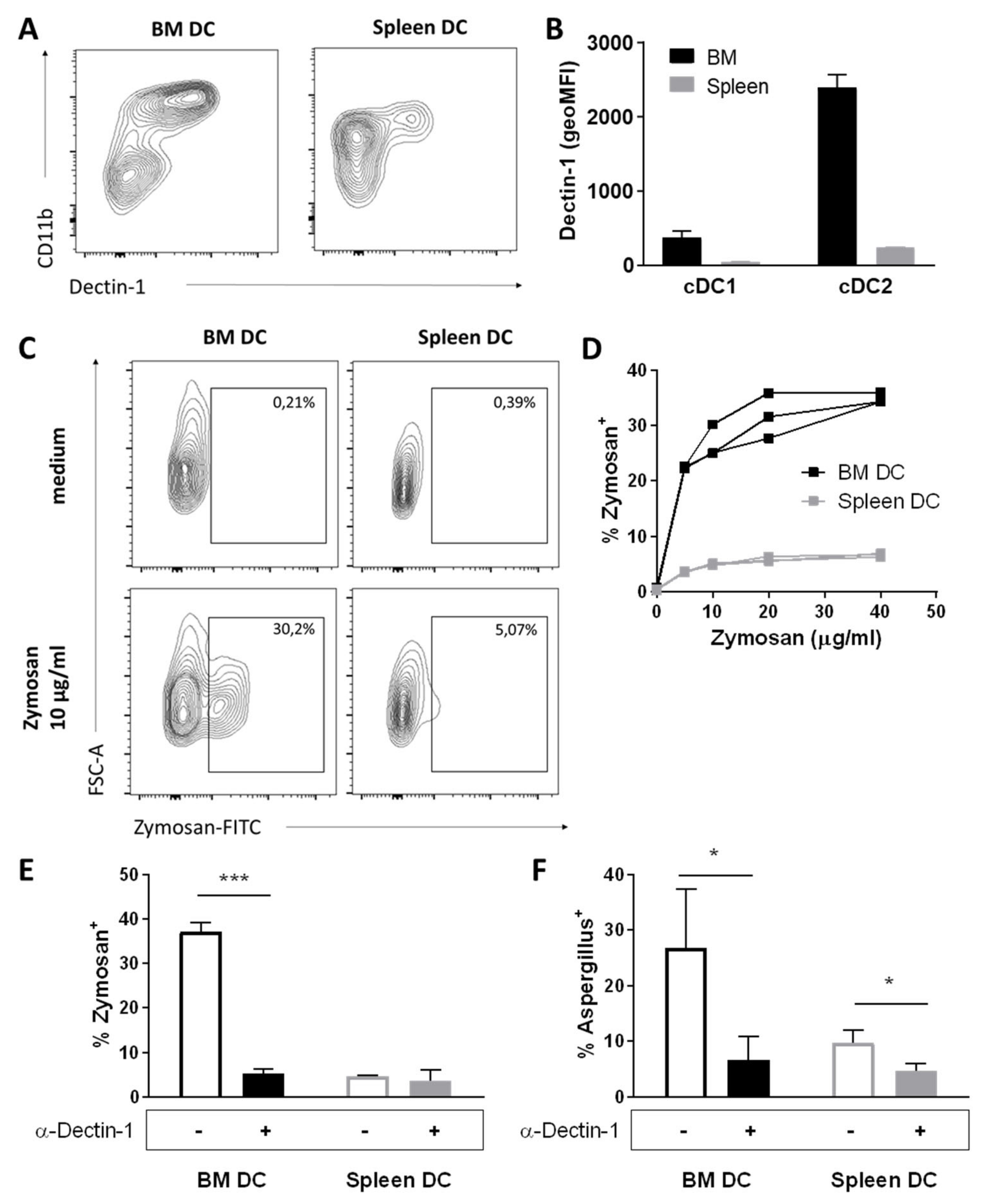
3.4. BM DCs Express High Levels of Dectin-1, Which Mediate Uptake of Fungal Antigens
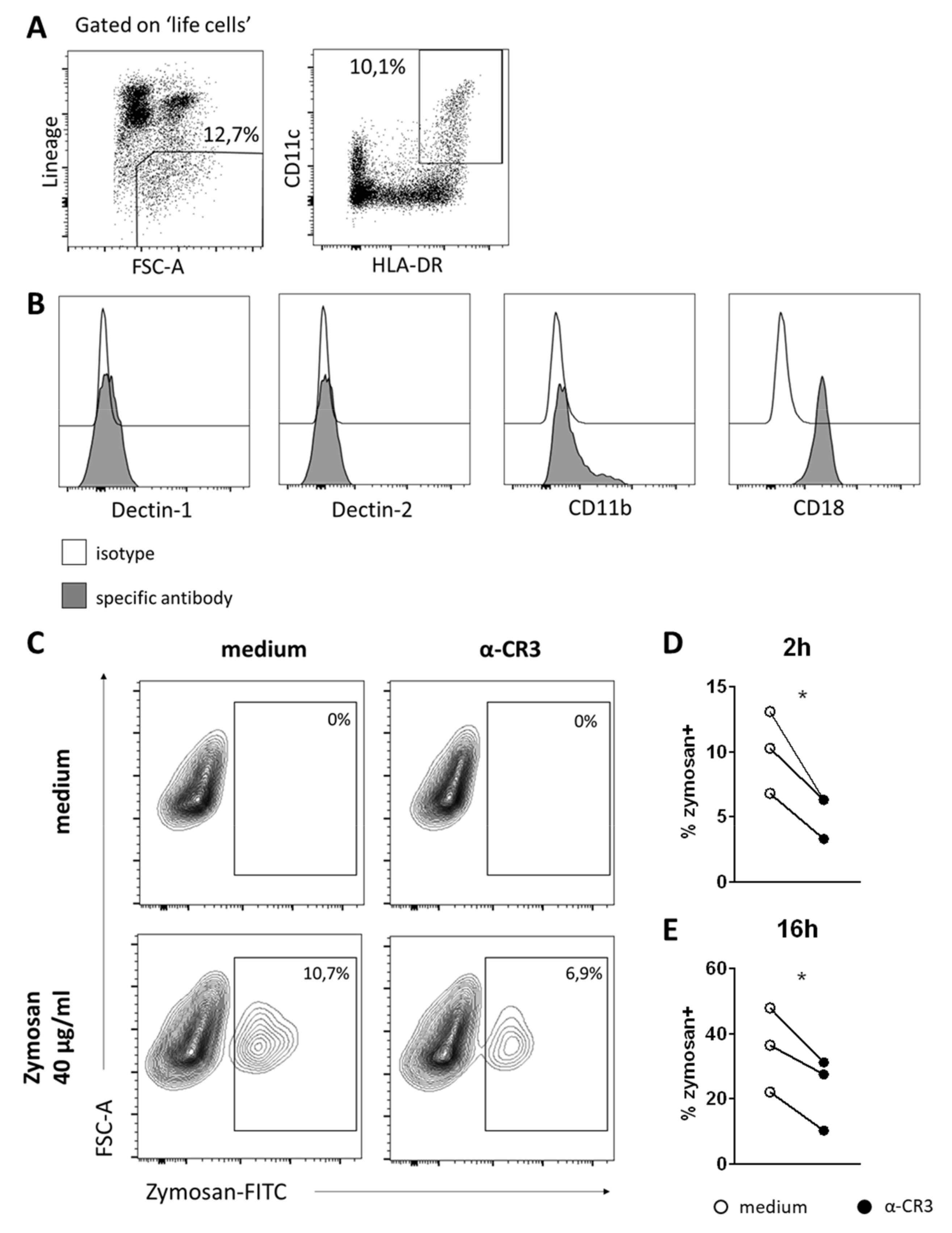
3.5. DCs in Human BM Also Take Up Zymosan, Which Is CR3-Mediated
3.6. Zymosan Is Taken Up by BM DCs after In Vivo Administration and Boosts the Number of Neutrophil Progenitors in BM
3.7. BM DCs Promote Neutrophil Formation in a G-CSF Dependent Manner upon Exposure to Zymosan
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.; McEver, R.P.; Koni, P.A.; et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Slamanig, S.A.; Nolte, M.A. The Bone Marrow as Sanctuary for Plasma Cells and Memory T-Cells: Implications for Adaptive Immunity and Vaccinology. Cells 2021, 10, 1508. [Google Scholar] [CrossRef] [PubMed]
- Milo, I.; Sapoznikov, A.; Kalchenko, V.; Tal, O.; Krauthgamer, R.; Van Rooijen, N.; Dudziak, D.; Jung, S.; Shakhar, G. Dynamic imaging reveals promiscuous crosspresentation of blood-borne antigens to naïve CD8+ T cells in the bone marrow. Blood 2013, 122, 193–208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Pewzner-Jung, Y.; Kalchenko, V.; Krauthgamer, R.; Shachar, I.; Jung, S. Perivascular clusters of dendritic cells provide critical survival signals to B cells in bone marrow niches. Nat. Immunol. 2008, 9, 388–395. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Endele, M.; Etzrodt, M.; Schroeder, T. Instruction of hematopoietic lineage choice by cytokine signaling. Exp. Cell Res. 2014, 329, 207–213. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, A.M.; Libregts, S.F.; Valkhof, M.; Boon, L.; Touw, I.P.; Nolte, M.A. IFNγ induces monopoiesis and inhibits neutrophil development during inflammation. Blood 2012, 119, 1543–1554. [Google Scholar] [CrossRef]
- De Bruin, A.M.; Buitenhuis, M.; van der Sluijs, K.F.; van Gisbergen, K.P.J.M.; Boon, L.; Nolte, M.A. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-gamma. Blood 2010, 116, 2559–2569. [Google Scholar] [CrossRef] [PubMed]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef]
- Libregts, S.F.; Gutiérrez, L.; de Bruin, A.M.; Wensveen, F.M.; Papadopoulos, P.; van Ijcken, W.; Ozgür, Z.; Philipsen, S.; Nolte, M.A. Chronic IFN-γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF-1/PU.1 axis. Blood 2011, 118, 2578–2588. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, B.; Yu, B.; Chen, T.; Hu, Q.; Peng, B.; Sheng, W. Poria cocos polysaccharide induced Th1-type immune responses to ovalbumin in mice. PLoS ONE 2021, 16, e0245207. [Google Scholar] [CrossRef]
- Wang, P.L.; Yim, A.K.; Kim, K.; Avey, D.; Czepielewski, R.S.; Colonna, M.; Milbrandt, J.; Randolph, G.J.; Project, T.I.G. Peripheral nerve resident macrophages are microglia-like cells with tissue-specific programming. bioRxiv 2019, 12, 3546. [Google Scholar] [CrossRef]
- MacLean, M.; Juranek, J.; Cuddapah, S.; López-Díez, R.; Ruiz, H.H.; Hu, J.; Frye, L.; Li, H.; Gugger, P.F.; Schmidt, A.M. Microglia RAGE exacerbates the progression of neurodegeneration within the SOD1G93A murine model of amyotrophic lateral sclerosis in a sex-dependent manner. J. Neuroinflamm. 2021, 18, 139. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Deutsch, M.; Alothman, S.; Alqunaibit, D.; Werba, G.; Pansari, M.; Pergamo, M.; Ochi, A.; Torres-Hernandez, A.; Levie, E.; et al. Dectin-1 Regulates Hepatic Fibrosis and Hepatocarcinogenesis by Suppressing TLR4 Signaling Pathways. Cell Rep. 2015, 13, 1909–1921. [Google Scholar] [CrossRef] [PubMed]
- Van Bruggen, R.; Drewniak, A.; Jansen, M.; van Houdt, M.; Roos, D.; Chapel, H.; Verhoeven, A.J.; Kuijpers, T.W. Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for β-glucan-bearing particles. Mol. Immunol. 2009, 47, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Bose, N.; Chan, A.S.H.; Guerrero, F.; Maristany, C.M.; Qiu, X.; Walsh, R.M.; Ertelt, K.E.; Jonas, A.B.; Gorden, K.B.; Dudney, C.M.; et al. Binding of soluble yeast β-glucan to human neutrophils and monocytes is complement-dependent. Front. Immunol. 2013, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Geerman, S.; Hickson, S.; Brasser, G.; Pascutti, M.F.; Nolte, M.A. Quantitative and qualitative analysis of bone marrow CD8+ T cells from different bones uncovers a major contribution of the bone marrow in the vertebrae. Front. Immunol. 2016, 6, 660. [Google Scholar] [CrossRef] [PubMed]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 Expression in Conventional Dendritic Cells Is Required for T Helper 2 Cell Responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Tailor, P.; Tamura, T.; Morse, H.C.; Ozato, K. The BXH2 mutation in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood 2008, 111, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.K.; Uronen-Hansson, H.; Semmrich, M.; Rivollier, A.; Hägerbrand, K.; Marsal, J.; Gudjonsson, S.; Håkansson, U.; Reizis, B.; Kotarsky, K.; et al. IRF4 Transcription-Factor-Dependent CD103+CD11b+ Dendritic Cells Drive Mucosal T Helper 17 Cell Differentiation. Immunity 2013, 38, 958–969. [Google Scholar] [CrossRef]
- Williams, J.W.; Tjota, M.Y.; Clay, B.S.; Vander Lugt, B.; Bandukwala, H.S.; Hrusch, C.L.; Decker, D.C.; Blaine, K.M.; Fixsen, B.R.; Singh, H.; et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat. Commun. 2013, 4, 2990. [Google Scholar] [CrossRef]
- Hoving, J.C.; Wilson, G.J.; Brown, G.D. Signalling C-type lectin receptors, microbial recognition and immunity. Cell. Microbiol. 2014, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Taylor, M.E.; Drickamer, K. The C-type lectin superfamily in the immune system. Immunol. Rev. 1998, 163, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Brown, G.D. The role of Dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 2011, 14, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.M.; Montoya, M.; Taylor, P.R.; Borrow, P.; Gordon, S.; Brown, G.D.; Wong, S.Y.C. Expression of the β-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J. Leukoc. Biol. 2004, 76, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.C.; Slack, E.C.; Edwards, A.D.; Nolte, M.A.; Schulz, O.; Schweighoffer, E.; Williams, D.L.; Gordon, S.; Tybulewicz, V.L.; Brown, G.D.; et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005, 22, 507–517. [Google Scholar] [CrossRef]
- Satake, S.; Hirai, H.; Hayashi, Y.; Shime, N.; Tamura, A.; Yao, H.; Yoshioka, S.; Miura, Y.; Inaba, T.; Fujita, N.; et al. C/EBPβ Is Involved in the Amplification of Early Granulocyte Precursors during Candidemia-Induced “Emergency” Granulopoiesis. J. Immunol. 2012, 189, 4546–4555. [Google Scholar] [CrossRef] [PubMed]
- Burgaleta, C.; Golde, D.W. Effect of Glucan on Granulopoiesis and Macrophage Genesis in Mice. Cancer Res. 1977, 37, 1739–1742. [Google Scholar] [PubMed]
- Hirai, H.; Zhang, P.; Dayaram, T.; Hetherington, C.J.; Mizuno, S.I.; Imanishi, J.; Akashi, K.; Tenen, D.G. C/EBPβ is required for “emergency” granulopoiesis. Nat. Immunol. 2006, 7, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [CrossRef]
- Zhang, J.; Supakorndej, T.; Krambs, J.R.; Rao, M.; Abou-Ezzi, G.; Ye, R.Y.; Li, S.; Trinkaus, K.; Link, D.C. Bone marrow dendritic cells regulate hematopoietic stem/progenitor cell trafficking. J. Clin. Invest. 2019, 129, 2920–2931. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yao, J.C.; Li, J.T.; Schmidt, A.P.; Link, D.C. TLR7/8 agonist treatment induces an increase in bone marrow resident dendritic cells and hematopoietic progenitor expansion and mobilization. Exp. Hematol. 2021, 96, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A.; Venkatesh, D.; Yun, S.H.; Mayadas, T.N. The β-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Griffin, J.D. Granulocyte colony-stimulating factor and its receptor. Blood 1991, 78, 2791–2808. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, S.; Gerosa, R.C.; Radpour, R.; Bauer, J.; Ampenberger, F.; Heikenwalder, M.; Kopf, M.; Manz, M.G. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 2014, 124, 1393–1403. [Google Scholar] [CrossRef]
- Stark, M.A.; Huo, Y.; Burcin, T.L.; Morris, M.A.; Olson, T.S.; Ley, K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 2005, 22, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Dragomir, A.-C.; Kocabayoglu, P.; Rahman, A.H.; Chow, A.; Hashimoto, D.; Leboeuf, M.; Kraus, T.; Moran, T.; Carrasco-Avino, G.; et al. Central Role of Conventional Dendritic Cells in Regulation of Bone Marrow Release and Survival of Neutrophils. J. Immunol. 2014, 192, 3374–3382. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, 427. [Google Scholar] [CrossRef]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, A.; Megías, J.; O’Connor, J.E.; Gozalbo, D.; Gil, M.L. Candida albicans induces selective development of macrophages and monocyte derived dendritic cells by a TLR2 dependent signalling. PLoS ONE 2011, 6, e24761. [Google Scholar] [CrossRef]
- Marr, K.A. Fungal infections in hematopoietic stem cell transplant recipients. Med. Mycol. 2008, 46, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Della Porta, M.; Rigolin, G.M.; Alessandrino, E.P.; Maiocchi, M.; Malcovati, L.; Vanelli, L.; Baratè, C.; Rumi, E.; Ciccone, M.; Cuneo, A.; et al. Dendritic cell recovery after allogeneic stem-cell transplantation in acute leukemia: Correlations with clinical and transplant characteristics. Eur. J. Haematol. 2004, 72, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Khatcheressian, J.; Lyman, G.H.; Ozer, H.; Armitage, J.O.; Balducci, L.; Bennett, C.L.; Cantor, S.B.; Crawford, J.; Cross, S.J.; et al. 2006 Update of recommendations for the use of white blood cell growth factors: An evidence-based clinical practice guideline. J. Clin. Oncol. 2006, 24, 3187–3205. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.; Martinez, S.; Corringham, S.; Medley, K.; Ball, E.D. Review and revision of clinical practice of using G-CSF after autologous and allogeneic hematopoietic stem cell transplantation at UCSD. J. Oncol. Pharm. Pract. 2011, 17, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.; Martinez, S.; Corringham, S.; Medley, K.; Ball, E.D. Optimal use of G-CSF administration after hematopoietic SCT. Bone Marrow Transplant. 2009, 43, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Remberger, M.; Naseh, N.; Aschan, J.; Barkholt, L.; LeBlanc, K.; Svennberg, P.; Ringdén, O. G-CSF given after haematopoietic stem cell transplantation using HLA-identical sibling donors is associated to a higher incidence of acute GVHD II-IV. Bone Marrow Transplant. 2003, 32, 217–223. [Google Scholar] [CrossRef]
- Teshima, T.; Reddy, P.; Lowler, K.P.; Kukuruga, M.A.; Liu, C.; Cooke, K.R.; Ferrara, J.L.M. Flt3 ligand therapy for recipients of allogeneic bone marrow transplants expands host CD8α+ dendritic cells and reduces experimental acute graft-versus-host disease. Blood 2002, 99, 1825–1832. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goedhart, M.; Slot, E.; Pascutti, M.F.; Geerman, S.; Rademakers, T.; Nota, B.; Huveneers, S.; Buul, J.D.v.; MacNamara, K.C.; Voermans, C.; et al. Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen. Cells 2022, 11, 55. https://doi.org/10.3390/cells11010055
Goedhart M, Slot E, Pascutti MF, Geerman S, Rademakers T, Nota B, Huveneers S, Buul JDv, MacNamara KC, Voermans C, et al. Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen. Cells. 2022; 11(1):55. https://doi.org/10.3390/cells11010055
Chicago/Turabian StyleGoedhart, Marieke, Edith Slot, Maria F. Pascutti, Sulima Geerman, Timo Rademakers, Benjamin Nota, Stephan Huveneers, Jaap D. van Buul, Katherine C. MacNamara, Carlijn Voermans, and et al. 2022. "Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen" Cells 11, no. 1: 55. https://doi.org/10.3390/cells11010055
APA StyleGoedhart, M., Slot, E., Pascutti, M. F., Geerman, S., Rademakers, T., Nota, B., Huveneers, S., Buul, J. D. v., MacNamara, K. C., Voermans, C., & Nolte, M. A. (2022). Bone Marrow Harbors a Unique Population of Dendritic Cells with the Potential to Boost Neutrophil Formation upon Exposure to Fungal Antigen. Cells, 11(1), 55. https://doi.org/10.3390/cells11010055