
Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment

 ,
, 
{kind=link}
Abstract
:1. Introduction
2. Tumor Microenvironment in Glioblastoma
2.1. Non-Immune Cellular Components
2.1.1. Vasculature
2.1.2. Glioma Stem Cells (GSC)
2.1.3. Glial Cells and Neurons
2.2. Immune Cellular Components
2.2.1. Microglia/Macrophages
2.2.2. Tumor-Infiltrating Lymphocytes (TILs)
2.2.3. Natural Killer Cells
2.2.4. Neutrophils
3. Genomic and Epigenomic Alterations in Glioblastoma
Correlation with Tumor Microenvironment
4. Metabolism in Glioblastoma
4.1. Metabolic Pathways
4.1.1. Aerobic Glycolysis
4.1.2. Amino Acids Metabolis
Tryptophan
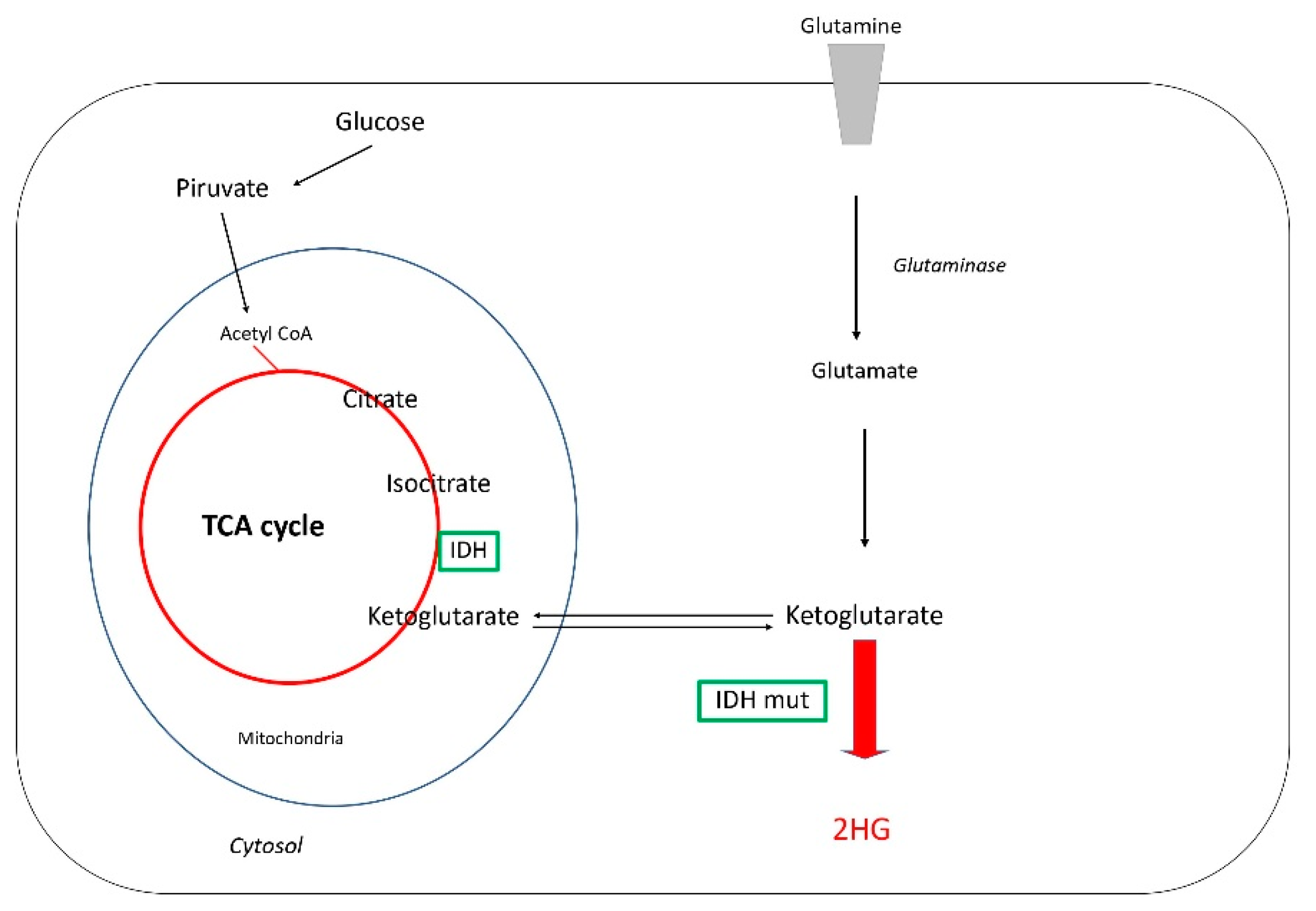
Glutamine and D2-Hydroxyglutarate Metabolism (2HG)
Other Amino Acids: Adenosine and Arginine
4.1.3. Lipid Metabolism
4.2. Correlation with Genomic Alterations
4.2.1. Receptor Tyrosine Kinase Amplification
4.2.2. Isocitrate Dehydrogenase (IDH) Mutations
4.2.3. Other Genomic Alterations
5. The Role of Metabolism in the GBM Microenvironment
5.1. Aerobic Glycolysis
5.2. Amino Acid Metabolism
5.3. Lipids
6. Therapeutic Opportunities
6.1. IDO Inhibitors
6.2. IDH Mutation Inhibitors
6.3. Pyruvate and Lactate Antagonist. Regulation of Hexokinase 2
6.4. Targeting Phospholipids
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Tan, A.C.; Ashley, D.M.; Lopez, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; Chapter 11; ISBN 9780994438126. [Google Scholar]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinto, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chioca, E.A.; et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro-Oncology 2020, 22, 1073–1113. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielke, H.R.; Zielke, C.L.; Baab, P.J. Direct measurement of oxidative metabolism in the living brain by microdialysis: A review. J. Neurochem. 2009, 109, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Medawar, P.B. Tests by tissue culture methods on the nature of immunity to transplanted skin. Q. J. Microsc. Sci. 1948, 89, 239–252. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualini, C.; Kozaki, T.; Bruschi, M.; Nguyen, T.H.H.; Minard-Colin, V.; Castel, D.; Grill, J.; Ginhoux, F. Modeling the Interaction between the Microenvironment and Tumor Cells in Brain Tumors. Neuron 2020, 108, 1025–1044. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S.; Bjorklund, A.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.M.; Cahan, P.; Shalek, A.K.; Satija, R.; DaleyKeyser, A.; Li, H.; Zhang, J.; Pardee, K.; Gennert, D.; Trombetta, J.J.; et al. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature 2014, 516, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Matersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Hu, Y.; Wu, F.; Guo, Q.; Qian, Z.; Hu, W.; Chen, J.; Wang, K.; Fan, X.; Wu, X.; et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl. Sci. Rev. 2020, 7, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Friebel, E.; Kapolou, K.; Unger, S.; Nunez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Caruso, F.P.; Garofano, L.; D’Angelo, F.; Yu, K.; Tang, F.; Yuan, J.; Zhang, J.; Cerulo, L.; Pagnotta, S.M.; Bedognetti, D.; et al. A map of tumor-host interactions in glioma at single-cell resolution. Gigascience 2020, 9, giaa109. [Google Scholar] [CrossRef]
- Wesseling, P.; Ruiter, D.J.; Burger, P.C. Angiogenesis in brain tumors; pathobiological and clinical aspects. J. Neurooncol. 1997, 32, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.R. The Role of Brain Vasculature in Glioblastoma. Mol. Neurobiol. 2019, 56, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martñinez-Saez, E.; Prudkin, L.; et al. TGF-beta Receptor Inhibitors Target the CD44(high)/Id1(high) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brenna, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Pietras, A.; Katz, A.M.; Ekstrom, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.l.; Amankulor, N.M.; Huse, J.T.; Hollando, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- da Fonseca, A.C.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Jiang, F.; Zheng, X.; Katakowski, M.; Buller, B.; To, S.S.; Chopp, M. TGF-beta1 promotes motility and invasiveness of glioma cells through activation of ADAM17. Oncol. Rep. 2011, 25, 1329–1335. [Google Scholar] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Lin, Y.C.; Yao, P.L.; Yuan, A.; Chen, H.Y.; Shun, C.T.; Tsai, M.F.; Chen, C.H.; Yang, P.C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiner, P.S.; Preusse, C.; Blank, A.E.; Zachskorn, C.; Baumgarten, P.; Caspary, L.; Braczynski, A.K.; Weissenberger, J.; Bratzke, H.; Reiss, S.; et al. MIF Receptor CD74 is Restricted to Microglia/Macrophages, Associated with a M1-Polarized Immune Milieu and Prolonged Patient Survival in Gliomas. Brain Pathol. 2015, 25, 491–504. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol. 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Ma, E.; Wang, X.; Yu, C.; Dong, T.; Zhan, W.; Wei, X.; Liang, G.; Feng, S. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol. Lett. 2016, 12, 2924–2929. [Google Scholar] [CrossRef] [Green Version]
- Di Cintio, F.; Dal Bo, M.; Baboci, L.; De Mattia, E.; Polano, M.; Toffoli, G. The Molecular and Microenvironmental Landscape of Glioblastomas: Implications for the Novel Treatment Choices. Front. Neurosci. 2020, 14, 603647. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiendl, H.; Mitsdoerffer, M.; Hofmeister, V.; Wischhusen, J.; Bornemann, A.; Meyermann, R.; Weiss, E.H.; Melms, A.; Weller, M. A functional role of HLA-G expression in human gliomas: An alternative strategy of immune escape. J. Immunol. 2002, 168, 4772–4780. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef]
- Mason, M.; Maurice, C.; McNamara, M.G.; Tieu, M.T.; Lwin, Z.; Millar, B.A.; Menard, C.; Laperriere, N.; Milosevic, M.; Atenafu, E.G.; et al. Neutrophil-lymphocyte ratio dynamics during concurrent chemo-radiotherapy for glioblastoma is an independent predictor for overall survival. J. Neurooncol. 2017, 132, 463–471. [Google Scholar] [CrossRef]
- Zadora, P.; Dabrowski, W.; Czarko, K.; Smolen, A.; Kotlinska-Hasiec, E.; Wiorkowski, K.; Sikora, A.; Jarosz, B.; Kura, K.; Rola, R.; et al. Preoperative neutrophil-lymphocyte count ratio helps predict the grade of glial tumor—A pilot study. Neurol. Neurochir. Pol. 2015, 49, 41–44. [Google Scholar] [CrossRef]
- Zhao, B.; Wang, Y.; Wang, Y.; Chen, W.; Liu, P.H.; Kong, Z.; Dai, C.; Wang, Y.; Ma, W. Systematic identification, development, and validation of prognostic biomarkers involving the tumor-immune microenvironment for glioblastoma. J. Cell. Physiol. 2021, 236, 507–522. [Google Scholar] [CrossRef]
- Haddad, A.F.; Chen, J.S.; Oh, T.; Pereira, M.P.; Joshi, R.S.; Aghi, M.K. Higher cytolytic score correlates with an immunosuppressive tumor microenvironment and reduced survival in glioblastoma. Sci. Rep. 2020, 10, 17580. [Google Scholar] [CrossRef]
- Schernberg, A.; Nivet, A.; Dhermain, F.; Ammari, S.; Escande, A.; Pallud, J.; Louvel, G.; Deutsch, E. Neutrophilia as a biomarker for overall survival in newly diagnosed high-grade glioma patients undergoing chemoradiation. Clin. Transl. Radiat. Oncol. 2018, 10, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, A.; Cederarv, M.; Wolmer-Solberg, N.; Tammik, C.; Stragliotto, G.; Peredo, I.; Fornara, O.; Xu, X.; Dzabic, M.; Taher, C.; et al. Enhanced neutrophil activity is associated with shorter time to tumor progression in glioblastoma patients. Oncoimmunology 2016, 5, e1075693. [Google Scholar] [CrossRef] [Green Version]
- Bertaut, A.; Truntzer, C.; Madkouri, R.; Kaderbhai, C.G.; Derangere, V.; Vincent, J.; Chauffert, B.; Aubriot-Lorton, M.H.; Farah, W.; Mourier, K.L.; et al. Blood baseline neutrophil count predicts bevacizumab efficacy in glioblastoma. Oncotarget 2016, 7, 70948–70958. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, J.M.; Misra, A.; Zhang, L.; Smirnov, I.; Colman, H.; Griffin, C.; Ozburn, N.; Chen, M.; Pan, E.; Koul, D.; et al. Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant molecular subtypes of glioblastoma. Cancer Res. 2005, 65, 1678–1686. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Nutt, C.L.; Mani, D.R.; Betensky, R.A.; Tamayo, P.; Cairncross, J.G.; Ladd, C.; Pohl, U.; Hartmann, C.; McLaughlin, M.E.; Batchelor, T.T.; et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003, 63, 1602–1607. [Google Scholar]
- Gravendeel, L.A.; Kouwenhoven, M.C.; Gevaert, O.; de Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Diehn, M.; Watson, N.; Bollen, A.W.; Aldape, K.D.; Nicholas, M.K.; Lamborn, K.R.; Berger, M.S.; Botstein, D.; Brown, P.O.; et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc. Natl. Acad. Sci. USA 2005, 102, 5814–5819. [Google Scholar] [CrossRef] [Green Version]
- Rickman, D.S.; Bobek, M.P.; Misek, D.E.; Kuick, R.; Blaivas, M.; Kurnit, D.M.; Taylor, J.; Hanash, S.M. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001, 61, 6885–6891. [Google Scholar]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, K.N.; Wang, Q.; Li, G.; Zeng, F.; Zhang, Y.; Wu, F.; Chai, R.; Wang, Z.; Zhang, C.; et al. Chinese Glioma Genome Atlas (CGGA): A Comprehensive Resource with Functional Genomic Data from Chinese Glioma Patients. Genom. Proteom. Bioinform. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Christensen, B.C.; Smith, A.A.; Zheng, S.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Wrensch, M.R.; et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. J. Natl. Cancer Inst. 2011, 103, 143–153. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.-C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, H.; Zhang, L.; Sulman, E.P.; McDonald, J.M.; Shooshtari, N.L.; Rivera, A.; Popoff, S.; Nutt, C.L.; Louis, D.N.; Cairncross, J.G.; et al. A multigene predictor of outcome in glioblastoma. Neuro-Oncology 2010, 12, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucette, T.; Rao, G.; Rao, A.; Shen, L.; Aldape, K.; Wei, J.; Dziurzynski, K.; Gilbert, M.; Heimberger, A.B. Immune heterogeneity of glioblastoma subtypes: Extrapolation from the cancer genome atlas. Cancer Immunol. Res. 2013, 1, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Carrato, C.; Alameda, F.; Esteve-Codina, A.; Pineda, E.; Arpi, O.; Martinez-Garcia, M.; Mallo, M.; Gut, M.; Lopez-Martos, R.; Barco, S.D.; et al. Glioblastoma TCGA Mesenchymal and IGS 23 Tumors are Identifiable by IHC and have an Immune-phenotype Indicating a Potential Benefit from Immunotherapy. Clin. Cancer Res. 2020, 26, 6600–6609. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes. Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahonero, C.; Sanchez-Gomez, P. EGFR-dependent mechanisms in glioblastoma: Towards a better therapeutic strategy. Cell. Mol. Life Sci. 2014, 71, 3465–3488. [Google Scholar] [CrossRef]
- Hao, Z.; Guo, D. EGFR mutation: Novel prognostic factor associated with immune infiltration in lower-grade glioma; an exploratory study. BMC Cancer 2019, 19, 1184. [Google Scholar] [CrossRef] [Green Version]
- Bruce, K.D.; Zsombok, A.; Eckel, R.H. Lipid Processing in the Brain: A Key Regulator of Systemic Metabolism. Front. Endocrinol. 2017, 8, 60. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Chinnaiyan, P.; Kensicki, E.; Bloom, G.; Prabhu, A.; Sarcar, B.; Kahali, S.; Eschrich, S.; Qu, X.; Forsyth, P.; Gillies, R. The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 2012, 72, 5878–5888. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Braidy, N.; Bessede, A.; Brew, B.J.; Grant, R.; Teo, C.; Guillemin, G.J. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. 2012, 72, 5649–5657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan. Res. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Falzoni, S.; Donvito, G.; Di Virgilio, F. Detecting adenosine triphosphate in the pericellular space. Interface Focus 2013, 3, 20120101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.A.; Tomaszewski, W.H.; Haskell-Mendoza, A.P.; Hotchkiss, K.M.; Singh, K.; Reedy, J.L.; Fecci, P.E.; Sampson, J.H.; Khasraw, M. Targeting Immunometabolism in Glioblastoma. Front. Oncol. 2021, 11, 696402. [Google Scholar] [CrossRef]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietschy, J.M. Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol. Chem. 2009, 390, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef] [PubMed]
- Riboni, L.; Abdel Hadi, L.; Navone, S.E.; Guarnaccia, L.; Campanella, R.; Marfia, G. Sphingosine-1-Phosphate in the Tumor Microenvironment: A Signaling Hub Regulating Cancer Hallmarks. Cells 2020, 9, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bien-Moller, S.; Lange, S.; Holm, T.; Bohm, A.; Paland, H.; Kupper, J.; Herzog, S.; Weitmann, C.; Havemann, C.; Vogelgesang, S.; et al. Expression of S1P metabolizing enzymes and receptors correlate with survival time and regulate cell migration in glioblastoma multiforme. Oncotarget 2016, 7, 13031–13046. [Google Scholar] [CrossRef] [Green Version]
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [Green Version]
- Mahajan-Thakur, S.; Bien-Moller, S.; Marx, S.; Schroeder, H.; Rauch, B.H. Sphingosine 1-phosphate (S1P) signaling in glioblastoma multiforme—A systematic review. Int. J. Mol. Sci. 2017, 18, 2448. [Google Scholar] [CrossRef] [Green Version]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Agostini, M.; Romeo, F.; Inoue, S.; Niklison-Chirou, M.V.; Elia, A.J.; Dinsdale, D.; Morone, N.; Knight, R.A.; Mak, T.W.; Melino, G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016, 23, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Masui, K.; Cavenee, W.K.; Mischel, P.S. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 2014, 25, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Grffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Osthus, R.C.; Shim, H.; Kim, S.; Li, Q.; Reddy, R.; Mukherjee, M.; Xu, Y.; Wonsey, D.; Lee, L.A.; Dang, C.V. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 2000, 275, 21797–21800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Kharbanda, S.; Pope, W.B.; Tran, A.; Solis, O.E.; Peale, F.; Forrest, W.F.; Pujara, K.; Carrillo, J.A.; Pandita, A.; et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J. Clin. Oncol. 2011, 29, 4482–4490. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggings, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Reitman, Z.J.; Parsons, D.W.; Yan, H. IDH1 and IDH2: Not your typical oncogenes. Cancer Cell 2010, 17, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Maus, A.; Peters, G.J. Glutamate and alpha-ketoglutarate: Key players in glioma metabolism. Amino Acids 2017, 49, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Latini, A.; Scussiato, K.; Rosa, R.B.; Llesuy, S.; Bello-Klein, A.; Dutra-Filho, C.S.; Wajner, M. D-2-hydroxyglutaric acid induces oxidative stress in cerebral cortex of young rats. Eur. J. Neurosci. 2003, 17, 2017–2022. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Jesionek-Kupnicka, D.; Szybka, M.; Malachowska, B.; Fendler, W.; Potemski, P.; Piaskowski, S.; Jaskolski, D.; Papierrz, W.; Skowronski, W.; Och, W.; et al. TP53 promoter methylation in primary glioblastoma: Relationship with TP53 mRNA and protein expression and mutation status. DNA Cell Biol. 2014, 33, 217–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puzio-Kuter, A.M. The Role of p53 in Metabolic Regulation. Genes Cancer 2011, 2, 385–391. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and Glucose Metabolism by Decreased Akt/mTORC1 Signaling Drives T Cell Impairment in B Cell Leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Philips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Rodriguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.; Lopez-Villegas, E.O.; Sanchez-Garcia, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platten, M.; Friedrich, M.; Wainwright, D.A.; Panitz, V.; Opitz, C.A. Tryptophan metabolism in brain tumors-IDO and beyond. Curr. Opin. Immunol. 2021, 70, 57–66. [Google Scholar] [CrossRef]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine receptors and cancer. Handb. Exp. Pharmacol. 2009, 399–441. [Google Scholar]
- Bunse, L.; Pusch, S.; Bunse, T.; Sahm, F.; Sanghvi, K.; Friedrich, M.; Alansary, D.; Sonner, J.K.; Green, E.; Deumelandt, K.; et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018, 24, 1192–1203. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Eslamadicy, A.A.; Dechant, C.A.; Kemery, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Nahr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Gomes, B.; Driessens, G.; Bartlett, D.; Cai, D.; Cauwenberghs, S.; Crosignani, S.; Dalvie, D.; Denies, S.; Dillon, C.P.; Fantin, V.R.; et al. Characterization of the Selective Indoleamine 2,3-Dioxygenase-1 (IDO1) Catalytic Inhibitor EOS200271/PF-06840003 Supports IDO1 as a Critical Resistance Mechanism to PD-(L)1 Blockade Therapy. Mol. Cancer Ther. 2018, 17, 2530–2542. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Desjardins, A.; Rixe, O.; Cloughesy, T.; Alekar, S.; Williams, J.H.; Li, R.; Taylor, C.T.; Lassman, A.B. A phase 1 study of PF-06840003, an oral indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor in patients with recurrent malignant glioma. Investig. New Drugs 2020, 38, 1784–1795. [Google Scholar] [CrossRef]
- Cady, S.G.; Sono, M. 1-Methyl-DL-tryptophan, beta-(3-benzofuranyl)-DL-alanine (the oxygen analog of tryptophan), and beta-[3-benzo(b)thienyl]-DL-alanine (the sulfur analog of tryptophan) are competitive inhibitors for indoleamine 2,3-dioxygenase. Arch. Biochem. Biophys. 1991, 291, 326–333. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [Green Version]
- El Sayed, S.M.; El-Magd, R.M.; Shishido, Y.; Yorita, K.; Chung, S.P.; Tran, D.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J. Bioenerg. Biomembr. 2012, 44, 513–523. [Google Scholar] [CrossRef]
- Ko, Y.H.; Smith, B.L.; Wang, Y.; Pomper, M.G.; Rini, D.A.; Torbenson, M.S.; Hullihen, J.; Pedersen, P.L. Advanced cancers: Eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem. Biophys. Res. Commun. 2004, 324, 269–275. [Google Scholar] [CrossRef]
- Chiasserini, D.; Davidescu, M.; Orvietani, P.L.; Susta, F.; Macchioni, L.; Petricciuolo, M.; Castigli, E.; Roberti, R.; Binaglia, L.; Corazzi, L. 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteom. 2017, 152, 329–338. [Google Scholar] [CrossRef]
- Davidescu, M.; Macchioni, L.; Scaramozzino, G.; Cristina Marchetti, M.; Migliorati, G.; Vitale, R.; Corcelli, A.; Roberti, R.; Castigli, E.; Corazzi, L. The energy blockers bromopyruvate and lonidamine lead GL15 glioblastoma cells to death by different p53-dependent routes. Sci. Rep. 2015, 5, 14343. [Google Scholar] [CrossRef] [Green Version]
- Reilly, P.T.; Mak, T.W. Molecular pathways: Tumor cells Co-opt the brain-specific metabolism gene CPT1C to promote survival. Clin. Cancer Res. 2012, 18, 5850–5855. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Fang, C.; Yi, K.; Liu, X.; Qi, H.; Tan, Y.; Zhou, J.; Li, Y.; Liu, M.; Zhang, Y.; et al. The role of PTRF/Cavin1 as a biomarker in both glioma and serum exosomes. Theranostics 2018, 8, 1540–1557. [Google Scholar] [CrossRef] [PubMed]
- Yi, K.; Zhan, Q.; Wang, Q.; Tan, Y.; Fang, C.; Wang, Y.; Zhou, J.; Yang, C.; Li, Y.; Kang, C. PTRF/cavin-1 remodels phospholipid metabolism to promote tumor proliferation and suppress immune responses in glioblastoma by stabilizing cPLA2. Neuro-Oncology 2021, 23, 387–399. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, A.; Domènech, M.; Muñoz-Mármol, A.M.; Carrato, C.; Balana, C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells 2021, 10, 3529. https://doi.org/10.3390/cells10123529
Hernández A, Domènech M, Muñoz-Mármol AM, Carrato C, Balana C. Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells. 2021; 10(12):3529. https://doi.org/10.3390/cells10123529
Chicago/Turabian StyleHernández, Ainhoa, Marta Domènech, Ana M. Muñoz-Mármol, Cristina Carrato, and Carmen Balana. 2021. "Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment" Cells 10, no. 12: 3529. https://doi.org/10.3390/cells10123529
APA StyleHernández, A., Domènech, M., Muñoz-Mármol, A. M., Carrato, C., & Balana, C. (2021). Glioblastoma: Relationship between Metabolism and Immunosuppressive Microenvironment. Cells, 10(12), 3529. https://doi.org/10.3390/cells10123529