
Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases

 , , ,
, , , 
_Haendeler.png) and
and 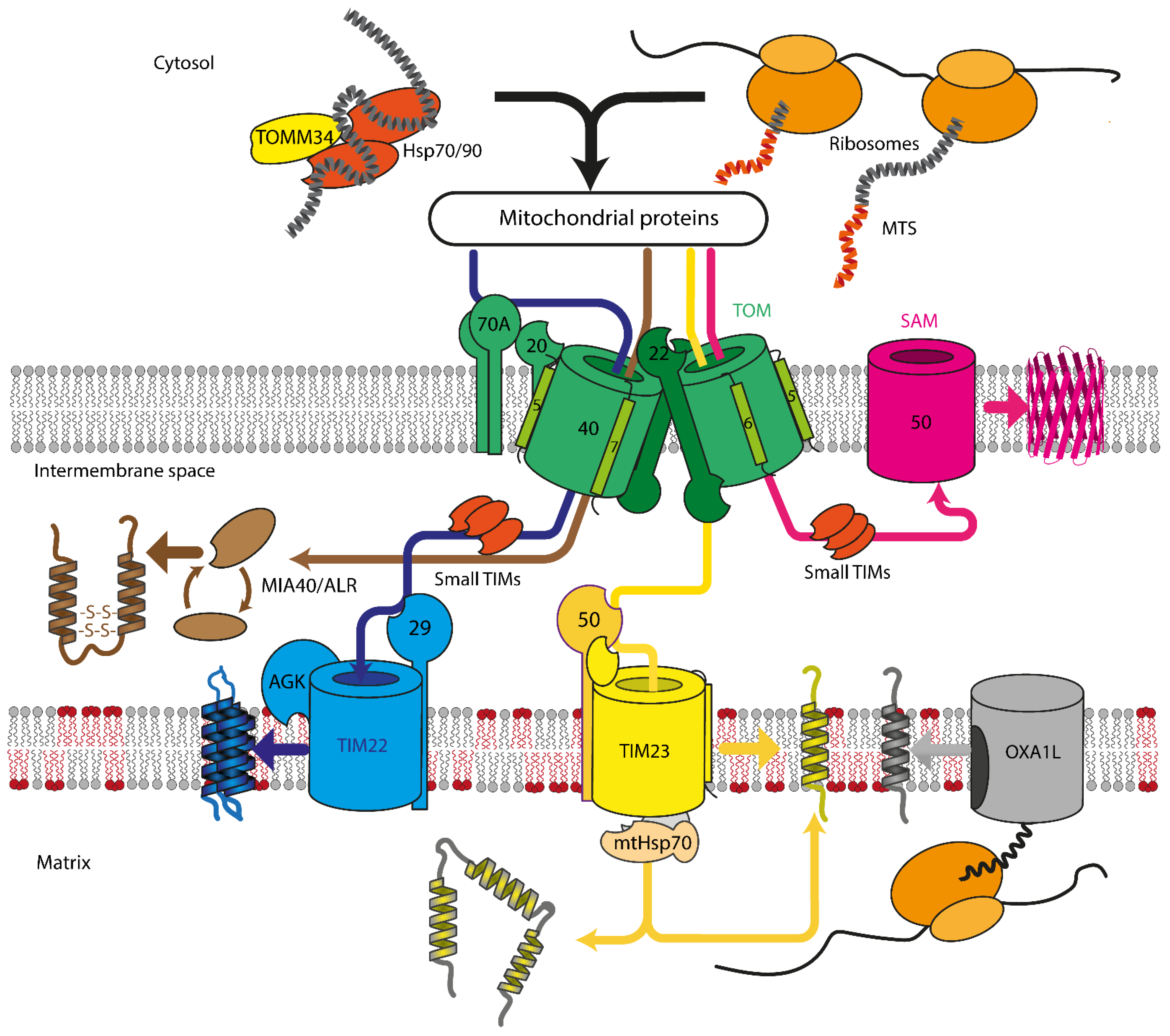{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mitochondrial Protein Import Machinery
2.1. Translocase of Outer Mitochondrial Membrane
2.2. Sorting and Assembly Machinery Component
2.3. Mitochondrial Intermembrane Space Assembly
2.4. Translocase of Inner Mitochondrial Membrane
2.4.1. TIM23
2.4.2. TIM22
2.5. Mitochondrial Lipids
3. Neurodegenerative Diseases
3.1. Parkinson’s Disease
3.2. Alzheimer’s Disease

4. Cardiovascular Diseases
4.1. Cardiolipin
4.2. Mitochondrial Telomerase Reverse Transcriptase
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Free Radical Theory of Aging. Antioxid. Redox Signal. 2003, 5, 557–561. [Google Scholar] [CrossRef]
- Park, C.B.; Larsson, N.-G. Mitochondrial DNA Mutations in Disease and Aging. J. Cell Biol. 2011, 193, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Age-Dependent Decline in the Cytochrome c Oxidase Activity in Rat Heart Mitochondria: Role of Cardiolipin. FEBS Lett. 1997, 406, 136–138. [Google Scholar] [CrossRef]
- Garcia-Euiz, C.; Mari, M.; Coiell, A.; Morales, A.; Caballero, F.; Montero, J.; Terrones, O.; Basañes, G.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Health and Disease. Histol. Histopathol. 2009, 24, 117–132. [Google Scholar] [CrossRef]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s Disease and Their Potential Role in Alzheimer’s Proteostasis. Exp. Neurol. 2020, 330, 113–321. [Google Scholar] [CrossRef]
- Voos, W. Chaperone–Protease Networks in Mitochondrial Protein Homeostasis. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 388–399. [Google Scholar] [CrossRef]
- Quirós, P.M.; Langer, T.; López-Otín, C. New Roles for Mitochondrial Proteases in Health, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2015, 16, 345–359. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Role of Mitochondrial Protein Quality Control in Oxidative Stress-Induced Neurodegenerative Diseases. Curr. Alzheimer Res. 2016, 13, 164–173. [Google Scholar] [CrossRef]
- Rüb, C.; Wilkening, A.; Voos, W. Mitochondrial Quality Control by the Pink1/Parkin System. Cell Tissue Res. 2017, 367, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Green, D.R.; Van Houten, B. SnapShot: Mitochondrial Quality Control. Cell 2011, 147, 950–950.e1. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Simon, D.K.; Johns, D.R. Mitochondrial Disorders: Clinical and Genetic Features. Annu. Rev. Med. 1999, 50, 111–127. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Palmer, C.S.; Anderson, A.J.; Stojanovski, D. Mitochondrial Protein Import Dysfunction: Mitochondrial Disease, Neurodegenerative Disease and Cancer. FEBS Lett. 2021, 595, 1107–1131. [Google Scholar] [CrossRef]
- Chacinska, A.; Koehler, C.M.; Milenkovic, D.; Lithgow, T.; Pfanner, N. Importing Mitochondrial Proteins: Machineries and Mechanisms. Cell 2009, 138, 628–644. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.P.; Muijsers, A.O.; Demol, H.; Dekker, H.L.; Van Beeumen, J.J. Human Heart Cytochrome c Oxidase Subunit VIII Purification and Determination of the Complete Amino Acid Sequence. FEBS Lett. 1988, 240, 127–132. [Google Scholar] [CrossRef]
- Pfanner, N.; Hoeben, P.; Tropschug, M.; Neupert, W. The Carboxyl-Terminal Two-Thirds of the ADP/ATP Carrier Polypeptide Contains Sufficient Information to Direct Translocation into Mitochondria. J. Biol. Chem. 1987, 262, 14851–14854. [Google Scholar] [CrossRef]
- Jores, T.; Klinger, A.; Groß, L.E.; Kawano, S.; Flinner, N.; Duchardt-Ferner, E.; Wöhnert, J.; Kalbacher, H.; Endo, T.; Schleiff, E.; et al. Characterization of the Targeting Signal in Mitochondrial β-Barrel Proteins. Nat. Commun. 2016, 7, 12036. [Google Scholar] [CrossRef]
- Becker, T.; Song, J.; Pfanner, N. Versatility of Preprotein Transfer from the Cytosol to Mitochondria. Trends Cell Biol. 2019, 29, 534–548. [Google Scholar] [CrossRef]
- Vögtle, F.-N.; Wortelkamp, S.; Zahedi, R.P.; Becker, D.; Leidhold, C.; Gevaert, K.; Kellermann, J.; Voos, W.; Sickmann, A.; Pfanner, N.; et al. Global Analysis of the Mitochondrial N-Proteome Identifies a Processing Peptidase Critical for Protein Stability. Cell 2009, 139, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Neupert, W.; Stuart, R.A. Oxa1p Acts as a General Membrane Insertion Machinery for Proteins Encoded by Mitochondrial DNA. EMBO J. 2001, 20, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Mai, N.; Oláhová, M.; Scialó, F.; Formosa, L.E.; Stroud, D.A.; Garrett, M.; Lax, N.Z.; Robertson, F.M.; Jou, C.; et al. OXA 1L Mutations Cause Mitochondrial Encephalopathy and a Combined Oxidative Phosphorylation Defect. EMBO Mol. Med. 2018, 10, e9060. [Google Scholar] [CrossRef]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Williams, C.C.; Jan, C.H.; Weissman, J.S. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Science 2014, 346, 748–751. [Google Scholar] [CrossRef]
- Bogorodskiy, A.; Okhrimenko, I.; Maslov, I.; Maliar, N.; Burkatovskii, D.; von Ameln, F.; Schulga, A.; Jakobs, P.; Altschmied, J.; Haendeler, J.; et al. Accessing Mitochondrial Protein Import in Living Cells by Protein Microinjection. Front. Cell Dev. Biol. 2021, 9, 1794. [Google Scholar] [CrossRef]
- Gao, J.; Schatton, D.; Martinelli, P.; Hansen, H.; Pla-Martin, D.; Barth, E.; Becker, C.; Altmueller, J.; Frommolt, P.; Sardiello, M.; et al. CLUH Regulates Mitochondrial Biogenesis by Binding MRNAs of Nuclear-Encoded Mitochondrial Proteins. J. Cell Biol. 2014, 207, 213–223. [Google Scholar] [CrossRef]
- Ravanidis, S.; Doxakis, E. RNA-Binding Proteins Implicated in Mitochondrial Damage and Mitophagy. Front. Cell Dev. Biol. 2020, 8, 372. [Google Scholar] [CrossRef]
- Qin, W.; Myers, S.A.; Carey, D.K.; Carr, S.A.; Ting, A.Y. Spatiotemporally-Resolved Mapping of RNA Binding Proteins via Functional Proximity Labeling Reveals a Mitochondrial MRNA Anchor Promoting Stress Recovery. Nat. Commun. 2021, 12, 4980. [Google Scholar] [CrossRef]
- Shiota, T.; Imai, K.; Qiu, J.; Hewitt, V.L.; Tan, K.; Shen, H.-H.; Sakiyama, N.; Fukasawa, Y.; Hayat, S.; Kamiya, M.; et al. Molecular Architecture of the Active Mitochondrial Protein Gate. Science 2015, 349, 1544–1548. [Google Scholar] [CrossRef]
- Guan, Z.; Yan, L.; Wang, Q.; Qi, L.; Hong, S.; Gong, Z.; Yan, C.; Yin, P. Structural Insights into Assembly of Human Mitochondrial Translocase TOM Complex. Cell Discov. 2021, 7, 22. [Google Scholar] [CrossRef]
- Kato, H.; Mihara, K. Identification of Tom5 and Tom6 in the Preprotein Translocase Complex of Human Mitochondrial Outer Membrane. Biochem. Biophys. Res. Commun. 2008, 369, 958–963. [Google Scholar] [CrossRef]
- Young, J.C.; Hoogenraad, N.J.; Hartl, F.U. Molecular Chaperones Hsp90 and Hsp70 Deliver Preproteins to the Mitochondrial Import Receptor Tom70. Cell 2003, 112, 41–50. [Google Scholar] [CrossRef]
- Faou, P.; Hoogenraad, N.J. Tom34: A Cytosolic Cochaperone of the Hsp90/Hsp70 Protein Complex Involved in Mitochondrial Protein Import. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 348–357. [Google Scholar] [CrossRef]
- Juszkiewicz, S.; Hegde, R.S. Quality Control of Orphaned Proteins. Mol. Cell 2018, 71, 443–457. [Google Scholar] [CrossRef]
- Armstrong, L.C.; Komiya, T.; Bergman, B.E.; Mihara, K.; Bornstein, P. Metaxin Is a Component of a Preprotein Import Complex in the Outer Membrane of the Mammalian Mitochondrion. J. Biol. Chem. 1997, 272, 6510–6518. [Google Scholar] [CrossRef]
- Armstrong, L.C.; Saenz, A.J.; Bornstein, P. Metaxin 1 Interacts with Metaxin 2, a Novel Related Protein Associated with the Mammalian Mitochondrial Outer Membrane. J. Cell. Biochem. 1999, 74, 11–22. [Google Scholar] [CrossRef]
- Kozjak-Pavlovic, V.; Ross, K.; Benlasfer, N.; Kimmig, S.; Karlas, A.; Rudel, T. Conserved Roles of Sam50 and Metaxins in VDAC Biogenesis. EMBO Rep. 2007, 8, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Dabir, D.V.; Leverich, E.P.; Kim, S.-K.; Tsai, F.D.; Hirasawa, M.; Knaff, D.B.; Koehler, C.M. A Role for Cytochrome c and Cytochrome c Peroxidase in Electron Shuttling from Erv1. EMBO J. 2007, 26, 4801–4811. [Google Scholar] [CrossRef]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Küttner, V.; Bien, M.; Kauff, F.; et al. Protein Import and Oxidative Folding in the Mitochondrial Intermembrane Space of Intact Mammalian Cells. Mol. Biol. Cell 2013, 24, 2160–2170. [Google Scholar] [CrossRef]
- Bauer, M.F.; Gempel, K.; Reichert, A.S.; Rappold, G.A.; Lichtner, P.; Gerbitz, K.-D.; Neupert, W.; Brunner, M.; Hofmann, S. Genetic and Structural Characterization of the Human Mitochondrial Inner Membrane Translocase 1 1Edited by J. Karn. J. Mol. Biol. 1999, 289, 69–82. [Google Scholar] [CrossRef]
- Sinha, D.; Srivastava, S.; Krishna, L.; D’Silva, P. Unraveling the Intricate Organization of Mammalian Mitochondrial Presequence Translocases: Existence of Multiple Translocases for Maintenance of Mitochondrial Function. Mol. Cell. Biol. 2014, 34, 1757–1775. [Google Scholar] [CrossRef]
- Sokol, A.M.; Sztolsztener, M.E.; Wasilewski, M.; Heinz, E.; Chacinska, A. Mitochondrial Protein Translocases for Survival and Wellbeing. FEBS Lett. 2014, 588, 2484–2495. [Google Scholar] [CrossRef]
- Curran, S.P.; Leuenberger, D.; Schmidt, E.; Koehler, C.M. The Role of the Tim8p–Tim13p Complex in a Conserved Import Pathway for Mitochondrial Polytopic Inner Membrane Proteins. J. Cell Biol. 2002, 158, 1017–1027. [Google Scholar] [CrossRef]
- Bauer, M.F.; Rothbauer, U.; Mühlenbein, N.; Smith, R.J.H.; Gerbitz, K.-D.; Neupert, W.; Brunner, M.; Hofmann, S. The Mitochondrial TIM22 Preprotein Translocase Is Highly Conserved throughout the Eukaryotic Kingdom. FEBS Lett. 1999, 464, 41–47. [Google Scholar] [CrossRef]
- Qi, L.; Wang, Q.; Guan, Z.; Wu, Y.; Shen, C.; Hong, S.; Cao, J.; Zhang, X.; Yan, C.; Yin, P. Cryo-EM Structure of the Human Mitochondrial Translocase TIM22 Complex. Cell Res. 2021, 31, 369–372. [Google Scholar] [CrossRef]
- Kang, Y.; Stroud, D.A.; Baker, M.J.; De Souza, D.P.; Frazier, A.E.; Liem, M.; Tull, D.; Mathivanan, S.; McConville, M.J.; Thorburn, D.R.; et al. Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex. Mol. Cell 2017, 67, 457–470.e5. [Google Scholar] [CrossRef]
- Kang, Y.; Baker, M.J.; Liem, M.; Louber, J.; McKenzie, M.; Atukorala, I.; Ang, C.S.; Keerthikumar, S.; Mathivanan, S.; Stojanovski, D. Tim29 Is a Novel Subunit of the Human TIM22 Translocase and Is Involved in Complex Assembly and Stability. eLife 2016, 5, e17463. [Google Scholar] [CrossRef]
- Mejia, E.M.; Hatch, G.M. Mitochondrial Phospholipids: Role in Mitochondrial Function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef]
- Gasanoff, E.S.; Yaguzhinsky, L.S.; Garab, G. Cardiolipin, Non-Bilayer Structures and Mitochondrial Bioenergetics: Relevance to Cardiovascular Disease. Cells 2021, 10, 1721. [Google Scholar] [CrossRef]
- Osman, C.; Voelker, D.R.; Langer, T. Making Heads or Tails of Phospholipids in Mitochondria. J. Cell Biol. 2011, 192, 7–16. [Google Scholar] [CrossRef]
- Gebert, N.; Joshi, A.S.; Kutik, S.; Becker, T.; McKenzie, M.; Guan, X.L.; Mooga, V.P.; Stroud, D.A.; Kulkarni, G.; Wenk, M.R.; et al. Mitochondrial Cardiolipin Involved in Outer-Membrane Protein Biogenesis: Implications for Barth Syndrome. Curr. Biol. 2009, 19, 2133–2139. [Google Scholar] [CrossRef]
- Haines, T.H.; Dencher, N.A. Cardiolipin: A Proton Trap for Oxidative Phosphorylation. FEBS Lett. 2002, 528, 35–39. [Google Scholar] [CrossRef]
- Malhotra, K.; Modak, A.; Nangia, S.; Daman, T.H.; Gunsel, U.; Robinson, V.L.; Mokranjac, D.; May, E.R.; Alder, N.N. Cardiolipin Mediates Membrane and Channel Interactions of the Mitochondrial TIM23 Protein Import Complex Receptor Tim50. Sci. Adv. 2017, 3, e1700532. [Google Scholar] [CrossRef]
- Elustondo, P.; Martin, L.A.; Karten, B. Mitochondrial Cholesterol Import. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2017, 1862, 90–101. [Google Scholar] [CrossRef]
- Martin, L.A.; Kennedy, B.E.; Karten, B. Mitochondrial Cholesterol: Mechanisms of Import and Effects on Mitochondrial Function. J. Bioenerg. Biomembr. 2016, 48, 137–151. [Google Scholar] [CrossRef]
- Bose, H.S.; Lingappa, V.R.; Miller, W.L. Rapid Regulation of Steroidogenesis by Mitochondrial Protein Import. Nature 2002, 417, 87–91. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21. [Google Scholar] [CrossRef]
- Pacelli, C.; Giguère, N.; Bourque, M.J.; Lévesque, M.; Slack, R.S.; Trudeau, L.É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. Monogenic Parkinson’s Disease and Parkinsonism: Clinical Phenotypes and Frequencies of Known Mutations. Parkinsonism Relat. Disord. 2013, 19, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial Processing Peptidase Regulates PINK1 Processing, Import and Parkin Recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Takatori, S.; Ito, G.; Iwatsubo, T. Cytoplasmic Localization and Proteasomal Degradation of N-Terminally Cleaved Form of PINK1. Neurosci. Lett. 2008, 430, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Uno, M.; Koyano, F.; Go, E.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. A Dimeric Pink1-Containing Complex on Depolarized Mitochondria Stimulates Parkin Recruitment. J. Biol. Chem. 2013, 288, 36372–36384. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 Phosphorylates Ubiquitin to Activate Parkin E3 Ubiquitin Ligase Activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 Import Regulation; a Fine System to Convey Mitochondrial Stress to the Cytosol. BMC Biol. 2018, 16, 1–13. [Google Scholar] [CrossRef]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-Content Genome-Wide RNAi Screens Identify Regulators of Parkin Upstream of Mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin Control Localized Translation of Respiratory Chain Component MRNAs on Mitochondria Outer Membrane. Physiol. Behav. 2018, 176, 139–148. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Fiesel, F.C.; Hudec, R.; Caulfield, T.R.; Ogaki, K.; Górka-Skoczylas, P.; Koziorowski, D.; Friedman, A.; Chen, L.; Dawson, V.L.; et al. The PINK1 p.I368N Mutation Affects Protein Stability and Ubiquitin Kinase Activity. Mol. Neurodegener. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Van Der Brug, M.; Ahmad, R.; Kesavapany, S.; Miller, D.W.; Petsko, G.A.; Cookson, M.R. Mutations in PTEN-Induced Putative Kinase 1 Associated with Recessive Parkinsonism Have Differential Effects on Protein Stability. Proc. Natl. Acad. Sci. USA 2005, 102, 5703–5708. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S Increases Risk of Parkinson’s Disease via a Dominant-Negative Mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.-J.; Corti, O.; Araujo, F.; Hampe, C.; Jacquier, S.; Lücking, C.B.; Abbas, N.; Duyckaerts, C.; Rooney, T.; Pradier, L.; et al. The C289G and C418R Missense Mutations Cause Rapid Sequestration of Human Parkin into Insoluble Aggregates. Neurobiol. Dis. 2003, 14, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Kitami, T.; Suzuki, T.; Mizuno, Y.; Hattori, N.; Tanaka, K. Diverse Effects of Pathogenic Mutations of Parkin That Catalyze Multiple Monoubiquitylation in Vitro. J. Biol. Chem. 2006, 281, 3204–3209. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein Binds to TOM20 and Inhibits Mitochondrial Protein Import in Parkinson’s Disease. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Desplats, P.; Spencer, B.; Rockenstein, E.; Adame, A.; Elstner, M.; Laub, C.; Mueller, S.; Koob, A.O.; Mante, M.; et al. TOM40 Mediates Mitochondrial Dysfunction Induced by α-Synuclein Accumulation in Parkinson’s Disease. PLoS ONE 2013, 8, e62277. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Rovini, A.; Gurnev, P.A.; Beilina, A.; Queralt-Martín, M.; Rosencrans, W.; Cookson, M.R.; Bezrukov, S.M.; Rostovtseva, T.K. Molecular Mechanism of Olesoxime-Mediated Neuroprotection through Targeting α-Synuclein Interaction with Mitochondrial VDAC. Cell. Mol. Life Sci. 2020, 77, 3611–3626. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage-Dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [CrossRef]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Uber Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Alzheimer’s Disease: The Amyloid Cascade Hypothesis: An Update and Reappraisal. J. Alzheimer’s Dis. 2006, 9, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Aβ42 Accumulation in Human Brain. Am. J. Pathol. 2000, 156, 15–20. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal β-Amyloid Accumulation and Synapse Pathology in Alzheimer’s Disease. Acta Neuropathol. 2010, 119, 523–541. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of Amyloid Plaque Formation Suggests an Intracellular Basis of Aβ Pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef]
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: Progress and Perspectives. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Narayan, P.; Holmström, K.M.; Kim, D.-H.; Whitcomb, D.J.; Wilson, M.R.; St. George-Hyslop, P.; Wood, N.W.; Dobson, C.M.; Cho, K.; Abramov, A.Y.; et al. Rare Individual Amyloid-β Oligomers Act on Astrocytes to Initiate Neuronal Damage. Biochemistry 2014, 53, 2442–2453. [Google Scholar] [CrossRef]
- Džinić, T.; Dencher, N.A. Oxygen Concentration and Oxidative Stress Modulate the Influence of Alzheimer’s Disease A β 1–42 Peptide on Human Cells. Oxid. Med. Cell. Longev. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dencher, N.A.; Bogorodskiy, A.O.; Borshchevskiy, V.I.; Gordeliy, V.I.; Malyar, N.L.; Maslov, I.V.; Okhrimenko, I.S.; Podolyak, E.Y.; Dani, D.D.V.; Dzinic, T.; et al. Challenge the “Free Radical Theory of Ageing” and the Aß Peptide Extracellular Plaque Hypothesis of Alzheimer’s Disease. In Proceedings of the Biomembranes 2018, Dolgoprudny, Russian, 15 December 2018; Volume 50, pp. 467–603. [Google Scholar]
- Devi, L. Accumulation of Amyloid Precursor Protein in the Mitochondrial Import Channels of Human Alzheimer’s Disease Brain Is Associated with Mitochondrial Dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.-A.; Avadhani, N.G. Mitochondrial Targeting and a Novel Transmembrane Arrest of Alzheimer’s Amyloid Precursor Protein Impairs Mitochondrial Function in Neuronal Cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.Κ.; Devi, L. Mitochondrial Translocation of Amyloid Precursor Protein and Its Cleaved Products: Relevance to Mitochondrial Dysfunction in Alzheimer’s Disease. Rev. Neurosci. 2007, 18, 343–354. [Google Scholar] [CrossRef]
- Cenini, G.; Rüb, C.; Bruderek, M.; Voos, W. Amyloid β-Peptides Interfere with Mitochondrial Preprotein Import Competence by a Coaggregation Process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.; Eckert, A. Amyloid-Beta Interaction with Mitochondria. Int. J. Alzheimers. Dis. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Harbauer, A.B.; Zahedi, R.P.; Sickmann, A.; Pfanner, N.; Meisinger, C. The Protein Import Machinery of Mitochondria—A Regulatory Hub in Metabolism, Stress, and Disease. Cell Metab. 2014, 19, 357–372. [Google Scholar] [CrossRef]
- Klingenberg, M.; Rrottenberg, H. Relation between the Gradient of the ATP/ADP Ratio and the Membrane Potential across the Mitochondrial Membrane. Eur. J. Biochem. 1977, 73, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Liberman, E.A.; Topaly, V.P.; Tsofina, L.M.; Jasaitis, A.A.; Skulachev, V.P. Mechanism of Coupling of Oxidative Phosphorylation and the Membrane Potential of Mitochondria. Nature 1969, 222, 1076–1078. [Google Scholar] [CrossRef]
- Wiedemann, N.; van der Laan, M.; Hutu, D.P.; Rehling, P.; Pfanner, N. Sorting Switch of Mitochondrial Presequence Translocase Involves Coupling of Motor Module to Respiratory Chain. J. Cell Biol. 2007, 179, 1115–1122. [Google Scholar] [CrossRef]
- Teo, E.; Ravi, S.; Barardo, D.; Kim, H.S.; Fong, S.; Gassiot, A.C.; Tan, T.Y.; Ching, J.; Kovalik, J.P.; Wenk, M.R.; et al. Metabolic Stress Is a Primary Pathogenic Event in Transgenic Caenorhabditis Elegans Expressing Pan-Neuronal Human Amyloid Beta. eLife 2019, 8, e50069. [Google Scholar] [CrossRef]
- Lehninger, A.L. Proton and Electric Charge Translocation in Mitochondrial Energy Transduction BT—Structure and Function Relationships in Biochemical Systems. In Structure and Function Relationships in Biochemical Systems; Bossa, F., Chiancone, E., Finazzi-Agrò, A., Strom, R., Eds.; Springer: Boston, MA, USA, 1982; pp. 171–186. ISBN 978-1-4615-9281-5. [Google Scholar]
- Skulachev, V.P. Mitochondrial Physiology and Pathology; Concepts of Programmed Death of Organelles, Cells and Organisms. Mol. Aspects Med. 1999, 20, 139–184. [Google Scholar] [CrossRef]
- Lejri, I.; Agapouda, A.; Grimm, A.; Eckert, A. Mitochondria- and Oxidative Stress-Targeting Substances in Cognitive Decline-Related Disorders: From Molecular Mechanisms to Clinical Evidence. Oxid. Med. Cell. Longev. 2019, 2019, 1–26. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oulès, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Bréchot, P.; et al. Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimer’s Dis. 2016, 55, 1549–1570. [Google Scholar] [CrossRef]
- Urban, A.S.; Pavlov, K.V.; Kamynina, A.V.; Okhrimenko, I.S.; Arseniev, A.S.; Bocharov, E.V. Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development. Molecules 2021, 26, 2897. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Yamazaki, T.; Ishiguro, K.; Shoji, M.; Nakazato, Y.; Hirai, S. Ultrastructural Localization of Alzheimer Amyloid Beta/A4 Protein Precursor in the Cytoplasm of Neurons and Senile Plaque-Associated Astrocytes. Acta Neuropathol. 1992, 85, 15–22. [Google Scholar] [CrossRef]
- Pavlov, P.F.; Wiehager, B.; Sakai, J.; Frykman, S.; Behbahani, H.; Winblad, B.; Ankarcrona, M. Mitochondrial Γ-secretase Participates in the Metabolism of Mitochondria-associated Amyloid Precursor Protein. FASEB J. 2011, 25, 78–88. [Google Scholar] [CrossRef]
- Hansson, C.A.; Frykman, S.; Farmery, M.R.; Tjernberg, L.O.; Nilsberth, C.; Pursglove, S.E.; Ito, A.; Winblad, B.; Cowburn, R.F.; Thyberg, J.; et al. Nicastrin, Presenilin, APH-1, and PEN-2 Form Active γ-Secretase Complexes in Mitochondria. J. Biol. Chem. 2004, 279, 51654–51660. [Google Scholar] [CrossRef]
- Nowicka, U.; Chroscicki, P.; Stroobants, K.; Sladowska, M.; Turek, M.; Uszczynska-Ratajczak, B.; Kundra, R.; Goral, T.; Perni, M.; Dobson, C.M.; et al. Cytosolic Aggregation of Mitochondrial Proteins Disrupts Cellular Homeostasis by Stimulating the Aggregation of Other Proteins. eLife 2021, 10, e65484. [Google Scholar] [CrossRef]
- Sirk, D.; Zhu, Z.; Wadia, J.S.; Shulyakova, N.; Phan, N.; Fong, J.; Mills, L.R. Chronic Exposure to Sub-Lethal Beta-Amyloid (Aβ) Inhibits the Import of Nuclear-Encoded Proteins to Mitochondria in Differentiated PC12 Cells. J. Neurochem. 2007, 103, 1989–2003. [Google Scholar] [CrossRef]
- Istrate, A.N.; Kozin, S.A.; Zhokhov, S.S.; Mantsyzov, A.B.; Kechko, O.I.; Pastore, A.; Makarov, A.A.; Polshakov, V.I. Interplay of Histidine Residues of the Alzheimer’s Disease Aβ Peptide Governs Its Zn-Induced Oligomerization. Sci. Rep. 2016, 6, 21734. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The Amyloid β-Peptide Is Imported into Mitochondria via the TOM Import Machinery and Localized to Mitochondrial Cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef]
- Tillement, L.; Lecanu, L.; Yao, W.; Greeson, J.; Papadopoulos, V. The Spirostenol (22R, 25R)-20α-Spirost-5-En-3β-Yl Hexanoate Blocks Mitochondrial Uptake of Aβ in Neuronal Cells and Prevents Aβ-Induced Impairment of Mitochondrial Function. Steroids 2006, 71, 725–735. [Google Scholar] [CrossRef]
- Falkevall, A.; Alikhani, N.; Bhushan, S.; Pavlov, P.F.; Busch, K.; Johnson, K.A.; Eneqvist, T.; Tjernberg, L.; Ankarcrona, M.; Glaser, E. Degradation of the Amyloid β-Protein by the Novel Mitochondrial Peptidasome, PreP. J. Biol. Chem. 2006, 281, 29096–29104. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Vögtle, F.-N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef]
- Pinho, C.M.; Teixeira, P.F.; Glaser, E. Mitochondrial Import and Degradation of Amyloid-β Peptide. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 1069–1074. [Google Scholar] [CrossRef]
- Fang, D.; Wang, Y.; Zhang, Z.; Du, H.; Yan, S.; Sun, Q.; Zhong, C.; Wu, L.; Vangavaragu, J.R.; Yan, S.; et al. Increased Neuronal PreP Activity Reduces Aβ Accumulation, Attenuates Neuroinflammation and Improves Mitochondrial and Synaptic Function in Alzheimer Disease’s Mouse Model. Hum. Mol. Genet. 2015, 24, 5198–5210. [Google Scholar] [CrossRef]
- Ostermann, J.; Voos, W.; Kang, P.J.; Craig, E.A.; Neupert, W.; Pfanner, N. Precursor Proteins in Transit through Mitochondrial Contact Sites Interact with Hsp70 in the Matrix. FEBS Lett. 1990, 277, 281–284. [Google Scholar] [CrossRef]
- Horst, M. Sequential Action of Two Hsp70 Complexes during Protein Import into Mitochondria. EMBO J. 1997, 16, 1842–1849. [Google Scholar] [CrossRef]
- Park, S.J.; Shin, J.H.; Jeong, J.I.; Song, J.H.; Jo, Y.K.; Kim, E.S.; Lee, E.H.; Hwang, J.J.; Lee, E.K.; Chung, S.J.; et al. Down-Regulation of Mortalin Exacerbates Aβ-Mediated Mitochondrial Fragmentation and Dysfunction. J. Biol. Chem. 2014, 289, 2195–2204. [Google Scholar] [CrossRef]
- Deocaris, C.C.; Kaul, S.C.; Wadhwa, R. From Proliferative to Neurological Role of an Hsp70 Stress Chaperone, Mortalin. Biogerontology 2008, 9, 391–403. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the Amyloid Precursor Protein by Human γ-Secretase. Science 2019, 363, 6428. [Google Scholar] [CrossRef]
- Nierzwicki, Ł.; Olewniczak, M.; Chodnicki, P.; Czub, J. Role of Cholesterol in Substrate Recognition by γ-Secretase. Sci. Rep. 2021, 11, 15213. [Google Scholar] [CrossRef]
- Langness, V.F.; van der Kant, R.; Das, U.; Wang, L.; dos Chaves, R.S.; Goldstein, L.S.B. Cholesterol-Lowering Drugs Reduce APP Processing to Aβ by Inducing APP Dimerization. Mol. Biol. Cell 2021, 32, 247–259. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef]
- Nadezhdin, K.D.; Bocharova, O.V.; Bocharov, E.V.; Arseniev, A.S. Structural and Dynamic Study of the Transmembrane Domain of the Amyloid Precursor Protein. Acta Nat. 2011, 3, 69–76. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Grimm, H.S.; Tomic, I.; Beyreuther, K.; Hartmann, T.; Bergmann, C. Independent Inhibition of Alzheimer Disease β- and γ-Secretase Cleavage by Lowered Cholesterol Levels. J. Biol. Chem. 2008, 283, 11302–11311. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Wang, C.; Holtzman, D.M.; Ferris, H.A.; Hansen, S.B. Regulation of Beta-Amyloid Production in Neurons by Astrocyte-Derived Cholesterol. Proc. Natl. Acad. Sci. USA 2021, 118, e2102191118. [Google Scholar] [CrossRef]
- Bodovitz, S.; Klein, W.L. Cholesterol Modulates α-Secretase Cleavage of Amyloid Precursor Protein. J. Biol. Chem. 1996, 271, 4436–4440. [Google Scholar] [CrossRef]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Yun, T.D.; Stavrovskaya, I.G.; Xu, Y.; Koo, S.Y.; et al. The Alzheimer’s Disease-associated C99 Fragment of APP Regulates Cellular Cholesterol Trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef]
- Kozin, S.A.; Barykin, E.P.; Mitkevich, V.A.; Makarov, A.A. Anti-Amyloid Therapy of Alzheimer’s Disease: Current State and Prospects. Biochemistry 2018, 83, 1057–1067. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Gremer, L.; Urban, A.S.; Okhrimenko, I.S.; Volynsky, P.E.; Nadezhdin, K.D.; Bocharova, O.V.; Kornilov, D.A.; Zagryadskaya, Y.A.; Kamynina, A.V.; et al. All-d-Enantiomeric Peptide D3 Designed for Alzheimer’s Disease Treatment Dynamically Interacts with Membrane-Bound Amyloid-β Precursors. J. Med. Chem. 2021, 64, 16464–16479. [Google Scholar] [CrossRef]
- Elfgen, A.; Hupert, M.; Bochinsky, K.; Tusche, M.; González de San Román Martin, E.; Gering, I.; Sacchi, S.; Pollegioni, L.; Huesgen, P.F.; Hartmann, R.; et al. Metabolic Resistance of the D-Peptide RD2 Developed for Direct Elimination of Amyloid-β Oligomers. Sci. Rep. 2019, 9, 5715. [Google Scholar] [CrossRef]
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European Society of Cardiology: Cardiovascular Disease Statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef]
- Zhao, F.; Zou, M.-H. Role of the Mitochondrial Protein Import Machinery and Protein Processing in Heart Disease. Front. Cardiovasc. Med. 2021, 8, 749756. [Google Scholar] [CrossRef]
- International Mouse Phenotyping Consortium. Available online: https://www.mousephenotype.org (accessed on 15 November 2021).
- Paradies, G.; Petrosillo, G.; Ruggiero, F.M. Cardiolipin-Dependent Decrease of Cytochrome c Oxidase Activity in Heart Mitochondria from Hypothyroid Rats. Biochim. Biophys. Acta-Bioenerg. 1997, 1319, 5–8. [Google Scholar] [CrossRef][Green Version]
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart. Circ. Res. 2004, 94, 53–59. [Google Scholar] [CrossRef]
- Maekawa, S.; Takada, S.; Nambu, H.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueyanagi, Y.; Kang, D.; Sabe, H.; Kinugawa, S. Linoleic Acid Improves Assembly of the CII Subunit and CIII2/CIV Complex of the Mitochondrial Oxidative Phosphorylation System in Heart Failure. Cell Commun. Signal. 2019, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- El-Hafidi, M.; Correa, F.; Zazueta, C. Mitochondrial Dysfunction in Metabolic and Cardiovascular Diseases Associated with Cardiolipin Remodeling. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165744. [Google Scholar] [CrossRef]
- Werner, C.; Hanhoun, M.; Widmann, T.; Kazakov, A.; Semenov, A.; Pöss, J.; Bauersachs, J.; Thum, T.; Pfreundschuh, M.; Müller, P.; et al. Effects of Physical Exercise on Myocardial Telomere-Regulating Proteins, Survival Pathways, and Apoptosis. J. Am. Coll. Cardiol. 2008, 52, 470–482. [Google Scholar] [CrossRef]
- Werner, C.; Fürster, T.; Widmann, T.; Pöss, J.; Roggia, C.; Hanhoun, M.; Scharhag, J.; Büchner, N.; Meyer, T.; Kindermann, W.; et al. Physical Exercise Prevents Cellular Senescence in Circulating Leukocytes and in the Vessel Wall. Circulation 2009, 120, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Richardson, G.D.; Breault, D.; Horrocks, G.; Cormack, S.; Hole, N.; Owens, W.A. Telomerase Expression in the Mammalian Heart. FASEB J. 2012, 26, 4832–4840. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial HTERT Exacerbates Free-Radical-Mediated MtDNA Damage. Aging Cell 2004, 3, 399–411. [Google Scholar] [CrossRef]
- Haendeler, J.; Dröse, S.; Büchner, N.; Jakob, S.; Altschmied, J.; Goy, C.; Spyridopoulos, I.; Zeiher, A.M.; Brandt, U.; Dimmeler, S. Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function From Damage. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 929–935. [Google Scholar] [CrossRef]
- Ahmed, S.; Passos, J.F.; Birket, M.J.; Beckmann, T.; Brings, S.; Peters, H.; Birch-Machin, M.A.; von Zglinicki, T.; Saretzki, G. Telomerase Does Not Counteract Telomere Shortening but Protects Mitochondrial Function under Oxidative Stress. J. Cell Sci. 2008, 121, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, O.A.; Caron, M.J.; Ulema, P.; Medrano, C.; Thomas, A.P.; Kimura, M.; Bonini, M.G.; Herbig, U.; Santos, J.H. A Mutant Telomerase Defective in Nuclear-Cytoplasmic Shuttling Fails to Immortalize Cells and Is Associated with Mitochondrial Dysfunction. Aging Cell 2010, 9, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; Freed, J.K.; Durand, M.J.; Riedel, M.; Ait-Aissa, K.; Green, P.; Hockenberry, J.C.; Morgan, R.G.; Donato, A.J.; Peleg, R.; et al. Critical Role for Telomerase in the Mechanism of Flow-Mediated Dilation in the Human Microcirculation. Circ. Res. 2016, 118, 856–866. [Google Scholar] [CrossRef]
- Oh, H.; Taffet, G.E.; Youker, K.A.; Entman, M.L.; Overbeek, P.A.; Michael, L.H.; Schneider, M.D. Telomerase Reverse Transcriptase Promotes Cardiac Muscle Cell Proliferation, Hypertrophy, and Survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Murasawa, S.; Llevadot, J.; Silver, M.; Isner, J.M.; Losordo, D.W.; Asahara, T. Constitutive Human Telomerase Reverse Transcriptase Expression Enhances Regenerative Properties of Endothelial Progenitor Cells. Circulation 2002, 106, 1133–1139. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Heisner, J.S.; Norwood Toro, L.E.; Bruemmer, D.; Doyon, G.; Harmann, L.; Geurts, A.; Camara, A.K.S.; Beyer, A.M. Telomerase Deficiency Predisposes to Heart Failure and Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 2019, 6, 31. [Google Scholar] [CrossRef]
- Haendeler, J.; Hoffmann, J.; Brandes, R.P.; Zeiher, A.M.; Dimmeler, S. Hydrogen Peroxide Triggers Nuclear Export of Telomerase Reverse Transcriptase via Src Kinase Family-Dependent Phosphorylation of Tyrosine 707. Mol. Cell. Biol. 2003, 23, 4598–4610. [Google Scholar] [CrossRef]
- Büchner, N.; Zschauer, T.-C.; Lukosz, M.; Altschmied, J.; Haendeler, J. Downregulation of Mitochondrial Telomerase Reverse Transcriptase Induced by H2O2 Is Src Kinase Dependent. Exp. Gerontol. 2010, 45, 558–562. [Google Scholar] [CrossRef]
- Hoffmann, J.; Richardson, G.; Haendeler, J.; Altschmied, J.; Andrés, V.; Spyridopoulos, I. Telomerase as a Therapeutic Target in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1047–1061. [Google Scholar] [CrossRef]
- Ale-Agha, N.; Jakobs, P.; Goy, C.; Zurek, M.; Rosen, J.; Dyballa-Rukes, N.; Metzger, S.; Greulich, J.; von Ameln, F.; Eckermann, O.; et al. Mitochondrial Telomerase Reverse Transcriptase Protects from Myocardial Ischemia/Reperfusion Injury by Improving Complex I Composition and Function. Circulation 2021, 144, 1876–1890. [Google Scholar] [CrossRef]
- Harley, C.B.; Liu, W.; Flom, P.L.; Raffaele, J.M. A Natural Product Telomerase Activator as Part of a Health Maintenance Program: Metabolic and Cardiovascular Response. Rejuvenation Res. 2013, 16, 386–395. [Google Scholar] [CrossRef]
- Maier, R.; Bawamia, B.; Bennaceur, K.; Dunn, S.; Marsay, L.; Amoah, R.; Kasim, A.; Filby, A.; Austin, D.; Hancock, H.; et al. Telomerase Activation to Reverse Immunosenescence in Elderly Patients With Acute Coronary Syndrome: Protocol for a Randomized Pilot Trial. JMIR Res. Protoc. 2020, 9, e19456. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogorodskiy, A.; Okhrimenko, I.; Burkatovskii, D.; Jakobs, P.; Maslov, I.; Gordeliy, V.; Dencher, N.A.; Gensch, T.; Voos, W.; Altschmied, J.; et al. Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases. Cells 2021, 10, 3528. https://doi.org/10.3390/cells10123528
Bogorodskiy A, Okhrimenko I, Burkatovskii D, Jakobs P, Maslov I, Gordeliy V, Dencher NA, Gensch T, Voos W, Altschmied J, et al. Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases. Cells. 2021; 10(12):3528. https://doi.org/10.3390/cells10123528
Chicago/Turabian StyleBogorodskiy, Andrey, Ivan Okhrimenko, Dmitrii Burkatovskii, Philipp Jakobs, Ivan Maslov, Valentin Gordeliy, Norbert A. Dencher, Thomas Gensch, Wolfgang Voos, Joachim Altschmied, and et al. 2021. "Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases" Cells 10, no. 12: 3528. https://doi.org/10.3390/cells10123528
APA StyleBogorodskiy, A., Okhrimenko, I., Burkatovskii, D., Jakobs, P., Maslov, I., Gordeliy, V., Dencher, N. A., Gensch, T., Voos, W., Altschmied, J., Haendeler, J., & Borshchevskiy, V. (2021). Role of Mitochondrial Protein Import in Age-Related Neurodegenerative and Cardiovascular Diseases. Cells, 10(12), 3528. https://doi.org/10.3390/cells10123528