
Human Induced Pluripotent Stem Cell as a Disease Modeling and Drug Development Platform—A Cardiac Perspective


Abstract
1. Introduction
2. Reprogramming Somatic Cells to iPSC
3. Delivery or Induction Methods of OSKM Factors
4. Derivation of CMs Subtypes
5. Challenges in iPSC-Derived-CMs Maturation
6. Metabolic Characterization of hiPSC-Derived-CMs
7. Applications of Genome Editing Technology in hiPSC
8. Strategies for Generating hiPSC for Heart Disease Modeling
9. Consideration of hiPSC for Use in Modeling Heart Disease
10. Drug Testing and Drugs Development Using hiPSC-Derived-CMs
11. Obtaining a Reproducible and Sufficient Number of hiPSC-Derived-CMs
12. Conclusions and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart, A.; et al. Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378. [Google Scholar] [CrossRef]
- Brodehl, A.; Ebbinghaus, H.; Deutsch, M.A.; Gummert, J.; Gartner, A.; Ratnavadivel, S.; Milting, H. Human Induced Pluripotent Stem-Cell-Derived Cardiomyocytes as Models for Genetic Cardiomyopathies. Int. J. Mol. Sci. 2019, 20, 4381–4437. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P.; Woo, K.; Statham, A.L.; Lundie, B.; Bagnall, R.D.; et al. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet. Med. 2019, 21, 650–662. [Google Scholar] [CrossRef]
- Halapas, A.; Papalois, A.; Stauropoulou, A.; Philippou, A.; Pissimissis, N.; Chatzigeorgiou, A.; Kamper, E.; Koutsilieris, M. In vivo models for heart failure research. Vivo 2008, 22, 767–780. [Google Scholar]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal Models of Heart Failure. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Tellez, N.; Greenway, S.C. Cellular models for human cardiomyopathy: What is the best option? World J. Cardiol. 2019, 11, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Mocanu, M.M.; Yellon, D.M. Retrograde heart perfusion: The Langendorff technique of isolated heart perfusion. J. Mol. Cell Cardiol. 2011, 50, 940–950. [Google Scholar] [CrossRef]
- Chlopcíková, S.; Psotová, J.; Miketová, P. Neonatal rat cardiomyocytes--a model for the study of morphological, biochemical and electrophysiological characteristics of the heart. Biomed. Pap. Palacky Univ. Olomouc 2001, 145, 49–55. [Google Scholar] [CrossRef]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Steinhelper, M.E.; Lanson, N.A.; Dresdner, K.P.; Delcarpio, J.B.; Wit, A.L.; Claycomb, W.C.; Field, L.J. Proliferation Invivo and in Culture of Differentiated Adult Atrial Cardiomyocytes from Transgenic Mice. Am. J. Physiol. 1990, 259, H1826–H1834. [Google Scholar]
- Delcarpio, J.B.; Lanson, N.A.; Field, L.J.; Claycomb, W.C. Morphological Characterization of Cardiomyocytes Isolated from a Transplantable Cardiac Tumor Derived from Transgenic Mouse Atria (at-1 Cells). Circ. Res. 1991, 69, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Jaffredo, T.; Chestier, A.; Bachnou, N.; Dieterlenlievre, F. Mc29-Immortalized Clonal Avian Heart Cell-Lines Can Partially Differentiate Invitro. Exp. Cell Res. 1991, 192, 481–491. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Milting, H.; Klauke, B.; Christensen, A.H.; Musebeck, J.; Walhorn, V.; Grannemann, S.; Munnich, T.; Saric, T.; Rasmussen, T.B.; Jensen, H.K.; et al. The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur. Heart J. 2015, 36, 872–881. [Google Scholar] [CrossRef]
- Stroud, M.J.; Fang, X.; Zhang, J.L.; Guimaraes-Camboa, N.; Veevers, J.; Dalton, N.D.; Gu, Y.; Bradford, W.H.; Peterson, K.L.; Evans, S.M.; et al. Luma is not essential for murine cardiac development and function. Cardiovasc. Res. 2018, 114, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.G.; Kho, C.; Hajjar, R.J.; Ishikawa, K. Experimental models of cardiac physiology and pathology. Heart Fail. Rev. 2019, 24, 601–615. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wilson, G.F.; Soerens, A.G.; Koonce, C.H.; Yu, J.; Palecek, S.P.; Thomson, J.A.; Kamp, T.J. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 2009, 104, e30–e41. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, S.P.; Malakhova, A.A.; Grigor’eva, E.V.; Shevchenko, A.I.; Dementyeva, E.V.; Sobolev, I.A.; Lebedev, I.N.; Shilov, A.G.; Zhimulev, I.F.; Zakian, S.M. Derivation of induced pluripotent stem cells from fetal human skin fibroblasts. Acta Nat. 2010, 2, 102–106. [Google Scholar] [CrossRef]
- Van Mil, A.; Balk, G.M.; Neef, K.; Buikema, J.W.; Asselbergs, F.W.; Wu, S.M.; Doevendans, P.A.; Sluijter, J.P.G. Modelling inherited cardiac disease using human induced pluripotent stem cell-derived cardiomyocytes: Progress, pitfalls, and potential. Cardiovasc. Res. 2018, 114, 1828–1842. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Wernig, M.; Lengner, C.J.; Hanna, J.; Lodato, M.A.; Steine, E.; Foreman, R.; Staerk, J.; Markoulaki, S.; Jaenisch, R. A drug-inducible transgenic system for direct reprogramming of multiple somatic cell types. Nat. Biotechnol. 2008, 26, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Nandan, M.O.; Yang, V.W. The role of Krüppel-like factors in the reprogramming of somatic cells to induced pluripotent stem cells. Histol. Histopathol. 2009, 24, 1343–1355. [Google Scholar] [CrossRef]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2011, 12, 253–265. [Google Scholar] [CrossRef]
- Jiang, J.; Chan, Y.S.; Loh, Y.H.; Cai, J.; Tong, G.Q.; Lim, C.A.; Robson, P.; Zhong, S.; Ng, H.H. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008, 10, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Chronis, C.; Soufi, A.; Bonora, G.; Edwards, M.; Smale, S.T.; Zaret, K.S.; Plath, K.; Pellegrini, M. Comparison of reprogramming factor targets reveals both species-specific and conserved mechanisms in early iPSC reprogramming. BMC Genom. 2018, 19, 956. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Ng, J.H.; Heng, J.C.; Ng, H.H. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell Stem Cell 2009, 4, 301–312. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Benda, C.; Duzinger, S.; Huang, Y.; Li, X.; Li, Y.; Guo, X.; Cao, G.; Chen, S.; Hao, L.; et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 2011, 22, 1221–1228. [Google Scholar] [CrossRef]
- Churko, J.M.; Burridge, P.W.; Wu, J.C. Generation of human iPSCs from human peripheral blood mononuclear cells using non-integrative Sendai virus in chemically defined conditions. Methods Mol. Biol. 2013, 1036, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Dadheech, N.; James Shapiro, A.M. Human Induced Pluripotent Stem Cells in the Curative Treatment of Diabetes and Potential Impediments Ahead. Adv. Exp. Med. Biol. 2019, 1144, 25–35. [Google Scholar] [CrossRef]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Petit, I.; Kesner, N.S.; Karry, R.; Robicsek, O.; Aberdam, E.; Müller, F.J.; Aberdam, D.; Ben-Shachar, D. Induced pluripotent stem cells from hair follicles as a cellular model for neurodevelopmental disorders. Stem Cell Res. 2012, 8, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.Y.; Lavoie, J.R.; Kwon, S.Y.; Chen, Z.; Manias, J.L.; Behbahani, J.; Ling, V.; Kandel, R.A.; Stewart, D.J.; Stanford, W.L. Feeder-independent derivation of induced-pluripotent stem cells from peripheral blood endothelial progenitor cells. Stem Cell Res. 2013, 10, 195–202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aoi, T.; Yae, K.; Nakagawa, M.; Ichisaka, T.; Okita, K.; Takahashi, K.; Chiba, T.; Yamanaka, S. Generation of pluripotent stem cells from adult mouse liver and stomach cells. Science 2008, 321, 699–702. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Zeng, F. Integration-free methods for generating induced pluripotent stem cells. Genom. Proteom. Bioinform. 2013, 11, 284–287. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef]
- Deng, X.Y.; Wang, H.; Wang, T.; Fang, X.T.; Zou, L.L.; Li, Z.Y.; Liu, C.B. Non-viral methods for generating integration-free, induced pluripotent stem cells. Curr. Stem Cell Res. Ther. 2015, 10, 153–158. [Google Scholar] [CrossRef]
- Kime, C.; Rand, T.A.; Ivey, K.N.; Srivastava, D.; Yamanaka, S.; Tomoda, K. Practical Integration-Free Episomal Methods for Generating Human Induced Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2015, 87, 21.22.1–21.22.21. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Hamada, Y.; Horiuchi, Y.; Matsuo-Takasaki, M.; Imoto, Y.; Satomi, K.; Arinami, T.; Hasegawa, M.; Fujioka, T.; Nakamura, Y.; et al. Generation of induced pluripotent stem cells from human nasal epithelial cells using a Sendai virus vector. PLoS ONE 2012, 7, e42855. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Fukuda, K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protoc. 2012, 7, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Macarthur, C.C.; Fontes, A.; Ravinder, N.; Kuninger, D.; Kaur, J.; Bailey, M.; Taliana, A.; Vemuri, M.C.; Lieu, P.T. Generation of human-induced pluripotent stem cells by a nonintegrating RNA Sendai virus vector in feeder-free or xeno-free conditions. Stem Cells Int. 2012, 2012, 564612. [Google Scholar] [CrossRef]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Hong, H.; Takahashi, K.; Yamanaka, S. Generation of mouse-induced pluripotent stem cells with plasmid vectors. Nat. Protoc. 2010, 5, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I., I; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Barragan Monasterio, M.; Tiscornia, G.; Montserrat Pulido, N.; Vassena, R.; Batlle Morera, L.; Rodriguez Piza, I.; Izpisua Belmonte, J.C. Generation of mouse-induced pluripotent stem cells by transient expression of a single nonviral polycistronic vector. Proc. Natl. Acad. Sci. USA 2009, 106, 8918–8922. [Google Scholar] [CrossRef] [PubMed]
- Meraviglia, V.; Zanon, A.; Lavdas, A.A.; Schwienbacher, C.; Silipigni, R.; Di Segni, M.; Chen, H.S.; Pramstaller, P.P.; Hicks, A.A.; Rossini, A. Generation of Induced Pluripotent Stem Cells from Frozen Buffy Coats using Non-integrating Episomal Plasmids. J. Vis. Exp. 2015, 5, 52885–52909. [Google Scholar] [CrossRef]
- Narsinh, K.H.; Jia, F.; Robbins, R.C.; Kay, M.A.; Longaker, M.T.; Wu, J.C. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011, 6, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Ni, Y.; Wang, J.; Guo, X. Feeder-free derivation of human induced pluripotent stem cells with messenger RNA. Sci Rep. 2012, 2, 657. [Google Scholar] [CrossRef] [PubMed]
- Card, D.A.; Hebbar, P.B.; Li, L.; Trotter, K.W.; Komatsu, Y.; Mishina, Y.; Archer, T.K. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell Biol. 2008, 28, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef]
- Han, M.J.; Kim, H.R.; O’Reilly, C.; Kim, C.H. Purification of functional reprogramming factors in mammalian cell using FLAG -Tag. Biochem. Biophys. Res. Commun. 2017, 492, 154–160. [Google Scholar] [CrossRef]
- Savić, N.; Schwank, G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl. Res. 2016, 168, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Weltner, J.; Balboa, D.; Katayama, S.; Bespalov, M.; Krjutškov, K.; Jouhilahti, E.M.; Trokovic, R.; Kere, J.; Otonkoski, T. Human pluripotent reprogramming with CRISPR activators. Nat. Commun. 2018, 9, 2643. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Mahas, A.; Neal Stewart, C., Jr.; Mahfouz, M.M. Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol. Adv. 2018, 36, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Müller-Werdan, U.; Klein, D.; Zander, M.; Werdan, K.; Hammer, C. Beating neonatal rat cardiomyocytes as a model to study the role of xenoreactive natural antibodies in xenotransplantation. Transplantation 1994, 58, 1403–1409. [Google Scholar]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Dias, P.; Desplantez, T.; El-Harasis, M.A.; Chowdhury, R.A.; Ullrich, N.D.; Cabestrero de Diego, A.; Peters, N.S.; Severs, N.J.; MacLeod, K.T.; Dupont, E. Characterisation of connexin expression and electrophysiological properties in stable clones of the HL-1 myocyte cell line. PLoS ONE 2014, 9, e90266. [Google Scholar] [CrossRef] [PubMed]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.H.; Gepstein, L. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef]
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE 2011, 6, e18293. [Google Scholar] [CrossRef]
- Hatani, T.; Miki, K.; Yoshida, Y. Induction of Human Induced Pluripotent Stem Cells to Cardiomyocytes Using Embryoid Bodies. Methods Mol. Biol. 2018, 1816, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Zhang, J.; Azarin, S.M.; Zhu, K.; Hazeltine, L.B.; Bao, X.; Hsiao, C.; Kamp, T.J.; Palecek, S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Horst, A.; Blinka, S.; Stamm, K.; Mahnke, D.; Schuman, J.; Gundry, R.; Tomita-Mitchell, A.; Lough, J. Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS ONE 2015, 10, e0118670. [Google Scholar] [CrossRef]
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes From Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 1482–1494. [Google Scholar] [CrossRef]
- Lee, J.H.; Protze, S.I.; Laksman, Z.; Backx, P.H.; Keller, G.M. Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 2017, 21, 179-194.e174. [Google Scholar] [CrossRef]
- Argenziano, M.; Lambers, E.; Hong, L.; Sridhar, A.; Zhang, M.; Chalazan, B.; Menon, A.; Savio-Galimberti, E.; Wu, J.C.; Rehman, J.; et al. Electrophysiologic Characterization of Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Atrial Cardiomyocytes. Stem Cell Rep. 2018, 10, 1867–1878. [Google Scholar] [CrossRef] [PubMed]
- Kleinsorge, M.; Cyganek, L. Subtype-Directed Differentiation of Human iPSCs into Atrial and Ventricular Cardiomyocytes. STAR Protoc. 2020, 1, 100026. [Google Scholar] [CrossRef] [PubMed]
- Protze, S.I.; Liu, J.; Nussinovitch, U.; Ohana, L.; Backx, P.H.; Gepstein, L.; Keller, G.M. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat. Biotechnol. 2017, 35, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Fang, Y.; Hou, X.; Yan, Y.; Xiao, H.; Zuo, D.; Wen, J.; Wang, L.; Zhou, Z.; Dang, X.; et al. Enrichment differentiation of human induced pluripotent stem cells into sinoatrial node-like cells by combined modulation of BMP, FGF, and RA signaling pathways. Stem Cell Res. Ther. 2020, 11, 284. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.Z.; Xie, Y.; Moyes, K.W.; Gold, J.D.; Askari, B.; Laflamme, M.A. Neuregulin/ErbB signaling regulates cardiac subtype specification in differentiating human embryonic stem cells. Circ. Res. 2010, 107, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Yassa, M.E.; Mansour, I.A.; Sewelam, N.I.; Hamza, H.; Gaafar, T. The impact of growth factors on human induced pluripotent stem cells differentiation into cardiomyocytes. Life Sci. 2018, 196, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rafatian, N.; Wang, E.Y.; Wu, Q.; Lai, B.F.L.; Lu, R.X.; Savoji, H.; Radisic, M. Towards chamber specific heart-on-a-chip for drug testing applications. Adv. Drug Deliv. Rev. 2020, 165–166, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, E.G.; Liang, P.; Lan, F.; Sanchez-Freire, V.; Simmons, C.; Gong, T.; Sharma, A.; Burridge, P.W.; Patlolla, B.; Lee, A.S.; et al. Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation 2013, 128, S3–S13. [Google Scholar] [CrossRef] [PubMed]
- Biendarra-Tiegs, S.M.; Li, X.; Ye, D.; Brandt, E.B.; Ackerman, M.J.; Nelson, T.J. Single-Cell RNA-Sequencing and Optical Electrophysiology of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Discordance Between Cardiac Subtype-Associated Gene Expression Patterns and Electrophysiological Phenotypes. Stem Cells Dev. 2019, 28, 659–673. [Google Scholar] [CrossRef]
- Cyganek, L.; Tiburcy, M.; Sekeres, K.; Gerstenberg, K.; Bohnenberger, H.; Lenz, C.; Henze, S.; Stauske, M.; Salinas, G.; Zimmermann, W.H.; et al. Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 2018, 3, 99941–99959. [Google Scholar] [CrossRef]
- Nafissi, N.A.; DeBenedittis, P.; Thomas, M.C.; Karra, R. Differentiation of Human Induced Pluripotent Stem Cells into Epicardial-Like Cells. Methods Mol. Biol. 2021, 2158, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Lalit, P.A.; Salick, M.R.; Nelson, D.O.; Squirrell, J.M.; Shafer, C.M.; Patel, N.G.; Saeed, I.; Schmuck, E.G.; Markandeya, Y.S.; Wong, R.; et al. Lineage Reprogramming of Fibroblasts into Proliferative Induced Cardiac Progenitor Cells by Defined Factors. Cell Stem Cell 2016, 18, 354–367. [Google Scholar] [CrossRef]
- Chen, W.; Bian, W.; Zhou, Y.; Zhang, J. Cardiac Fibroblasts and Myocardial Regeneration. Front. Bioeng. Biotechnol. 2021, 9, 599928. [Google Scholar] [CrossRef]
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10, 2238. [Google Scholar] [CrossRef]
- Nachlas, A.L.Y.; Li, S.; Jha, R.; Singh, M.; Xu, C.; Davis, M.E. Human iPSC-derived mesenchymal stem cells encapsulated in PEGDA hydrogels mature into valve interstitial-like cells. Acta Biomater. 2018, 71, 235–246. [Google Scholar] [CrossRef]
- Dash, B.C.; Jiang, Z.; Suh, C.; Qyang, Y. Induced pluripotent stem cell-derived vascular smooth muscle cells: Methods and application. Biochem. J. 2015, 465, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Quertermous, T.; Fischbein, M.P.; Wu, J.C. Generation of Vascular Smooth Muscle Cells From Induced Pluripotent Stem Cells: Methods, Applications, and Considerations. Circ. Res. 2021, 128, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Craft, A.M.; Sharma, P.; Elliott, D.A.; Stanley, E.G.; Elefanty, A.G.; Gramolini, A.; Keller, G. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS ONE 2011, 6, e23657. [Google Scholar] [CrossRef] [PubMed]
- Kita-Matsuo, H.; Barcova, M.; Prigozhina, N.; Salomonis, N.; Wei, K.; Jacot, J.G.; Nelson, B.; Spiering, S.; Haverslag, R.; Kim, C.; et al. Lentiviral vectors and protocols for creation of stable hESC lines for fluorescent tracking and drug resistance selection of cardiomyocytes. PLoS ONE 2009, 4, e5046. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High purity human-induced pluripotent stem cell-derived cardiomyocytes: Electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2006-2017. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef]
- Bedada, F.B.; Wheelwright, M.; Metzger, J.M. Maturation status of sarcomere structure and function in human iPSC-derived cardiac myocytes. Biochim. Biophys. Acta 2016, 1863, 1829–1838. [Google Scholar] [CrossRef]
- Bekhite, M.M.; Gonzalez Delgado, A.; Menz, F.; Kretzschmar, T.; Wu, J.M.F.; Bekfani, T.; Nietzsche, S.; Wartenberg, M.; Westermann, M.; Greber, B.; et al. Longitudinal metabolic profiling of cardiomyocytes derived from human-induced pluripotent stem cells. Basic Res. Cardiol. 2020, 115, 37. [Google Scholar] [CrossRef] [PubMed]
- Zuppinger, C.; Gibbons, G.; Dutta-Passecker, P.; Segiser, A.; Most, H.; Suter, T.M. Characterization of cytoskeleton features and maturation status of cultured human iPSC-derived cardiomyocytes. Eur. J. Histochem. 2017, 61, 2763. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W.T. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef]
- Kim, H.D.; Kim, D.J.; Lee, I.J.; Rah, B.J.; Sawa, Y.; Schaper, J. Human fetal heart development after mid-term: Morphometry and ultrastructural study. J. Mol. Cell Cardiol. 1992, 24, 949–965. [Google Scholar] [CrossRef]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.S.; Severs, N.J.; Rothery, S.M.; Lincoln, C.; Yacoub, M.H.; Green, C.R. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation 1994, 90, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Skoglund, G.; Lainé, J.; Darabi, R.; Fournier, E.; Perlingeiro, R.; Tabti, N. Physiological and ultrastructural features of human induced pluripotent and embryonic stem cell-derived skeletal myocytes in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 8275–8280. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, J.; Klein, L.J.; van der Bijl, M.; Huybregts, M.A.; Stooker, W.; Witkop, J.; Eijsman, L.; Visser, C.A.; Visser, F.C.; Stienen, G.J. Force production in mechanically isolated cardiac myocytes from human ventricular muscle tissue. Cardiovasc. Res. 1998, 38, 414–423. [Google Scholar] [CrossRef]
- Lundy, S.D.; Zhu, W.Z.; Regnier, M.; Laflamme, M.A. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef]
- Mills, R.J.; Titmarsh, D.M.; Koenig, X.; Parker, B.L.; Ryall, J.G.; Quaife-Ryan, G.A.; Voges, H.K.; Hodson, M.P.; Ferguson, C.; Drowley, L.; et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA 2017, 114, E8372–E8381. [Google Scholar] [CrossRef]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef]
- Jansen, J.A.; van Veen, T.A.B.; de Bakker, J.M.T.; van Rijen, H.V.M. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Hunkeler, N.M.; Kullman, J.; Murphy, A.M. Troponin I isoform expression in human heart. Circ. Res. 1991, 69, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Saggin, L.; Gorza, L.; Ausoni, S.; Schiaffino, S. Troponin I switching in the developing heart. J. Biol. Chem. 1989, 264, 16299–16302. [Google Scholar] [CrossRef]
- Mahdavi, V.; Lompre, A.M.; Chambers, A.P.; Nadal-Ginard, B. Cardiac myosin heavy chain isozymic transitions during development and under pathological conditions are regulated at the level of mRNA availability. Eur. Heart J. 1984, 5 (Suppl. F), 181–191. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A.; et al. Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E. Beta 1- and beta 2-adrenoceptors in the human heart: Properties, function, and alterations in chronic heart failure. Pharm. Rev. 1991, 43, 203–242. [Google Scholar] [PubMed]
- Engelhardt, S.; Böhm, M.; Erdmann, E.; Lohse, M.J. Analysis of beta-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: Progressive reduction of beta 1-adrenergic receptor mRNA in heart failure. J. Am. Coll Cardiol. 1996, 27, 146–154. [Google Scholar] [CrossRef]
- Drouin, E.; Charpentier, F.; Gauthier, C.; Laurent, K.; Le Marec, H. Electrophysiologic characteristics of cells spanning the left ventricular wall of human heart: Evidence for presence of M cells. J. Am. Coll Cardiol. 1995, 26, 185–192. [Google Scholar] [CrossRef]
- Goversen, B.; van der Heyden, M.A.G.; van Veen, T.A.B.; de Boer, T.P. The immature electrophysiological phenotype of iPSC-CMs still hampers in vitro drug screening: Special focus on I(K1). Pharm. Ther. 2018, 183, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-K.; Ng, K.-M.; Lai, W.-H.; Chan, Y.-C.; Lau, Y.-M.; Lian, Q.; Tse, H.-F.; Siu, C.-W. Calcium Homeostasis in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rev. Rep. 2011, 7, 976–986. [Google Scholar] [CrossRef]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human iPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef]
- Li, S.; Chen, G.; Li, R.A. Calcium signalling of human pluripotent stem cell-derived cardiomyocytes. J. Physiol. 2013, 591, 5279–5290. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.; Couch, L.; Terracciano, C.M.N. Excitation-contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes. Front. Cell Dev. Biol. 2015, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Machiraju, P.; Greenway, S.C. Current methods for the maturation of induced pluripotent stem cell-derived cardiomyocytes. World J. Stem Cells 2019, 11, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.S.; Aasum, E. Metabolic (in)flexibility of the diabetic heart. Cardiovasc Drugs Ther. 2008, 22, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Batten, B.E.; Albertini, D.F.; Ducibella, T. Patterns of organelle distribution in mouse embryos during preimplantation development. Am. J. Anat. 1987, 178, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Wilding, M.; Dale, B.; Marino, M.; di Matteo, L.; Alviggi, C.; Pisaturo, M.L.; Lombardi, L.; De Placido, G. Mitochondrial aggregation patterns and activity in human oocytes and preimplantation embryos. Hum. Reprod. 2001, 16, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Facucho-Oliveira, J.M.; Alderson, J.; Spikings, E.C.; Egginton, S.; St John, J.C. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J. Cell Sci. 2007, 120, 4025–4034. [Google Scholar] [CrossRef]
- Prigione, A.; Adjaye, J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int. J. Dev. Biol. 2010, 54, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Jaswal, J.S. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J. Cardiovasc. Pharm. 2010, 56, 130–140. [Google Scholar] [CrossRef]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Hu, D.; Linders, A.; Yamak, A.; Correia, C.; Kijlstra, J.D.; Garakani, A.; Xiao, L.; Milan, D.J.; van der Meer, P.; Serra, M.; et al. Metabolic Maturation of Human Pluripotent Stem Cell-Derived Cardiomyocytes by Inhibition of HIF1alpha and LDHA. Circ. Res. 2018, 123, 1066–1079. [Google Scholar] [CrossRef]
- Ulmer, B.M.; Stoehr, A.; Schulze, M.L.; Patel, S.; Gucek, M.; Mannhardt, I.; Funcke, S.; Murphy, E.; Eschenhagen, T.; Hansen, A. Contractile Work Contributes to Maturation of Energy Metabolism in hiPSC-Derived Cardiomyocytes. Stem Cell Rep. 2018, 10, 834–847. [Google Scholar] [CrossRef] [PubMed]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Bassett, A.R. Editing the genome of hiPSC with CRISPR/Cas9: Disease models. Mamm. Genome 2017, 28, 348–364. [Google Scholar] [CrossRef] [PubMed]
- De Masi, C.; Spitalieri, P.; Murdocca, M.; Novelli, G.; Sangiuolo, F. Application of CRISPR/Cas9 to human-induced pluripotent stem cells: From gene editing to drug discovery. Hum. Genom. 2020, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Maeder, M.L.; Mali, P.; Pruett-Miller, S.M.; Thibodeau-Beganny, S.; Chou, B.K.; Chen, G.; Ye, Z.; Park, I.H.; Daley, G.Q.; et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 2009, 5, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Siksnys, V. RNA-dependent DNA endonuclease Cas9 of the CRISPR system: Holy Grail of genome editing? Trends Microbiol. 2013, 21, 562–567. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Scesa, G.; Adami, R.; Bottai, D. iPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells 2021, 10, 1470–1489. [Google Scholar] [CrossRef] [PubMed]
- Carcamo-Orive, I.; Hoffman, G.E.; Cundiff, P.; Beckmann, N.D.; D’Souza, S.L.; Knowles, J.W.; Patel, A.; Papatsenko, D.; Abbasi, F.; Reaven, G.M.; et al. Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity. Cell Stem Cell 2017, 20, 518-532.e519. [Google Scholar] [CrossRef]
- DeBoever, C.; Li, H.; Jakubosky, D.; Benaglio, P.; Reyna, J.; Olson, K.M.; Huang, H.; Biggs, W.; Sandoval, E.; D’Antonio, M.; et al. Large-Scale Profiling Reveals the Influence of Genetic Variation on Gene Expression in Human Induced Pluripotent Stem Cells. Cell Stem Cell 2017, 20, 533-546.e537. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Marczenke, M.; Piccini, I.; Mengarelli, I.; Fell, J.; Röpke, A.; Seebohm, G.; Verkerk, A.O.; Greber, B. Cardiac Subtype-Specific Modeling of K(v)1.5 Ion Channel Deficiency Using Human Pluripotent Stem Cells. Front. Physiol. 2017, 8, 469. [Google Scholar] [CrossRef]
- Brandão, K.O.; van den Brink, L.; Miller, D.C.; Grandela, C.; van Meer, B.J.; Mol, M.P.H.; de Korte, T.; Tertoolen, L.G.J.; Mummery, C.L.; Sala, L.; et al. Isogenic Sets of hiPSC-CMs Harboring Distinct KCNH2 Mutations Differ Functionally and in Susceptibility to Drug-Induced Arrhythmias. Stem Cell Rep. 2020, 15, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Sun, Y.; Wang, X.; Wang, H.; Wang, J.; Gong, T.; Chen, X.; Zhang, P.; Su, L.; Fu, G.; et al. Patient-Specific and Gene-Corrected Induced Pluripotent Stem Cell-Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Short QT Syndrome. Circ. Res. 2019, 124, 66–78. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Musunuru, K. Genome Editing for the Study of Cardiovascular Diseases. Curr. Cardiol. Rep. 2017, 19, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal. Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Motta, B.M.; Pramstaller, P.P.; Hicks, A.A.; Rossini, A. The Impact of CRISPR/Cas9 Technology on Cardiac Research: From Disease Modelling to Therapeutic Approaches. Stem Cells Int. 2017, 2017, 8960236. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Dixon, J.E.; Medway, C.; Georgiou, O.; Patel, M.J.; Morgan, K.; Kemp, P.J.; Staniforth, A.; Mellor, I.; Denning, C. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur. Heart J. 2014, 35, 1078–1087. [Google Scholar] [CrossRef]
- Kelly, M.; Semsarian, C. Multiple mutations in genetic cardiovascular disease: A marker of disease severity? Circ. Cardiovasc. Genet. 2009, 2, 182–190. [Google Scholar] [CrossRef]
- Mullen, M.; Zhang, A.; Lui, G.K.; Romfh, A.W.; Rhee, J.-W.; Wu, J.C. Race and Genetics in Congenital Heart Disease: Application of iPSCs, Omics, and Machine Learning Technologies. Front. Cardiovasc. Med. 2021, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.T.; Chandy, M.; Wu, J.C. Patient and Disease-Specific Induced Pluripotent Stem Cells for Discovery of Personalized Cardiovascular Drugs and Therapeutics. Pharm. Rev. 2020, 72, 320–342. [Google Scholar] [CrossRef]
- Musunuru, K.; Sheikh, F.; Gupta, R.M.; Houser, S.R.; Maher, K.O.; Milan, D.J.; Terzic, A.; Wu, J.C. Induced Pluripotent Stem Cells for Cardiovascular Disease Modeling and Precision Medicine: A Scientific Statement From the American Heart Association. Circ. Genom Precis. Med. 2018, 11, e000043. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.W.; Durbin, M.D.; Hong, C.C. Genome Editing and Induced Pluripotent Stem Cell Technologies for Personalized Study of Cardiovascular Diseases. Curr. Cardiol. Rep. 2018, 20, 38. [Google Scholar] [CrossRef]
- Pourrier, M.; Fedida, D. The Emergence of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes (hiPSC-CMs) as a Platform to Model Arrhythmogenic Diseases. Int. J. Mol. Sci. 2020, 21, 657–684. [Google Scholar] [CrossRef]
- Prajapati, C.; Ojala, M.; Lappi, H.; Aalto-Setälä, K.; Pekkanen-Mattila, M. Electrophysiological evaluation of human induced pluripotent stem cell-derived cardiomyocytes obtained by different methods. Stem Cell Res. 2021, 51, 102176. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; D’Souza, S.L.; Ang, Y.S.; Schaniel, C.; Lee, D.F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Pacileo, G.; Russo, M.G.; Sarkozy, A.; Felicetti, M.; Di Salvo, G.; Morelli, C.; Calabrò, P.; Paladini, D.; Marino, B.; et al. Severe, early onset hypertrophic cardiomyopathy in a family with LEOPARD syndrome. J. Prenat. Med. 2008, 2, 24–26. [Google Scholar]
- Adamcova, M.; Skarkova, V.; Seifertova, J.; Rudolf, E. Cardiac Troponins are Among Targets of Doxorubicin-Induced Cardiotoxicity in hiPCS-CMs. Int. J. Mol. Sci. 2019, 20, 2638–2651. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Bu, M.; Yun, H. Sevoflurane prevents hypoxia/reoxygenation-induced cardiomyocyte apoptosis by inhibiting PI3KC3-mediated autophagy. Hum. Cell 2019, 32, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Naftali-Shani, N.; Molotski, N.; Nevo-Caspi, Y.; Arad, M.; Kuperstein, R.; Amit, U.; Huber, I.; Zeltzer, L.A.; Levich, A.; Abbas, H.; et al. Modeling Peripartum Cardiomyopathy With Human Induced Pluripotent Stem Cells Reveals Distinctive Abnormal Function of Cardiomyocytes. Circulation 2018, 138, 2721–2723. [Google Scholar] [CrossRef] [PubMed]
- Bekhite, M.; Gonzalez-Delgado, A.; Hubner, S.; Haxhikadrija, P.; Kretzschmar, T.; Muller, T.; Wu, J.M.F.; Bekfani, T.; Franz, M.; Wartenberg, M.; et al. The role of ceramide accumulation in human induced pluripotent stem cell-derived cardiomyocytes on mitochondrial oxidative stress and mitophagy. Free Radic. Biol. Med. 2021, 167, 66–80. [Google Scholar] [CrossRef]
- Granéli, C.; Hicks, R.; Brolén, G.; Synnergren, J.; Sartipy, P. Diabetic Cardiomyopathy Modelling Using Induced Pluripotent Stem Cell Derived Cardiomyocytes: Recent Advances and Emerging Models. Stem Cell Rev. Rep. 2019, 15, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Drawnel, F.M.; Boccardo, S.; Prummer, M.; Delobel, F.; Graff, A.; Weber, M.; Gérard, R.; Badi, L.; Kam-Thong, T.; Bu, L.; et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014, 9, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.M.; Lau, Y.M.; Dhandhania, V.; Cai, Z.J.; Lee, Y.K.; Lai, W.H.; Tse, H.F.; Siu, C.W. Empagliflozin Ammeliorates High Glucose Induced-Cardiac Dysfuntion in Human iPSC-Derived Cardiomyocytes. Sci. Rep. 2018, 8, 14872. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Marceau, C.; Hamaguchi, R.; Burridge, P.W.; Rajarajan, K.; Churko, J.M.; Wu, H.; Sallam, K.I.; Matsa, E.; Sturzu, A.C.; et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 2014, 115, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, A.; Sayed, N.; Matsa, E.; Sass, G.; Neofytou, E.; Clemons, K.V.; Correa-Oliveira, R.; Stevens, D.A.; Wu, J.C. Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes as a Model to Study Trypanosoma cruzi Infection. Stem Cell Rep. 2019, 12, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Vatta, M.; Towbin, J.A. Mutations in KCNE1 in long QT syndrome (LQTS): Insights into mechanism of LQTS and drug sensitivity? Heart Rhythm 2006, 3, 1041–1042. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Schwartz, P.J. 25th anniversary of the International Long-QT Syndrome Registry: An ongoing quest to uncover the secrets of long-QT syndrome. Circulation 2005, 111, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT Syndrome: Genetics and Future Perspective. Pediatr. Cardiol. 2019, 40, 1419–1430. [Google Scholar] [CrossRef]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef]
- Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules 2020, 10, 1144–1159. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Wilde, A.A.M.; Ackerman, M.J. The genetic architecture of long QT syndrome: A critical reappraisal. Trends Cardiovasc. Med. 2018, 28, 453–464. [Google Scholar] [CrossRef]
- Singh, M.; Morin, D.P.; Link, M.S. Sudden cardiac death in Long QT syndrome (LQTS), Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (CPVT). Prog. Cardiovasc. Dis. 2019, 62, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Eckardt, L.; Franz, M.R.; Mönnig, G.; Loh, P.; Wedekind, H.; Schulze-Bahr, E.; Breithardt, G.; Haverkamp, W. Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J. Cardiovasc. Electrophysiol. 2003, 14, 1027–1033. [Google Scholar] [CrossRef]
- Ma, D.; Wei, H.; Lu, J.; Huang, D.; Liu, Z.; Loh, L.J.; Islam, O.; Liew, R.; Shim, W.; Cook, S.A. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2015, 6, 39. [Google Scholar] [CrossRef]
- Wei, H.; Wu, J.; Liu, Z. Studying KCNQ1 Mutation and Drug Response in Type 1 Long QT Syndrome Using Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Methods Mol. Biol. 2018, 1684, 7–28. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, M.; Hsueh, B.; Jia, X.; Pasca, A.M.; Bernstein, J.A.; Hallmayer, J.; Dolmetsch, R.E. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 2011, 471, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Park, S.E.; Isseroff, Y.; Morikawa, K.; Yazawa, M. Inhibition of CDK5 Alleviates the Cardiac Phenotypes in Timothy Syndrome. Stem Cell Rep. 2017, 9, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, M.; Sala, L.; Dreizehnter, L.; Crotti, L.; Sinnecker, D.; Mura, M.; Pane, L.S.; Altomare, C.; Torre, E.; Mostacciuolo, G.; et al. Elucidating arrhythmogenic mechanisms of long-QT syndrome CALM1-F142L mutation in patient-specific induced pluripotent stem cell-derived cardiomyocytes. Cardiovasc. Res. 2017, 113, 531–541. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Makiyama, T.; Harita, T.; Sasaki, K.; Wuriyanghai, Y.; Hayano, M.; Nishiuchi, S.; Kohjitani, H.; Hirose, S.; Chen, J.; et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum. Mol. Genet. 2017, 26, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Hayano, M.; Makiyama, T.; Kamakura, T.; Watanabe, H.; Sasaki, K.; Funakoshi, S.; Wuriyanghai, Y.; Nishiuchi, S.; Harita, T.; Yamamoto, Y.; et al. Development of a Patient-Derived Induced Pluripotent Stem Cell Model for the Investigation of SCN5A-D1275N-Related Cardiac Sodium Channelopathy. Circ. J. 2017, 81, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Veerman, C.C.; Mengarelli, I.; Guan, K.; Stauske, M.; Barc, J.; Tan, H.L.; Wilde, A.A.M.; Verkerk, A.O.; Bezzina, C.R. hiPSC-derived cardiomyocytes from Brugada Syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Sci. Rep. 2016, 6, 30967. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Makiyama, T.; Yoshida, Y.; Wuriyanghai, Y.; Kamakura, T.; Nishiuchi, S.; Hayano, M.; Harita, T.; Yamamoto, Y.; Kohjitani, H.; et al. Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a Potential Therapeutic Agent for Catecholaminergic Polymorphic Ventricular Tachycardia. PLoS ONE 2016, 11, e0164795. [Google Scholar] [CrossRef]
- Seidman, C.E.; Seidman, J.G. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ. Res. 2011, 108, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.G.W.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef]
- Phelan, D.G.; Anderson, D.J.; Howden, S.E.; Wong, R.C.; Hickey, P.F.; Pope, K.; Wilson, G.R.; Pébay, A.; Davis, A.M.; Petrou, S.; et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur. Heart J. 2016, 37, 2586–2590. [Google Scholar] [CrossRef] [PubMed]
- Josowitz, R.; Mulero-Navarro, S.; Rodriguez, N.A.; Falce, C.; Cohen, N.; Ullian, E.M.; Weiss, L.A.; Rauen, K.A.; Sobie, E.A.; Gelb, B.D. Autonomous and Non-autonomous Defects Underlie Hypertrophic Cardiomyopathy in BRAF-Mutant hiPSC-Derived Cardiomyocytes. Stem Cell Rep. 2016, 7, 355–369. [Google Scholar] [CrossRef]
- Klein, S.; Dvornik, J.L.; Yarrabothula, A.R.; Schaniel, C. A Marfan syndrome human induced pluripotent stem cell line with a heterozygous FBN1 c.4082G>A mutation, ISMMSi002-B, for disease modeling. Stem Cell Res. 2017, 23, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Hick, A.; Wattenhofer-Donzé, M.; Chintawar, S.; Tropel, P.; Simard, J.P.; Vaucamps, N.; Gall, D.; Lambot, L.; André, C.; Reutenauer, L.; et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich’s ataxia. Dis. Model. Mech 2013, 6, 608–621. [Google Scholar] [CrossRef]
- Chou, S.J.; Yu, W.C.; Chang, Y.L.; Chen, W.Y.; Chang, W.C.; Chien, Y.; Yen, J.C.; Liu, Y.Y.; Chen, S.J.; Wang, C.Y.; et al. Energy utilization of induced pluripotent stem cell-derived cardiomyocyte in Fabry disease. Int. J. Cardiol. 2017, 232, 255–263. [Google Scholar] [CrossRef]
- Li, S.; Pan, H.; Tan, C.; Sun, Y.; Song, Y.; Zhang, X.; Yang, W.; Wang, X.; Li, D.; Dai, Y.; et al. Mitochondrial Dysfunctions Contribute to Hypertrophic Cardiomyopathy in Patient iPSC-Derived Cardiomyocytes with MT-RNR2 Mutation. Stem Cell Rep. 2018, 10, 808–821. [Google Scholar] [CrossRef]
- Wu, H.; Yang, H.; Rhee, J.W.; Zhang, J.Z.; Lam, C.K.; Sallam, K.; Chang, A.C.Y.; Ma, N.; Lee, J.; Zhang, H.; et al. Modelling diastolic dysfunction in induced pluripotent stem cell-derived cardiomyocytes from hypertrophic cardiomyopathy patients. Eur. Heart J. 2019, 40, 3685–3695. [Google Scholar] [CrossRef] [PubMed]
- Dementyeva, E.V.; Medvedev, S.P.; Kovalenko, V.R.; Vyatkin, Y.V.; Kretov, E.I.; Slotvitsky, M.M.; Shtokalo, D.N.; Pokushalov, E.A.; Zakian, S.M. Applying Patient-Specific Induced Pluripotent Stem Cells to Create a Model of Hypertrophic Cardiomyopathy. Biochemistry 2019, 84, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Dambrot, C.; Braam, S.R.; Tertoolen, L.G.; Birket, M.; Atsma, D.E.; Mummery, C.L. Serum supplemented culture medium masks hypertrophic phenotypes in human pluripotent stem cell derived cardiomyocytes. J. Cell Mol. Med. 2014, 18, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Birket, M.J.; Ribeiro, M.C.; Kosmidis, G.; Ward, D.; Leitoguinho, A.R.; van de Pol, V.; Dambrot, C.; Devalla, H.D.; Davis, R.P.; Mastroberardino, P.G.; et al. Contractile Defect Caused by Mutation in MYBPC3 Revealed under Conditions Optimized for Human PSC-Cardiomyocyte Function. Cell Rep. 2015, 13, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.; Ross, S.B.; Lim, S.; Semsarian, C. Generation of an induced pluripotent stem cell line from a hypertrophic cardiomyopathy patient with a pathogenic myosin binding protein C (MYBPC3) p.Arg502Trp mutation. Stem Cell Res. 2018, 33, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Seeger, T.; Shrestha, R.; Lam, C.K.; Chen, C.; McKeithan, W.L.; Lau, E.; Wnorowski, A.; McMullen, G.; Greenhaw, M.; Lee, J.; et al. A Premature Termination Codon Mutation in MYBPC3 Causes Hypertrophic Cardiomyopathy via Chronic Activation of Nonsense-Mediated Decay. Circulation 2019, 139, 799–811. [Google Scholar] [CrossRef]
- Ojala, M.; Prajapati, C.; Pölönen, R.P.; Rajala, K.; Pekkanen-Mattila, M.; Rasku, J.; Larsson, K.; Aalto-Setälä, K. Mutation-Specific Phenotypes in hiPSC-Derived Cardiomyocytes Carrying Either Myosin-Binding Protein C Or α-Tropomyosin Mutation for Hypertrophic Cardiomyopathy. Stem Cells Int. 2016, 2016, 1684792. [Google Scholar] [CrossRef]
- Prajapati, C.; Ojala, M.; Aalto-Setälä, K. Divergent effects of adrenaline in human induced pluripotent stem cell-derived cardiomyocytes obtained from hypertrophic cardiomyopathy. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Müller, O.J.; Münch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.K.; Puckelwartz, M.J.; Mehta, A.; Ramachandra, C.J.A.; Jagadeesan, A.; Fritsche-Danielson, R.; Bhat, R.V.; Wong, P.; Kandoi, S.; Schwanekamp, J.A.; et al. Association of Cardiomyopathy With MYBPC3 D389V and MYBPC3Δ25bpIntronic Deletion in South Asian Descendants. JAMA Cardiol. 2018, 3, 481–488. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Fraser, S.T.; Nowak, N.; Semsarian, C. Generation of induced pluripotent stem cells (iPSCs) from a hypertrophic cardiomyopathy patient with the pathogenic variant p.Val698Ala in beta-myosin heavy chain (MYH7) gene. Stem Cell Res. 2017, 20, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.C.; Breitbart, A.; De Lange, W.J.; Hofsteen, P.; Futakuchi-Tsuchida, A.; Xu, J.; Schopf, C.; Razumova, M.V.; Jiao, A.; Boucek, R.; et al. Novel Adult-Onset Systolic Cardiomyopathy Due to MYH7 E848G Mutation in Patient-Derived Induced Pluripotent Stem Cells. JACC Basic Transl. Sci. 2018, 3, 728–740. [Google Scholar] [CrossRef]
- Pioner, J.M.; Racca, A.W.; Klaiman, J.M.; Yang, K.C.; Guan, X.; Pabon, L.; Muskheli, V.; Zaunbrecher, R.; Macadangdang, J.; Jeong, M.Y.; et al. Isolation and Mechanical Measurements of Myofibrils from Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2016, 6, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Zhou, W.; Bos, J.M.; Ye, D.; Tester, D.J.; Hrstka, S.; Maleszewski, J.J.; Ommen, S.R.; Nishimura, R.A.; Schaff, H.V.; Kim, C.S.; et al. Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with MYL2-R58Q-Mediated Apical Hypertrophic Cardiomyopathy Show Hypertrophy, Myofibrillar Disarray, and Calcium Perturbations. J. Cardiovasc. Transl. Res. 2019, 12, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, J.Z.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the Pathogenicity of a Genomic Variant of Uncertain Significance Using CRISPR/Cas9 and Human-Induced Pluripotent Stem Cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef] [PubMed]
- Ben Jehuda, R.; Eisen, B.; Shemer, Y.; Mekies, L.N.; Szantai, A.; Reiter, I.; Cui, H.; Guan, K.; Haron-Khun, S.; Freimark, D.; et al. CRISPR correction of the PRKAG2 gene mutation in the patient’s induced pluripotent stem cell-derived cardiomyocytes eliminates electrophysiological and structural abnormalities. Heart Rhythm 2018, 15, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Lowe, A.; Sheng, C.C.; Gupta, R.M.; Kuppusamy, R.; O’Sullivan, J.; Rowe, G.; Wakimoto, H.; Gorham, J.; et al. Integrative Analysis of PRKAG2 Cardiomyopathy iPS and Microtissue Models Identifies AMPK as a Regulator of Metabolism, Survival, and Fibrosis. Cell Rep. 2017, 19, 2410. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Baskfield, A.; Lin, Y.; Beers, J.; Zou, J.; Liu, C.; Jaffré, F.; Roberts, A.E.; Ottinger, E.A.; Kontaridis, M.I.; et al. Generation of an induced pluripotent stem cell line (TRNDi003-A) from a Noonan syndrome with multiple lentigines (NSML) patient carrying a p.Q510P mutation in the PTPN11 gene. Stem Cell Res. 2019, 34, 101374. [Google Scholar] [CrossRef] [PubMed]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. Cell Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Karakikes, I.; Termglinchan, V.; Cepeda, D.A.; Lee, J.; Diecke, S.; Hendel, A.; Itzhaki, I.; Ameen, M.; Shrestha, R.; Wu, H.; et al. A Comprehensive TALEN-Based Knockout Library for Generating Human-Induced Pluripotent Stem Cell-Based Models for Cardiovascular Diseases. Circ. Res. 2017, 120, 1561–1571. [Google Scholar] [CrossRef]
- Garg, P.; Garg, V.; Shrestha, R.; Sanguinetti, M.C.; Kamp, T.J.; Wu, J.C. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes as Models for Cardiac Channelopathies: A Primer for Non-Electrophysiologists. Circ. Res. 2018, 123, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Ciarambino, T.; Menna, G.; Sansone, G.; Giordano, M. Cardiomyopathies: An Overview. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Lakdawala, N.K.; Winterfield, J.R.; Funke, B.H. Dilated cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2013, 6, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L.; Towbin, J.A. Dilated cardiomyopathy. Lancet 2010, 375, 752–762. [Google Scholar] [CrossRef]
- Kellermayer, D.; Smith, J.E., 3rd; Granzier, H. Titin mutations and muscle disease. Pflug. Arch. 2019, 471, 673–682. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Morales, A. LMNA-Related Dilated Cardiomyopathy. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhao, Y.; Feng, Y.; Zhang, Y.M.; Ding, X.X.; Song, Y.Z.; Zhang, A.M.; Liu, L.; Zhang, H.; Ding, J.H.; Xia, X.S. Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int. J. Mol. Med. 2015, 36, 1479–1486. [Google Scholar] [CrossRef]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef]
- Ehlermann, P.; Weichenhan, D.; Zehelein, J.; Steen, H.; Pribe, R.; Zeller, R.; Lehrke, S.; Zugck, C.; Ivandic, B.T.; Katus, H.A. Adverse events in families with hypertrophic or dilated cardiomyopathy and mutations in the MYBPC3 gene. BMC Med. Genet. 2008, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, H.; Song, Y.; Feng, Y.; Ding, X.; Pang, M.; Zhang, Y.; Zhang, H.; Ding, J.; Xia, X. Identification of novel mutations including a double mutation in patients with inherited cardiomyopathy by a targeted sequencing approach using the Ion Torrent PGM system. Int. J. Mol. Med. 2016, 37, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Tse, H.F.; Ho, J.C.; Choi, S.W.; Lee, Y.K.; Butler, A.W.; Ng, K.M.; Siu, C.W.; Simpson, M.A.; Lai, W.H.; Chan, Y.C.; et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum. Mol. Genet. 2013, 22, 1395–1403. [Google Scholar] [CrossRef]
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Li, X.; Yücel, G.; Lang, S.; Sattler, K.; Schünemann, J.D.; Zimmermann, W.H.; Cyganek, L.; et al. Ion Channel Dysfunctions in Dilated Cardiomyopathy in Limb-Girdle Muscular Dystrophy. Circ. Genom Precis. Med. 2018, 11, e001893. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Virtanen, L.; Prajapati, C.; Kiamehr, M.; Gullmets, J.; West, G.; Kreutzer, J.; Pekkanen-Mattila, M.; Heliö, T.; Kallio, P.; et al. Modeling of LMNA-Related Dilated Cardiomyopathy Using Human Induced Pluripotent Stem Cells. Cells 2019, 8, 594–615. [Google Scholar] [CrossRef]
- Ho, J.C.; Zhou, T.; Lai, W.H.; Huang, Y.; Chan, Y.C.; Li, X.; Wong, N.L.; Li, Y.; Au, K.W.; Guo, D.; et al. Generation of induced pluripotent stem cell lines from 3 distinct laminopathies bearing heterogeneous mutations in lamin A/C. Aging 2011, 3, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Siu, C.W.; Lee, Y.K.; Ho, J.C.; Lai, W.H.; Chan, Y.C.; Ng, K.M.; Wong, L.Y.; Au, K.W.; Lau, Y.M.; Zhang, J.; et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging 2012, 4, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Lau, Y.M.; Cai, Z.J.; Lai, W.H.; Wong, L.Y.; Tse, H.F.; Ng, K.M.; Siu, C.W. Modeling Treatment Response for Lamin A/C Related Dilated Cardiomyopathy in Human Induced Pluripotent Stem Cells. J. Am. Heart Assoc. 2017, 6, 5677–5712. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef]
- Ceholski, D.K.; Turnbull, I.C.; Kong, C.W.; Koplev, S.; Mayourian, J.; Gorski, P.A.; Stillitano, F.; Skodras, A.A.; Nonnenmacher, M.; Cohen, N.; et al. Functional and transcriptomic insights into pathogenesis of R9C phospholamban mutation using human induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 2018, 119, 147–154. [Google Scholar] [CrossRef]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef] [PubMed]
- Stillitano, F.; Turnbull, I.C.; Karakikes, I.; Nonnenmacher, M.; Backeris, P.; Hulot, J.S.; Kranias, E.G.; Hajjar, R.J.; Costa, K.D. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. Eur. Heart J. 2016, 37, 3282–3284. [Google Scholar] [CrossRef] [PubMed]
- Streckfuss-Bömeke, K.; Tiburcy, M.; Fomin, A.; Luo, X.; Li, W.; Fischer, C.; Özcelik, C.; Perrot, A.; Sossalla, S.; Haas, J.; et al. Severe DCM phenotype of patient harboring RBM20 mutation S635A can be modeled by patient-specific induced pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell Cardiol. 2017, 113, 9–21. [Google Scholar] [CrossRef]
- Wyles, S.P.; Li, X.; Hrstka, S.C.; Reyes, S.; Oommen, S.; Beraldi, R.; Edwards, J.; Terzic, A.; Olson, T.M.; Nelson, T.J. Modeling structural and functional deficiencies of RBM20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum. Mol. Genet. 2016, 25, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra147. [Google Scholar] [CrossRef]
- Broughton, K.M.; Li, J.; Sarmah, E.; Warren, C.M.; Lin, Y.H.; Henze, M.P.; Sanchez-Freire, V.; Solaro, R.J.; Russell, B. A myosin activator improves actin assembly and sarcomere function of human-induced pluripotent stem cell-derived cardiomyocytes with a troponin T point mutation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H107–H117. [Google Scholar] [CrossRef] [PubMed]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef]
- Schick, R.; Mekies, L.N.; Shemer, Y.; Eisen, B.; Hallas, T.; Ben Jehuda, R.; Ben-Ari, M.; Szantai, A.; Willi, L.; Shulman, R.; et al. Functional abnormalities in induced Pluripotent Stem Cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE 2018, 13, e0205719. [Google Scholar] [CrossRef] [PubMed]
- Camman, M.; Joanne, P.; Agbulut, O.; Hélary, C. 3D models of dilated cardiomyopathy: Shaping the chemical, physical and topographical properties of biomaterials to mimic the cardiac extracellular matrix. Bioact. Mater. 2022, 7, 275–291. [Google Scholar] [CrossRef]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Era, T.; Kimura, S.; Eto, Y.; Ida, H.; Ohashi, T. Disease modeling and lentiviral gene transfer in patient-specific induced pluripotent stem cells from late-onset Pompe disease patient. Mol. Ther. Methods Clin. Dev. 2015, 2, 15023. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.M.; Green, K.J. Desmosomes in the heart: A review of clinical and mechanistic analyses. Cell Commun. Adhes 2014, 21, 109–128. [Google Scholar] [CrossRef]
- Horikoshi, Y.; Yan, Y.; Terashvili, M.; Wells, C.; Horikoshi, H.; Fujita, S.; Bosnjak, Z.J.; Bai, X. Fatty Acid-Treated Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes Exhibit Adult Cardiomyocyte-Like Energy Metabolism Phenotypes. Cells 2019, 8, 1095–1116. [Google Scholar] [CrossRef]
- Feyen, D.A.M.; McKeithan, W.L.; Bruyneel, A.A.N.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.; Pabon, L.; Fischer, K.A.; Reinecke, H.; Regnier, M.; Sniadecki, N.J.; Ruohola-Baker, H.; Murry, C.E. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2014, 72, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, A.; Ojamaa, K. Thyroid Hormone Stimulates Protein Synthesis in the Cardiomyocyte by Activating the Akt-mTOR and p70S6K Pathways. J. Biol. Chem. 2006, 281, 20666–20672. [Google Scholar] [CrossRef]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.-C.; Ting, S.; Lee, Y.-K.; Ng, K.-M.; Zhang, J.; Chen, Z.; Siu, C.-W.; Oh, S.K.W.; Tse, H.-F. Electrical Stimulation Promotes Maturation of Cardiomyocytes Derived from Human Embryonic Stem Cells. J. Cardiovasc. Transl. Res. 2013, 6, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Kroll, K.; Chabria, M.; Wang, K.; Häusermann, F.; Schuler, F.; Polonchuk, L. Electro-mechanical conditioning of human iPSC-derived cardiomyocytes for translational research. Prog. Biophys. Mol. Biol. 2017, 130, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kong, C.W.; Tong, M.H.; Chooi, W.H.; Huang, N.; Li, R.A.; Chan, B.P. Maturation of human embryonic stem cell-derived cardiomyocytes (hESC-CMs) in 3D collagen matrix: Effects of niche cell supplementation and mechanical stimulation. Acta Biomater. 2017, 49, 204–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Peres Moreno Maia-Joca, R.; Ong, C.S.; Wilson, I.; DiSilvestre, D.; Tomaselli, G.F.; Reich, D.H. Enhancement of human iPSC-derived cardiomyocyte maturation by chemical conditioning in a 3D environment. J. Mol. Cell. Cardiol. 2020, 138, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sacchetto, C.; Vitiello, L.; de Windt, L.J.; Rampazzo, A.; Calore, M. Modeling Cardiovascular Diseases with hiPSC-Derived Cardiomyocytes in 2D and 3D Cultures. Int. J. Mol. Sci. 2020, 21, 3404–3436. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xu, Z.; Xiao, L.; Shi, T.; Xiao, H.; Wang, Y.; Li, Y.; Xue, F.; Zeng, W. Review on the Vascularization of Organoids and Organoids-on-a-Chip. Front. Bioeng. Biotechnol. 2021, 9, 1–10. [Google Scholar] [CrossRef]
- Thomas, D.; Cunningham, N.J.; Shenoy, S.; Wu, J.C. Human-induced pluripotent stem cells in cardiovascular research: Current approaches in cardiac differentiation, maturation strategies, and scalable production. Cardiovasc. Res. 2021, Online ahead of print, 1–17. [Google Scholar] [CrossRef]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-culture of hiPSC-Derived Cardiomyocytes With Cardiac Fibroblasts Improves Tissue-Like Features of Cardiac Spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Decarli, M.C.; do Amaral, R.L.F.; Dos Santos, D.P.; Tofani, L.B.; Katayama, E.; Rezende, R.A.; Silva, J.; Swiech, K.; Suazo, C.A.T.; Mota, C.; et al. Cell spheroids as a versatile research platform: Formation mechanisms, high throughput production, characterization and applications. Biofabrication 2021, 13, 032002. [Google Scholar] [CrossRef]
- Cho, S.; Lee, C.; Skylar-Scott, M.A.; Heilshorn, S.C.; Wu, J.C. Reconstructing the heart using iPSCs: Engineering strategies and applications. J. Mol. Cell Cardiol. 2021, 157, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Broguiere, N.; Miyamoto, M.; Boni, A.; Guiet, R.; Girgin, M.; Kelly, R.G.; Kwon, C.; Lutolf, M.P. Capturing Cardiogenesis in Gastruloids. Cell Stem Cell 2021, 28, 230-240.e236. [Google Scholar] [CrossRef]
- Zhao, D.; Lei, W.; Hu, S. Cardiac organoid — a promising perspective of preclinical model. Stem Cell Res. Ther. 2021, 12, 272. [Google Scholar] [CrossRef] [PubMed]
- Drakhlis, L.; Biswanath, S.; Farr, C.-M.; Lupanow, V.; Teske, J.; Ritzenhoff, K.; Franke, A.; Manstein, F.; Bolesani, E.; Kempf, H.; et al. Human heart-forming organoids recapitulate early heart and foregut development. Nat. Biotechnol. 2021, 39, 737–746. [Google Scholar] [CrossRef]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Aguirre, A. Heart Organoids and Engineered Heart Tissues: Novel Tools for Modeling Human Cardiac Biology and Disease. Biomolecules 2021, 11, 1277. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. NeuroReport 2018, 29, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Kitsuka, T.; Itoh, M.; Amamoto, S.; Arai, K.I.; Oyama, J.; Node, K.; Toda, S.; Morita, S.; Nishida, T.; Nakayama, K. 2-Cl-C.OXT-A stimulates contraction through the suppression of phosphodiesterase activity in human induced pluripotent stem cell-derived cardiac organoids. PLoS ONE 2019, 14, e0213114. [Google Scholar] [CrossRef]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.-J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.-Y.; Lombroso, A.P.; Hwang, S.-M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383-398.e387. [Google Scholar] [CrossRef]
- Mannhardt, I.; Breckwoldt, K.; Letuffe-Brenière, D.; Schaaf, S.; Schulz, H.; Neuber, C.; Benzin, A.; Werner, T.; Eder, A.; Schulze, T.; et al. Human Engineered Heart Tissue: Analysis of Contractile Force. Stem Cell Rep. 2016, 7, 29–42. [Google Scholar] [CrossRef]
- Weinberger, F.; Mannhardt, I.; Eschenhagen, T. Engineering Cardiac Muscle Tissue. Circ. Res. 2017, 120, 1487–1500. [Google Scholar] [CrossRef]
- Tadevosyan, K.; Iglesias-García, O.; Mazo, M.M.; Prósper, F.; Raya, A. Engineering and Assessing Cardiac Tissue Complexity. Int. J. Mol. Sci. 2021, 22, 1479. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Fink, C.; Remmers, U.; Scholz, H.; Wattchow, J.; Weil, J.; Zimmermann, W.; Dohmen, H.H.; Schäfer, H.; Bishopric, N.; et al. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: A new heart muscle model system. FASEB J. 1997, 11, 683–694. [Google Scholar] [CrossRef]
- Majid, Q.A.; Fricker, A.T.R.; Gregory, D.A.; Davidenko, N.; Hernandez Cruz, O.; Jabbour, R.J.; Owen, T.J.; Basnett, P.; Lukasiewicz, B.; Stevens, M.; et al. Natural Biomaterials for Cardiac Tissue Engineering: A Highly Biocompatible Solution. Front. Cardiovasc. Med. 2020, 7, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Roshandel, M.; Dorkoosh, F. Cardiac tissue engineering, biomaterial scaffolds, and their fabrication techniques. Polym. Adv. Technol. 2021, 32, 2290–2305. [Google Scholar] [CrossRef]
- Liu, H.; Bolonduro, O.A.; Hu, N.; Ju, J.; Rao, A.A.; Duffy, B.M.; Huang, Z.; Black, L.D.; Timko, B.P. Heart-on-a-Chip Model with Integrated Extra- and Intracellular Bioelectronics for Monitoring Cardiac Electrophysiology under Acute Hypoxia. Nano Lett. 2020, 20, 2585–2593. [Google Scholar] [CrossRef] [PubMed]
- Martewicz, S.; Michielin, F.; Serena, E.; Zambon, A.; Mongillo, M.; Elvassore, N. Reversible alteration of calcium dynamics in cardiomyocytes during acute hypoxia transient in a microfluidic platform. Integr. Biol. 2012, 4, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Mousavi Shaegh, S.A.; De Ferrari, F.; Zhang, Y.S.; Nabavinia, M.; Binth Mohammad, N.; Ryan, J.; Pourmand, A.; Laukaitis, E.; Banan Sadeghian, R.; Nadhman, A.; et al. A microfluidic optical platform for real-time monitoring of pH and oxygen in microfluidic bioreactors and organ-on-chip devices. Biomicrofluidics 2016, 10, 044111. [Google Scholar] [CrossRef] [PubMed]
- Sticker, D.; Rothbauer, M.; Ehgartner, J.; Steininger, C.; Liske, O.; Liska, R.; Neuhaus, W.; Mayr, T.; Haraldsson, T.; Kutter, J.P.; et al. Oxygen Management at the Microscale: A Functional Biochip Material with Long-Lasting and Tunable Oxygen Scavenging Properties for Cell Culture Applications. ACS Appl. Mater. Interfaces 2019, 11, 9730–9739. [Google Scholar] [CrossRef] [PubMed]
- Schneider, O.; Zeifang, L.; Fuchs, S.; Sailer, C.; Loskill, P. User-Friendly and Parallelized Generation of Human Induced Pluripotent Stem Cell-Derived Microtissues in a Centrifugal Heart-on-a-Chip. Tissue Eng. Part. A 2019, 25, 786–798. [Google Scholar] [CrossRef]
- Vollert, I.; Seiffert, M.; Bachmair, J.; Sander, M.; Eder, A.; Conradi, L.; Vogelsang, A.; Schulze, T.; Uebeler, J.; Holnthoner, W.; et al. In vitro perfusion of engineered heart tissue through endothelialized channels. Tissue Eng. Part A 2014, 20, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Engineered Tissue Patch for Cardiac Cell Therapy. Curr. Treat. Options Cardiovasc. Med. 2015, 17, 399. [Google Scholar] [CrossRef]
- Shadrin, I.Y.; Allen, B.W.; Qian, Y.; Jackman, C.P.; Carlson, A.L.; Juhas, M.E.; Bursac, N. Cardiopatch platform enables maturation and scale-up of human pluripotent stem cell-derived engineered heart tissues. Nat. Commun. 2017, 8, 1825. [Google Scholar] [CrossRef] [PubMed]
- Noor, N.; Shapira, A.; Edri, R.; Gal, I.; Wertheim, L.; Dvir, T. 3D Printing of Personalized Thick and Perfusable Cardiac Patches and Hearts. Adv. Sci. 2019, 6, 1900344. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Inglese, J.; Walters, M.A. Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discov. 2015, 14, 279–294. [Google Scholar] [CrossRef]
- Raposo, V.L. Safe Drugs Versus Innovative Drugs (Can We Have Both?). Adv. Pharm. Bull. 2020, 10, 334–337. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharm. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Abrams, R.M.; Babiarz, J.E.; Cohen, J.D.; Kameoka, S.; Sanders, M.J.; Chiao, E.; Kolaja, K.L. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2011, 123, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Weaver, R.J.; Valentin, J.P. Today’s Challenges to De-Risk and Predict Drug Safety in Human “Mind-the-Gap“. Toxicol. Sci. 2019, 167, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Braam, S.R.; Passier, R.; Mummery, C.L. Cardiomyocytes from human pluripotent stem cells in regenerative medicine and drug discovery. Trends Pharm. Sci 2009, 30, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, J.K.; Tsaioun, K.; Hajjar, R.J. Cardionomics: A new integrative approach for screening cardiotoxicity of drug candidates. Expert Opin. Drug Metab. Toxicol. 2009, 5, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Vargas, H.M.; Bass, A.S.; Koerner, J.; Matis-Mitchell, S.; Pugsley, M.K.; Skinner, M.; Burnham, M.; Bridgland-Taylor, M.; Pettit, S.; Valentin, J.P. Evaluation of drug-induced QT interval prolongation in animal and human studies: A literature review of concordance. Br. J. Pharm. 2015, 172, 4002–4011. [Google Scholar] [CrossRef]
- Toplak, Ž.; Hendrickx, L.A.; Abdelaziz, R.; Shi, X.; Peigneur, S.; Tomašič, T.; Tytgat, J.; Peterlin-Mašič, L.; Pardo, L.A. Overcoming challenges of HERG potassium channel liability through rational design: Eag1 inhibitors for cancer treatment. Med. Res. Rev. 2021, 42, 183–226. [Google Scholar] [CrossRef] [PubMed]
- Cubeddu, L.X. Drug-induced Inhibition and Trafficking Disruption of ion Channels: Pathogenesis of QT Abnormalities and Drug-induced Fatal Arrhythmias. Curr. Cardiol. Rev. 2016, 12, 141–154. [Google Scholar] [CrossRef]
- Johannesen, L.; Vicente, J.; Gray, R.A.; Galeotti, L.; Loring, Z.; Garnett, C.E.; Florian, J.; Ugander, M.; Stockbridge, N.; Strauss, D.G. Improving the assessment of heart toxicity for all new drugs through translational regulatory science. Clin. Pharm. Ther. 2014, 95, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.B.; Moretti, A.; Mederos y Schnitzler, M.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Witchel, H.J. Drug-induced hERG block and long QT syndrome. Cardiovasc. Ther. 2011, 29, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Rampe, D.; Brown, A.M. A history of the role of the hERG channel in cardiac risk assessment. J. Pharm. Toxicol. Methods 2013, 68, 13–22. [Google Scholar] [CrossRef]
- Pointon, A.; Harmer, A.R.; Dale, I.L.; Abi-Gerges, N.; Bowes, J.; Pollard, C.; Garside, H. Assessment of cardiomyocyte contraction in human-induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2015, 144, 227–237. [Google Scholar] [CrossRef]
- Sirenko, O.; Cromwell, E.F.; Crittenden, C.; Wignall, J.A.; Wright, F.A.; Rusyn, I. Assessment of beating parameters in human induced pluripotent stem cells enables quantitative in vitro screening for cardiotoxicity. Toxicol. Appl. Pharm. 2013, 273, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Blinova, K.; Stohlman, J.; Vicente, J.; Chan, D.; Johannesen, L.; Hortigon-Vinagre, M.P.; Zamora, V.; Smith, G.; Crumb, W.J.; Pang, L.; et al. Comprehensive Translational Assessment of Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes for Evaluating Drug-Induced Arrhythmias. Toxicol. Sci. 2017, 155, 234–247. [Google Scholar] [CrossRef]
- Reynolds, J.G.; Geretti, E.; Hendriks, B.S.; Lee, H.; Leonard, S.C.; Klinz, S.G.; Noble, C.O.; Lücker, P.B.; Zandstra, P.W.; Drummond, D.C.; et al. HER2-targeted liposomal doxorubicin displays enhanced anti-tumorigenic effects without associated cardiotoxicity. Toxicol. Appl. Pharm. 2012, 262, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hnatiuk, A.P.; Briganti, F.; Staudt, D.W.; Mercola, M. Human iPSC modeling of heart disease for drug development. Cell Chem. Biol. 2021, 28, 271–282. [Google Scholar] [CrossRef]
- Maizels, L.; Huber, I.; Arbel, G.; Tijsen, A.J.; Gepstein, A.; Khoury, A.; Gepstein, L. Patient-Specific Drug Screening Using a Human Induced Pluripotent Stem Cell Model of Catecholaminergic Polymorphic Ventricular Tachycardia Type 2. Circ. Arrhythmia Electrophysiol. 2017, 10, e004725. [Google Scholar] [CrossRef] [PubMed]
- Knottnerus, S.J.G.; Mengarelli, I.; Wüst, R.C.I.; Baartscheer, A.; Bleeker, J.C.; Coronel, R.; Ferdinandusse, S.; Guan, K.; IJlst, L.; Li, W.; et al. Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. Int. J. Mol. Sci. 2020, 21, 2589. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Muscle and cardiac therapeutic strategies for Duchenne muscular dystrophy: Past, present, and future. Pharm. Rep. 2020, 72, 1227–1263. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Andrysiak, K.; Stępniewski, J.; Dulak, J. Human-induced pluripotent stem cell-derived cardiomyocytes, 3D cardiac structures, and heart-on-a-chip as tools for drug research. Pflügers Arch. Eur. J. Physiol. 2021, 473, 1061–1085. [Google Scholar] [CrossRef] [PubMed]
- Gharanei, M.; Shafaattalab, S.; Sangha, S.; Gunawan, M.; Laksman, Z.; Hove-Madsen, L.; Tibbits, G.F. Atrial-specific hiPSC-derived cardiomyocytes in drug discovery and disease modeling. Methods 2021, 06, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; McKeithan, W.L.; Serrano, R.; Kitani, T.; Burridge, P.W.; Del Álamo, J.C.; Mercola, M.; Wu, J.C. Use of human induced pluripotent stem cell-derived cardiomyocytes to assess drug cardiotoxicity. Nat. Protoc. 2018, 13, 3018–3041. [Google Scholar] [CrossRef]
- Le, M.N.T.; Hasegawa, K. Expansion Culture of Human Pluripotent Stem Cells and Production of Cardiomyocytes. Bioengineering 2019, 6, 48–72. [Google Scholar] [CrossRef]
- Kami, D.; Watakabe, K.; Yamazaki-Inoue, M.; Minami, K.; Kitani, T.; Itakura, Y.; Toyoda, M.; Sakurai, T.; Umezawa, A.; Gojo, S. Large-scale cell production of stem cells for clinical application using the automated cell processing machine. BMC Biotechnol. 2013, 13, 102. [Google Scholar] [CrossRef]
- Zweigerdt, R.; Olmer, R.; Singh, H.; Haverich, A.; Martin, U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat. Protoc. 2011, 6, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Kempf, H.; Kropp, C.; Olmer, R.; Martin, U.; Zweigerdt, R. Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nat. Protoc. 2015, 10, 1345–1361. [Google Scholar] [CrossRef]
- Olmer, R.; Haase, A.; Merkert, S.; Cui, W.; Palecek, J.; Ran, C.; Kirschning, A.; Scheper, T.; Glage, S.; Miller, K.; et al. Long term expansion of undifferentiated human iPS and ES cells in suspension culture using a defined medium. Stem Cell Res. 2010, 5, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chou, B.K.; Dowey, S.; He, C.; Gerecht, S.; Cheng, L. Scalable expansion of human induced pluripotent stem cells in the defined xeno-free E8 medium under adherent and suspension culture conditions. Stem Cell Res. 2013, 11, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Olmer, R.; Lange, A.; Selzer, S.; Kasper, C.; Haverich, A.; Martin, U.; Zweigerdt, R. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng. Part. C Methods 2012, 18, 772–784. [Google Scholar] [CrossRef]
- Yang, H.S.; Jeon, O.; Bhang, S.H.; Lee, S.H.; Kim, B.S. Suspension culture of mammalian cells using thermosensitive microcarrier that allows cell detachment without proteolytic enzyme treatment. Cell Transplant. 2010, 19, 1123–1132. [Google Scholar] [CrossRef]
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling expansion and cardiomyogenic differentiation of human pluripotent stem cells in scalable suspension culture. Stem Cell Rep. 2014, 3, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Konagaya, S.; Ando, T.; Yamauchi, T.; Suemori, H.; Iwata, H. Long-term maintenance of human induced pluripotent stem cells by automated cell culture system. Sci. Rep. 2015, 5, 16647. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, H.; Zhang, M.; Bartosek, T.; Murry, C.E. Survival, Integration, and Differentiation of Cardiomyocyte Grafts. Circulation 1999, 100, 193–202. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
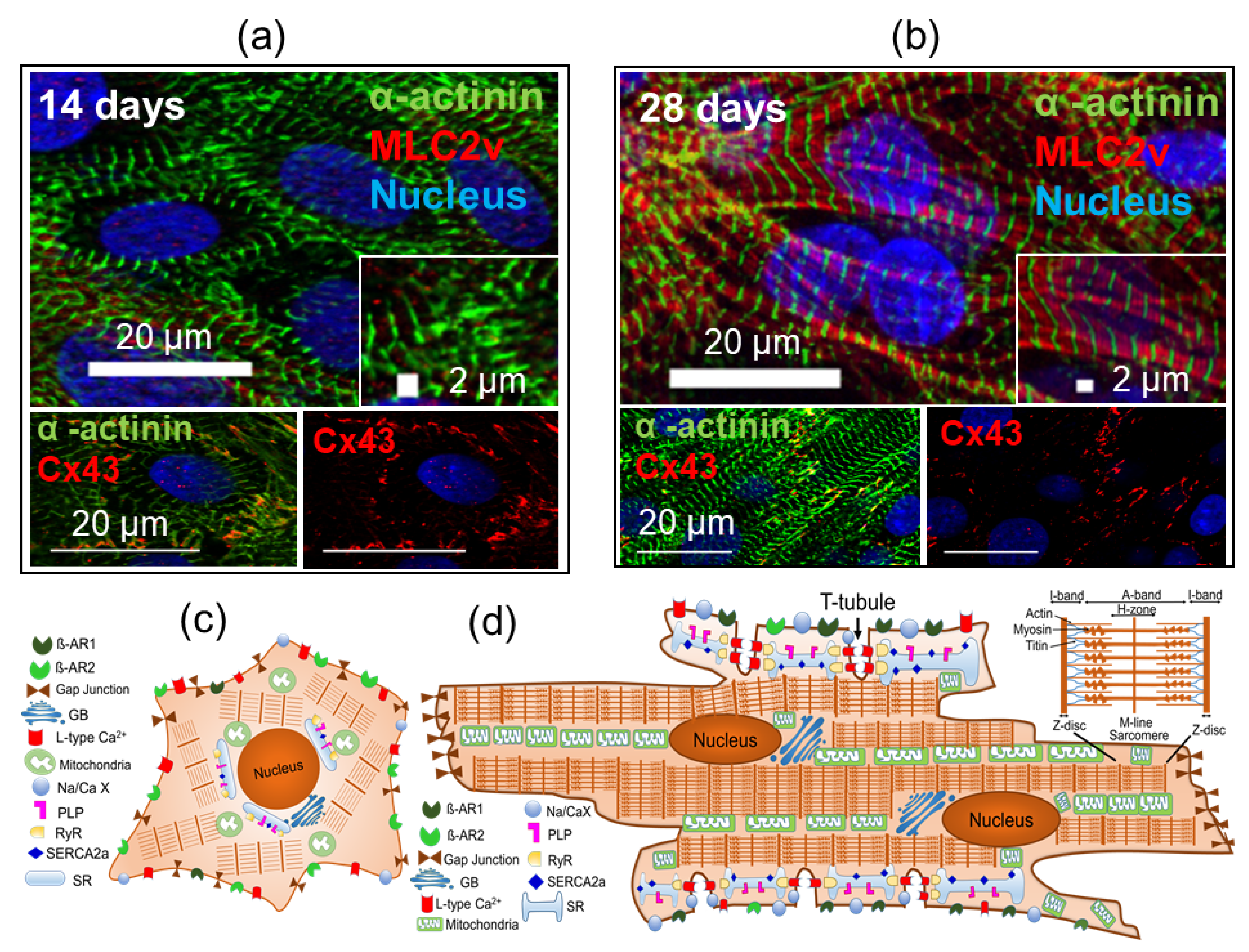{kind=link}
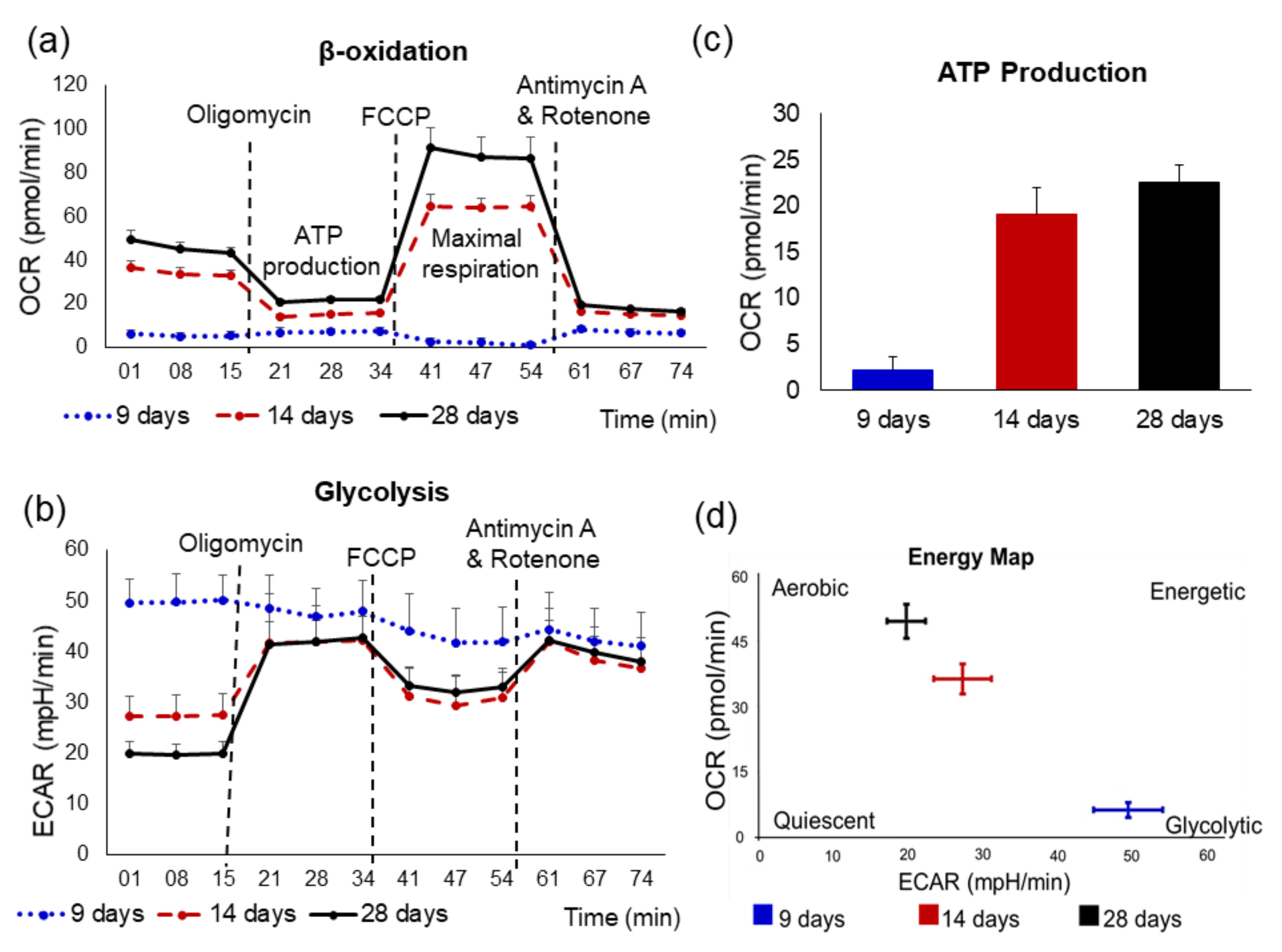{kind=link}
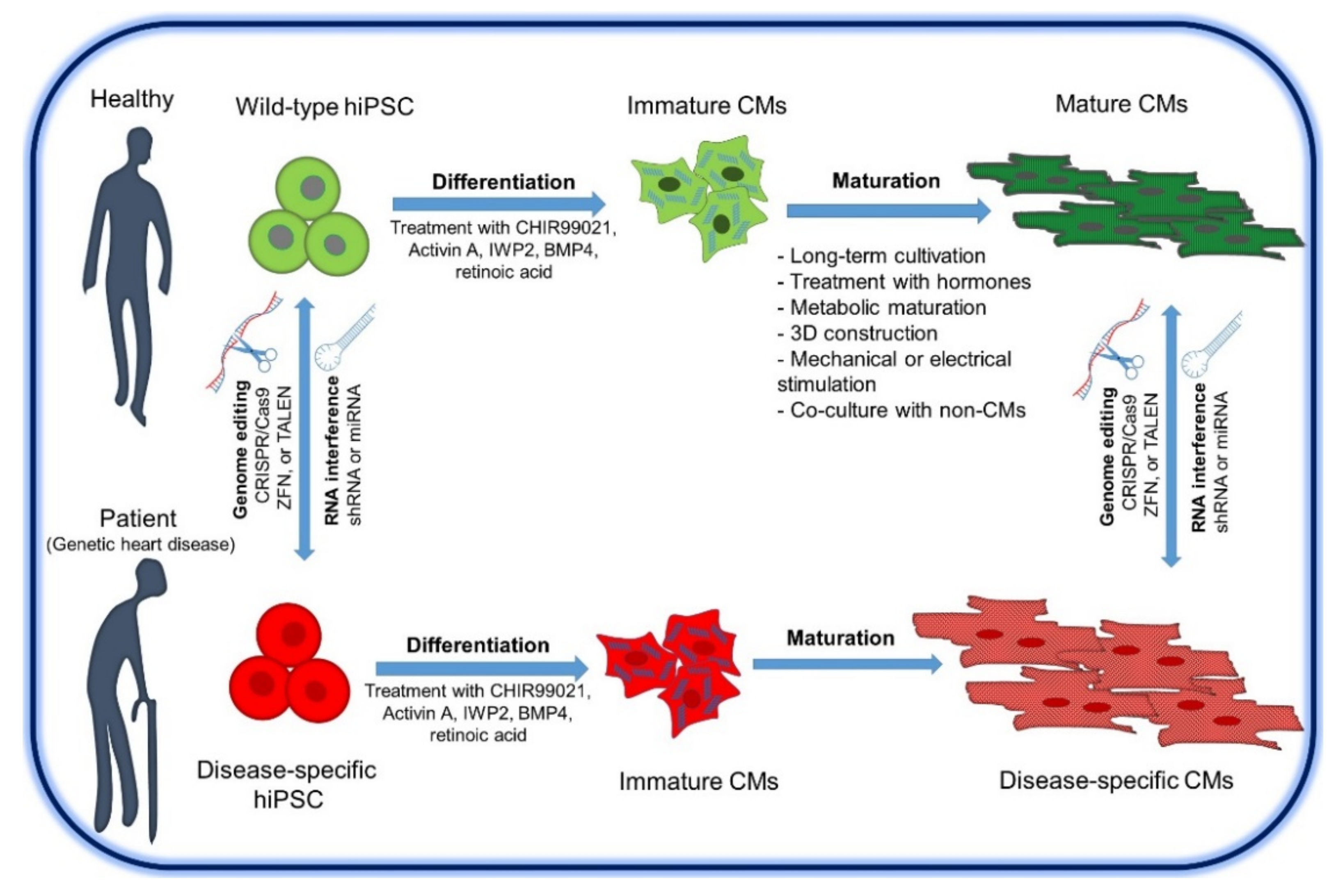{kind=link}
| Parameter | Immature CMs | Mature CMs |
|---|---|---|
| Cell shape | Irregular | Rod-shaped |
| Cell area | 480 ± 32 µm2 | 1716 ± 150 µm2 |
| Sarcomere structure | Disorganized and less-developed | Organized and M-line developed |
| Sarcomere length | ≈1.65 µm | ≈1.81−2.3 µm |
| Cardiac troponin I | ssTnI | cTnI |
| Titin | N2BA isoform (N2B and N2A) | N2B |
| MHC | β-MHC ≤ α-MHC | β-MHC >> α-MHC |
| T-tubules | Deficient | Abundant |
| Mitochondria | Round and cristae are less mature | Elongated and more mature cristae (up to 40% cell volume) |
| Sarcoplasmic reticulum network | Underdeveloped | Well-developed |
| Metabolic | Glycolysis (80%) | Fatty acid β-oxidation 70–80%) |
| Nucleation | Mononuclear | Dinuclear (~35.3%) |
| Upstroke velocity | ≈44 to 50 V/s | ≈188.7 to 250 V/s |
| Resting membrane potential | ≈ −50 to −60 mV | ≈ −90 mV |
| Gap junction | Circumferentially | Periphery at the intercalated disc |
| Adrenergic receptors | ≈β1 ≤ β2 | ≈70 β1:30 β2 and response to β-adrenergic stimulation |
| SERCA2a | Low expressed | Highly expressed |
| Contraction | Asynchronous | Synchronous |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bekhite, M.M.; Schulze, P.C. Human Induced Pluripotent Stem Cell as a Disease Modeling and Drug Development Platform—A Cardiac Perspective. Cells 2021, 10, 3483. https://doi.org/10.3390/cells10123483
Bekhite MM, Schulze PC. Human Induced Pluripotent Stem Cell as a Disease Modeling and Drug Development Platform—A Cardiac Perspective. Cells. 2021; 10(12):3483. https://doi.org/10.3390/cells10123483
Chicago/Turabian StyleBekhite, Mohamed M., and P. Christian Schulze. 2021. "Human Induced Pluripotent Stem Cell as a Disease Modeling and Drug Development Platform—A Cardiac Perspective" Cells 10, no. 12: 3483. https://doi.org/10.3390/cells10123483
APA StyleBekhite, M. M., & Schulze, P. C. (2021). Human Induced Pluripotent Stem Cell as a Disease Modeling and Drug Development Platform—A Cardiac Perspective. Cells, 10(12), 3483. https://doi.org/10.3390/cells10123483