
Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Genetic Studies and Review of the Literature
2.2. Coherent Anti-Stokes Raman Scattering (CARS) and SHG Microscopy and Immunofluorescence
2.3. Statistical Evaluation of Muscle Fibre Calibers
2.4. Extraction of Spectra for the Determined Features
2.5. Fluorescent Labelling of Cryo-Embedded Muscle Sections
2.6. Confocal Laser Scanning Microscopy (CLSM) and Image Processing of Fluorescent-Labelled Muscle Sections
3. Results
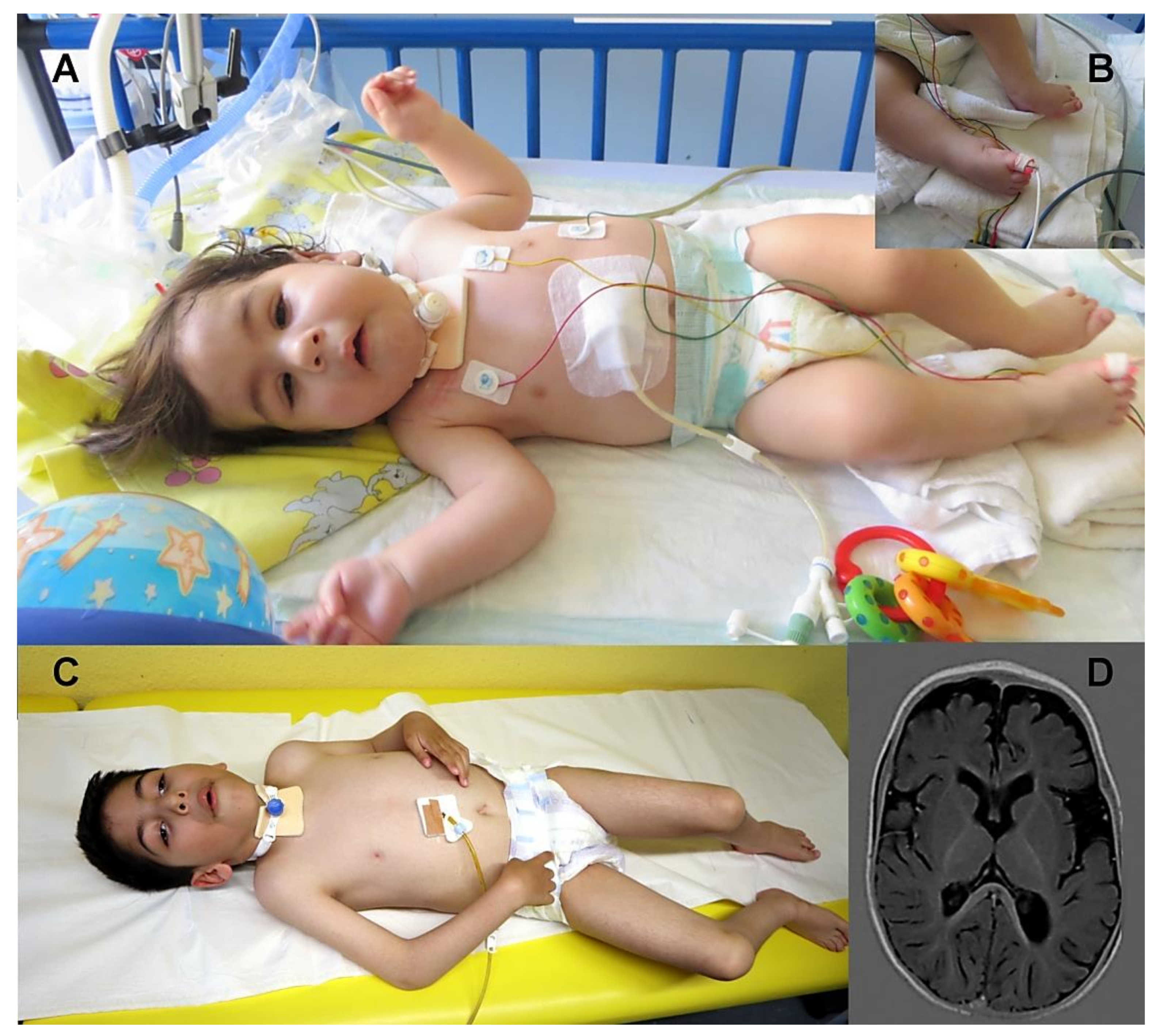
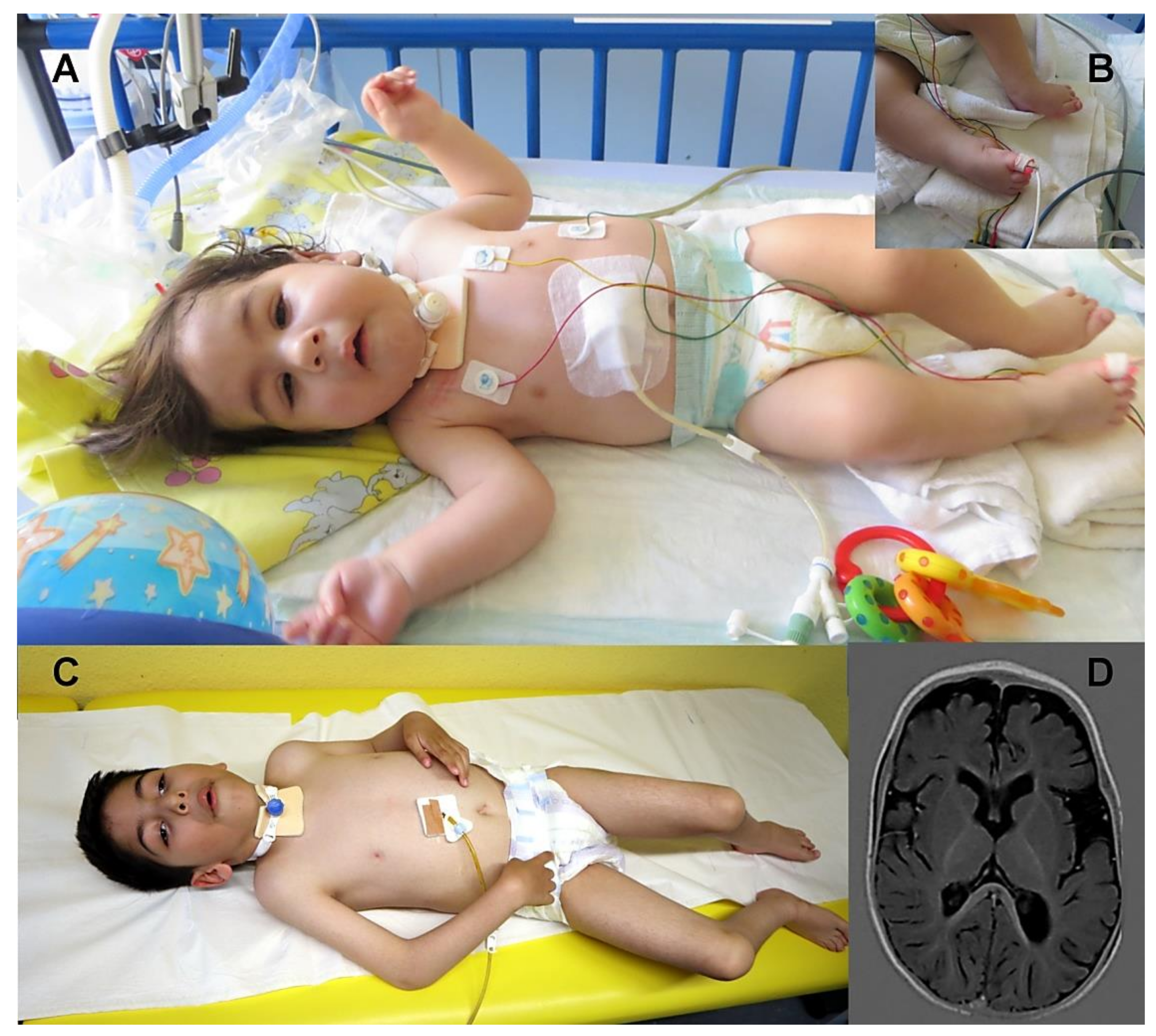
3.1. Clinical Findings
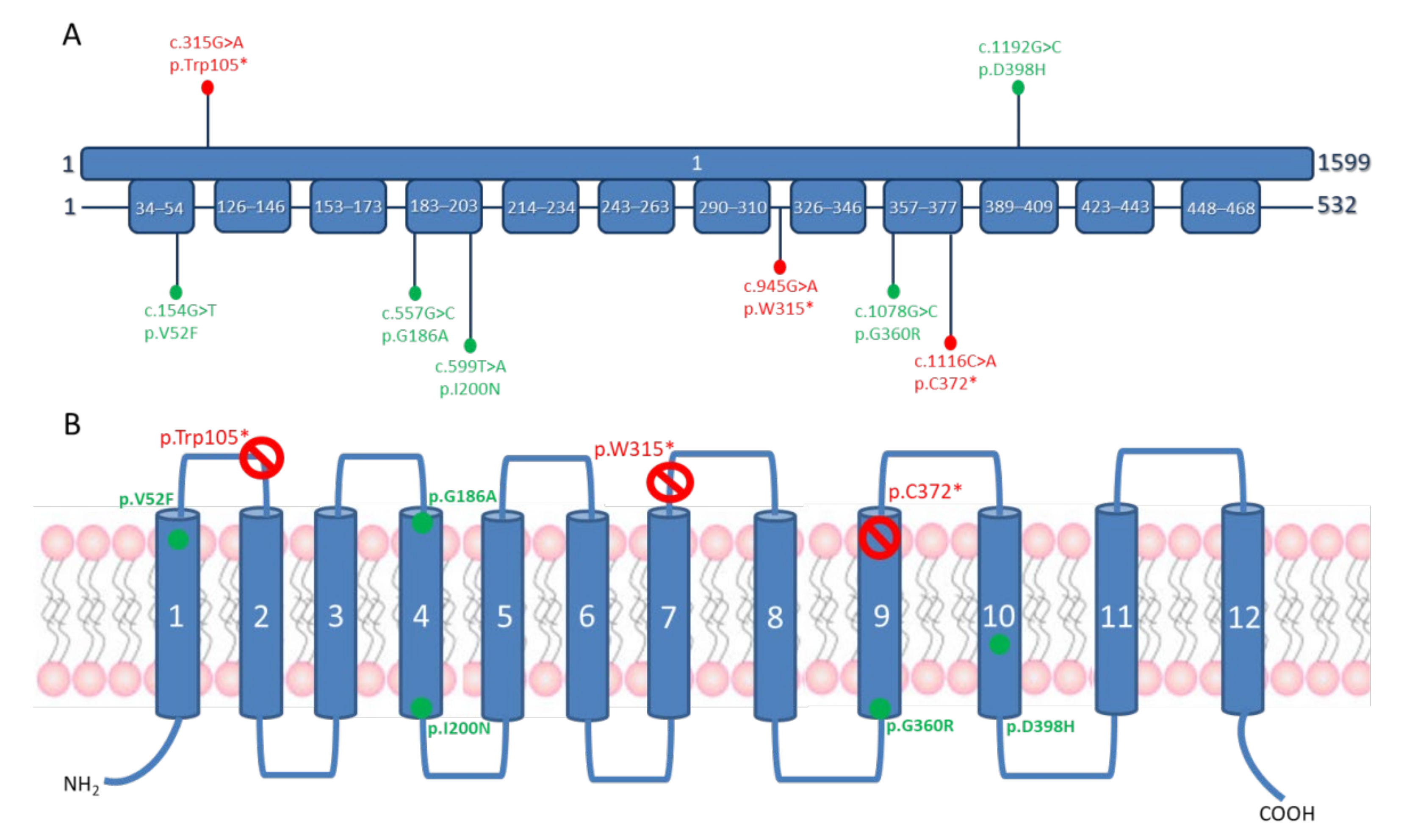
3.2. Molecular Genetics Findings
3.3. Review of the Patients Reported in the Literature
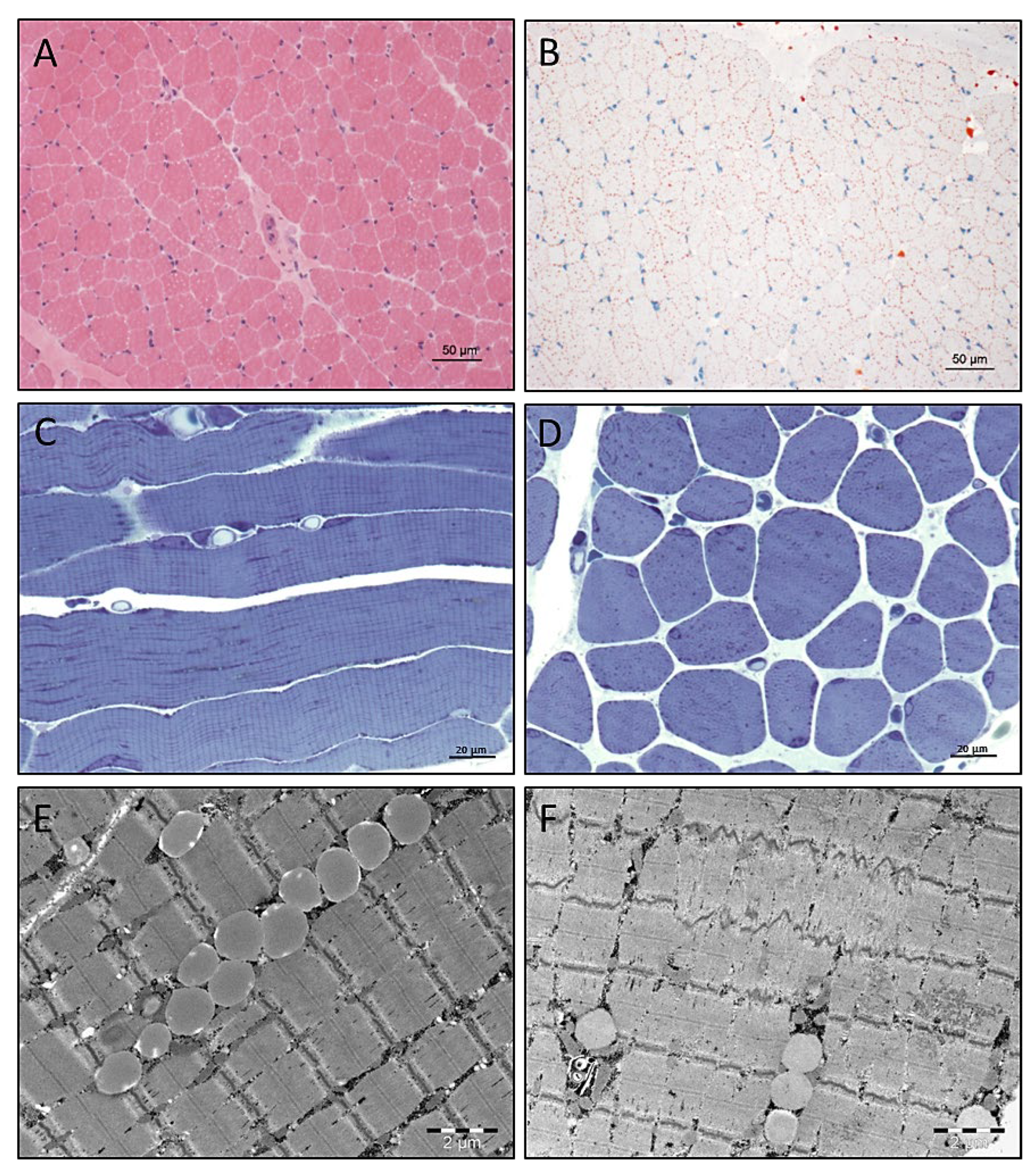
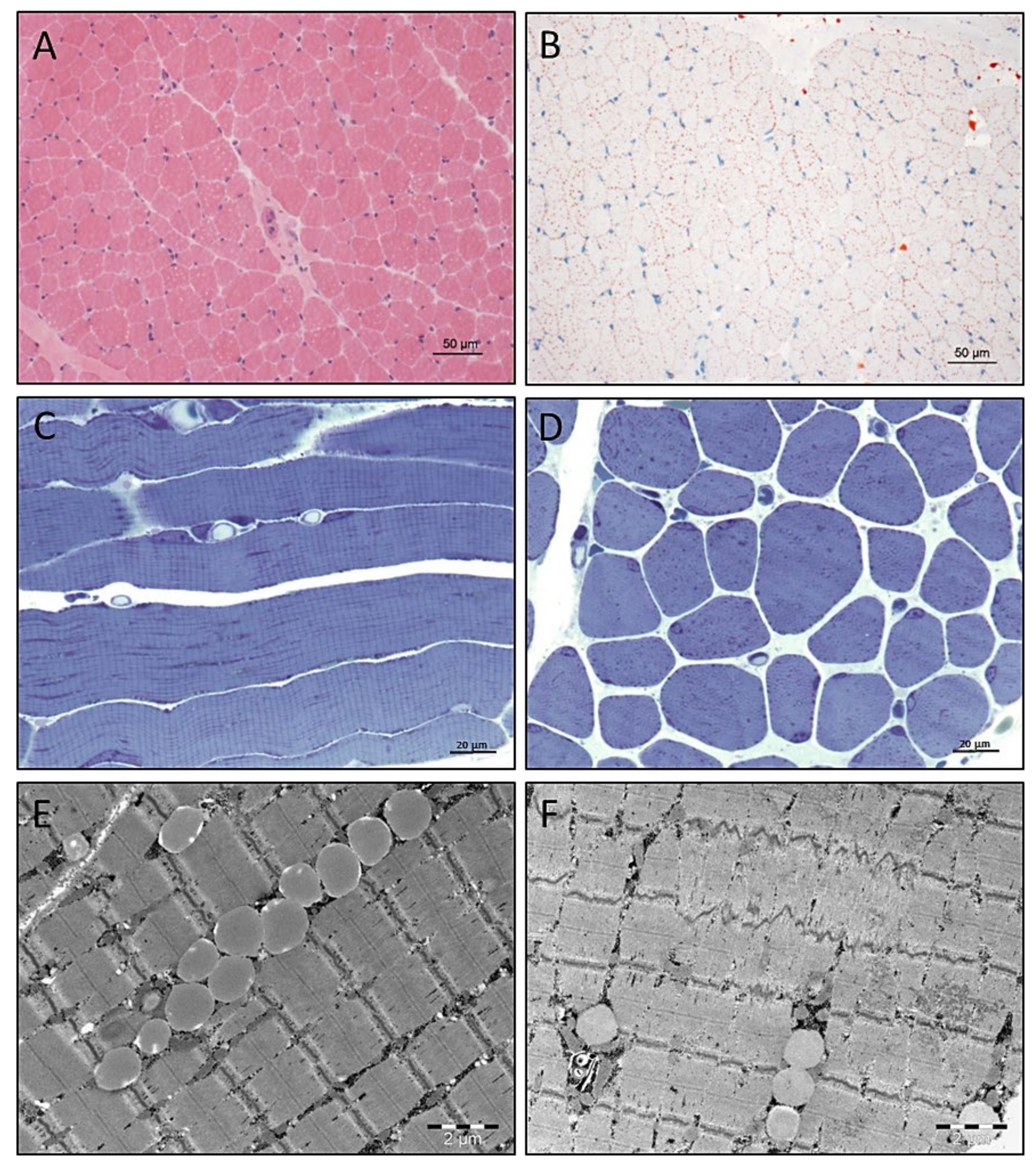
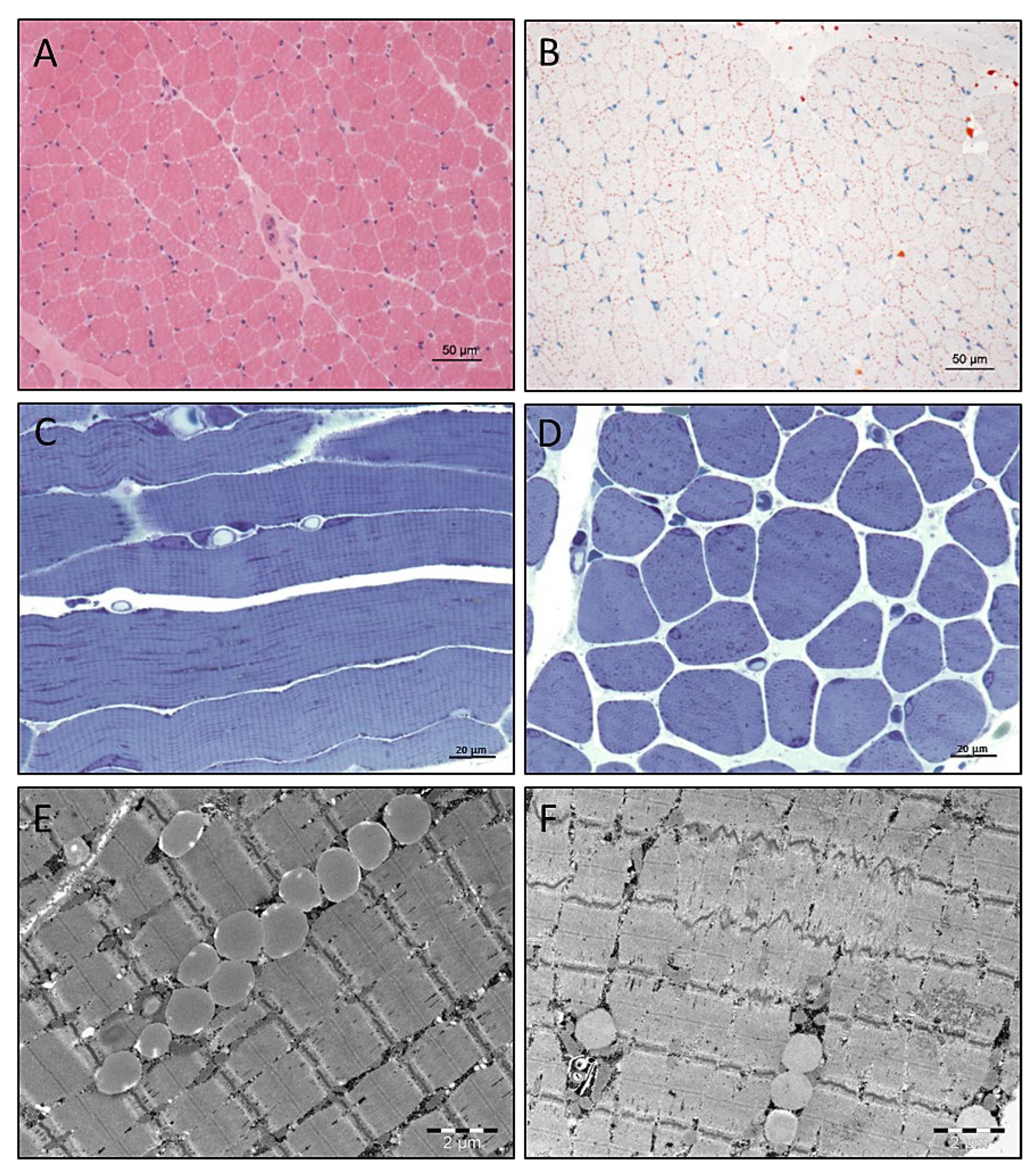
3.4. Light and Electron Microscopic Studies on Quadriceps Muscle Biopsy
3.5. Electron Microscopy
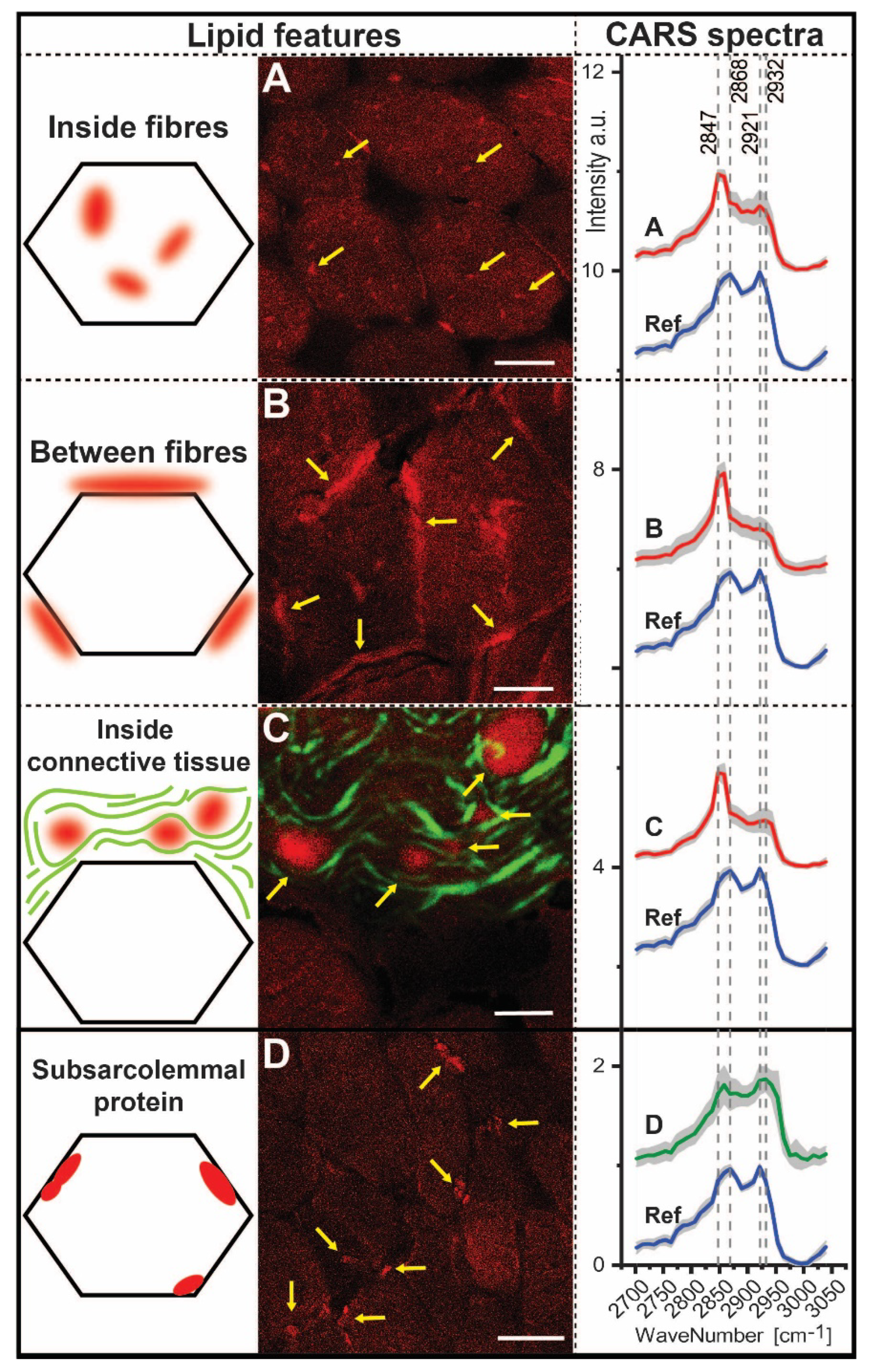
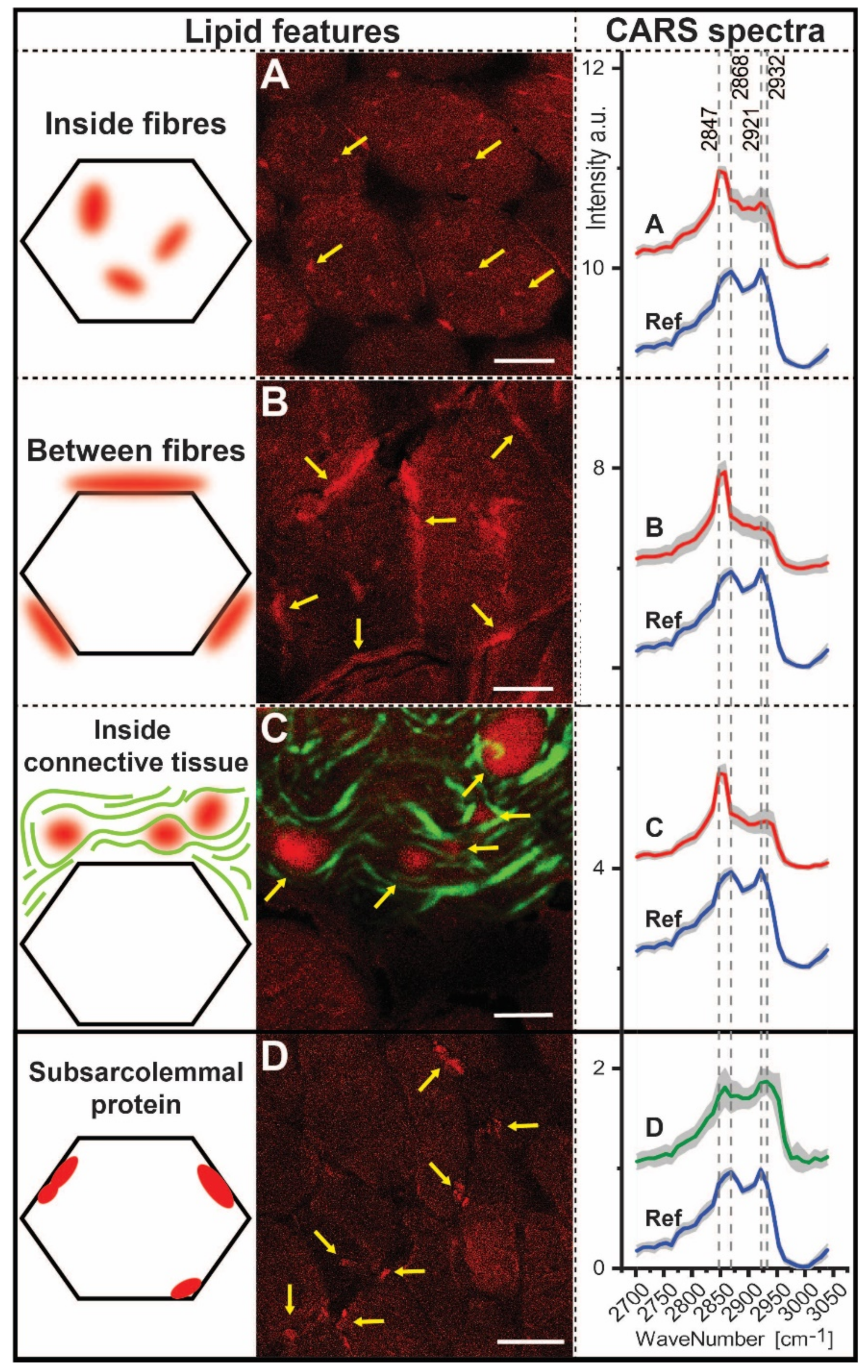
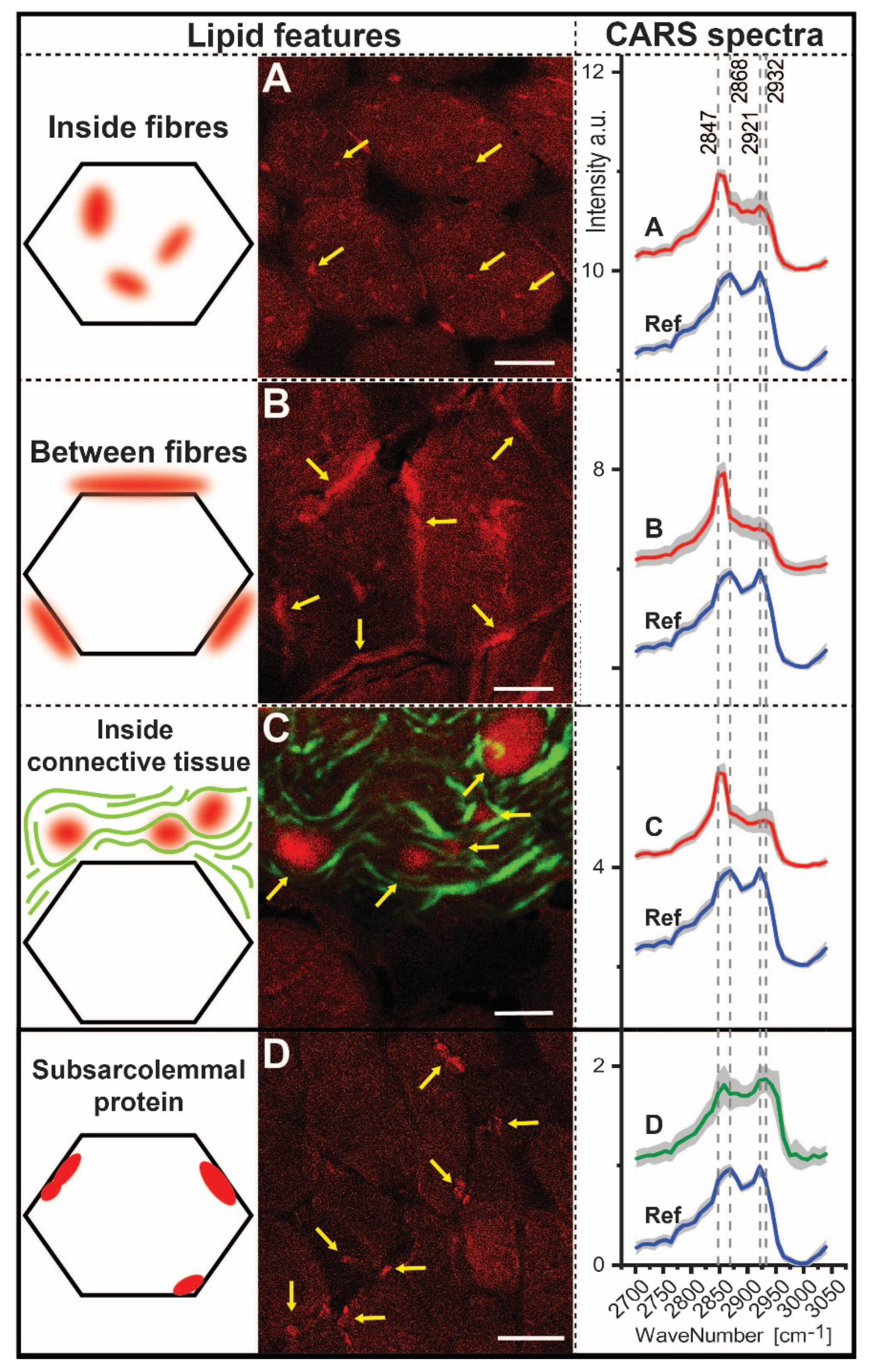
3.6. CARS Microscopic Studies on Quadriceps Muscle Biopsy
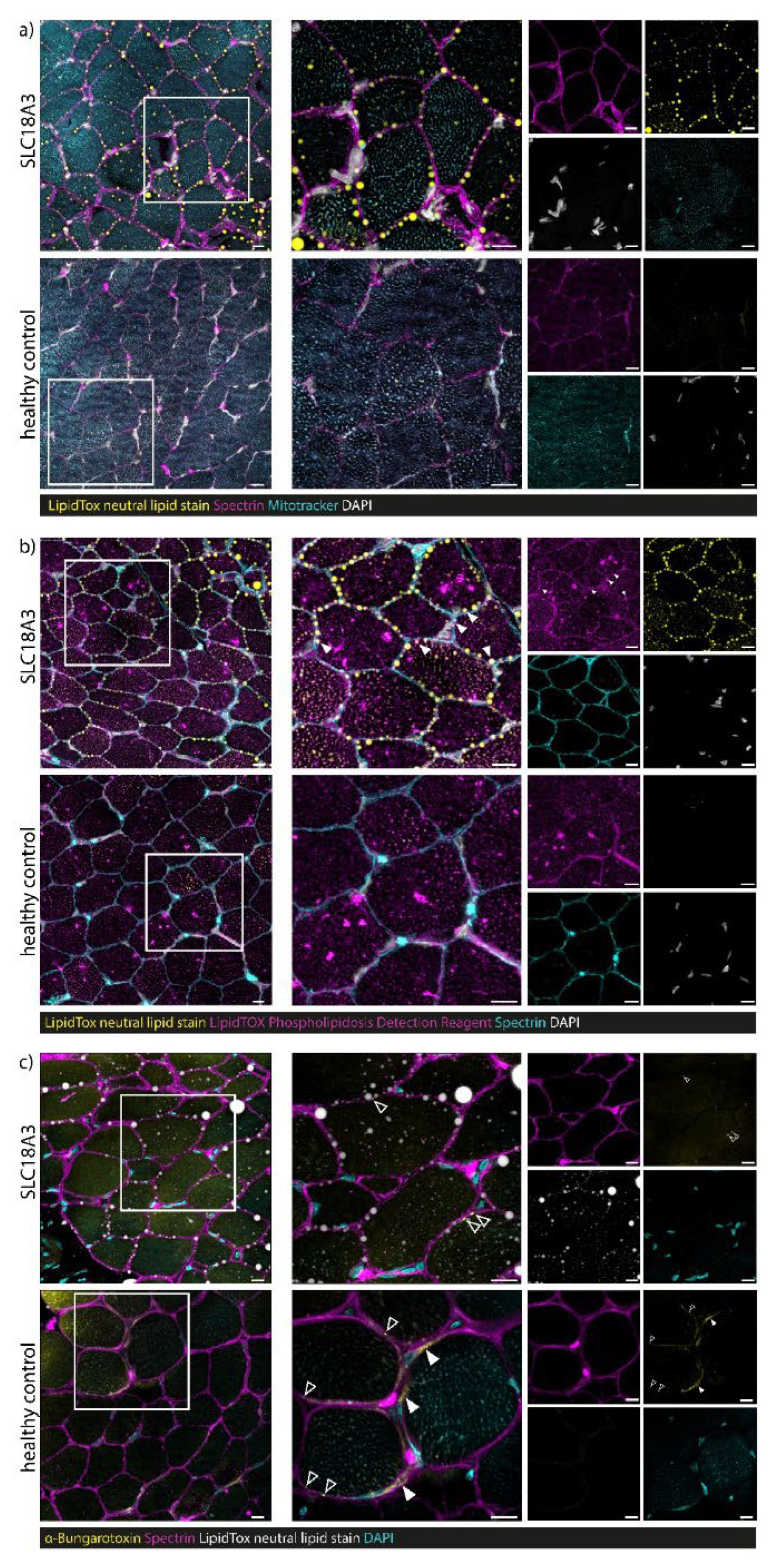
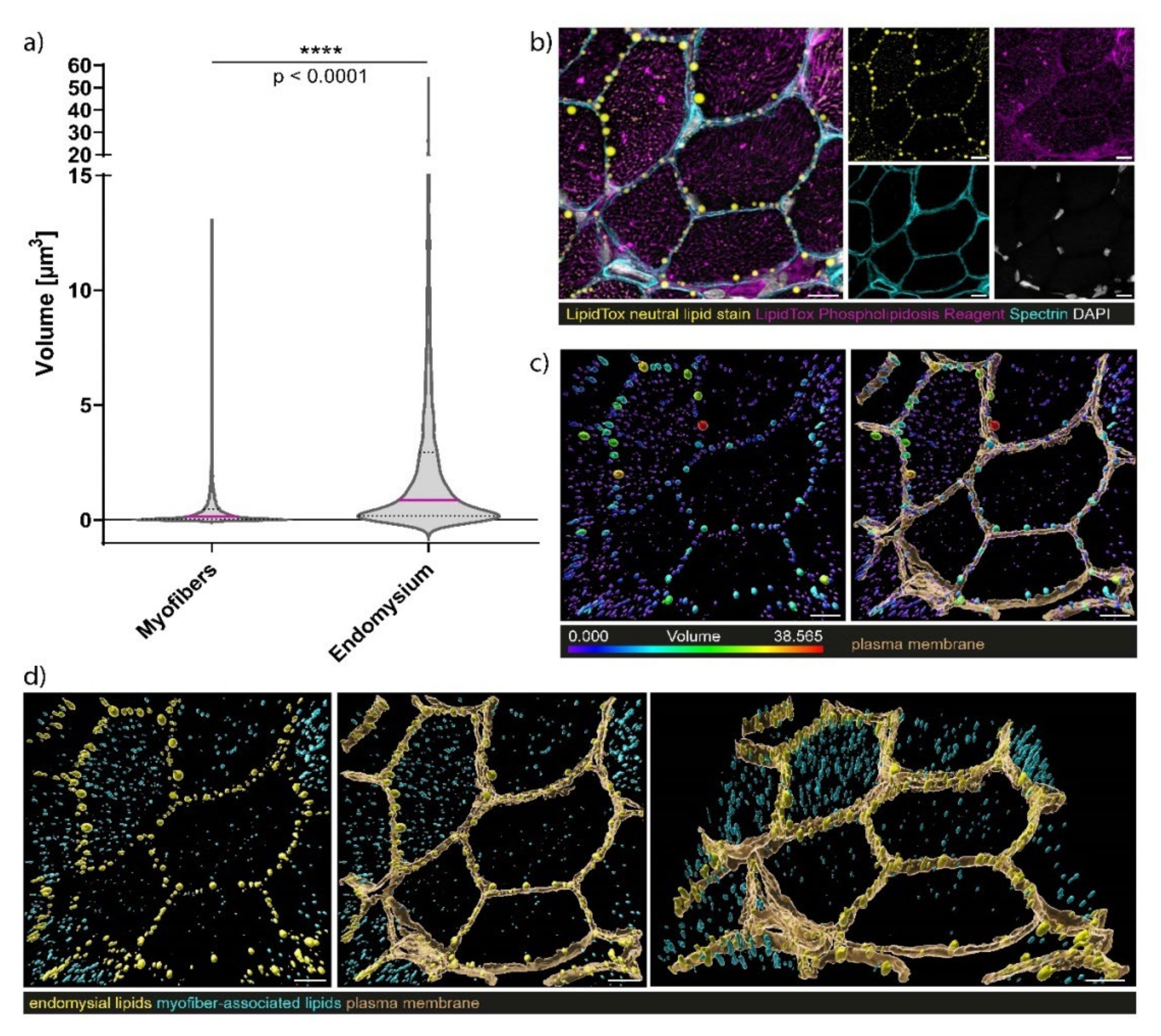
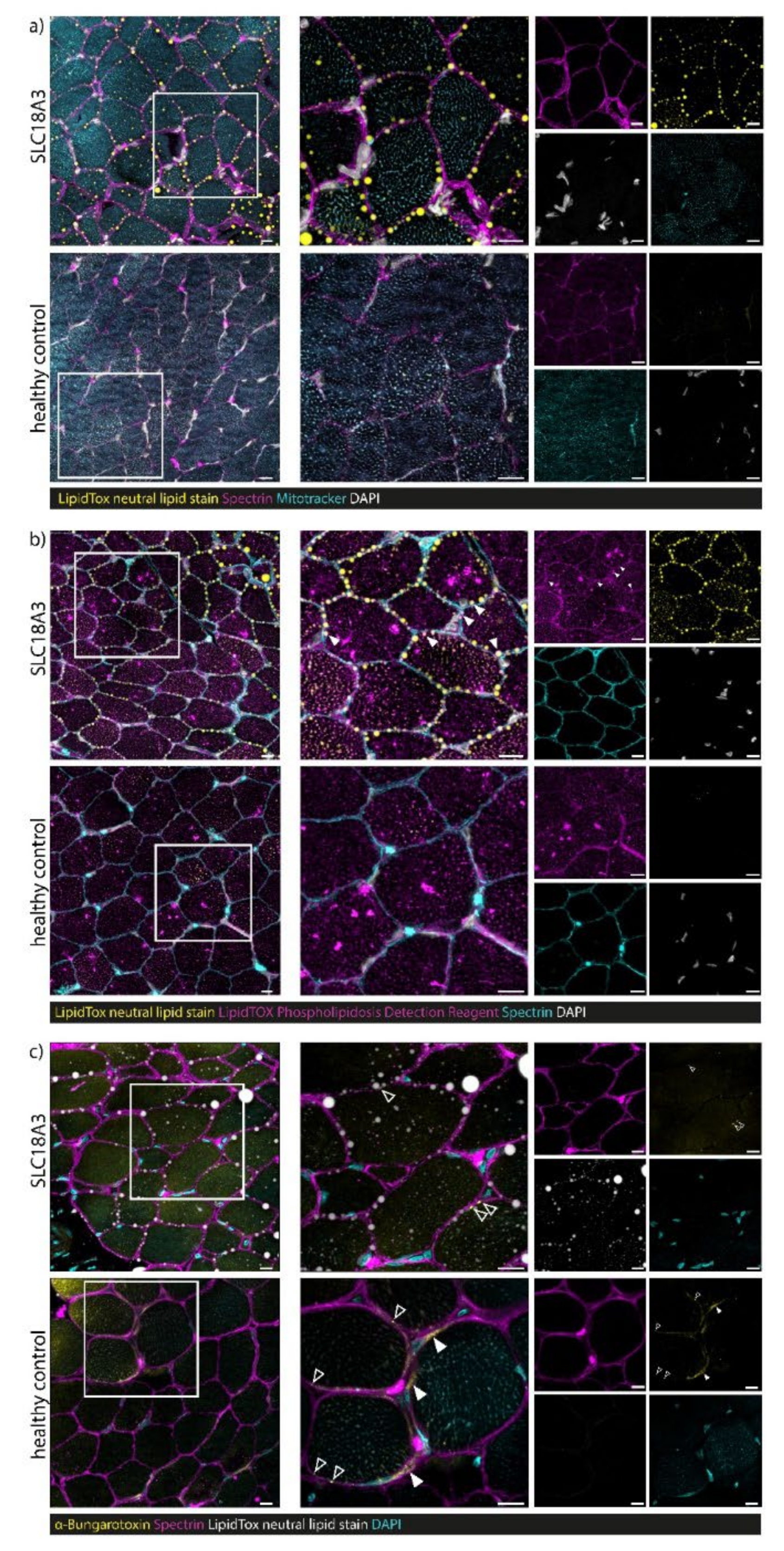
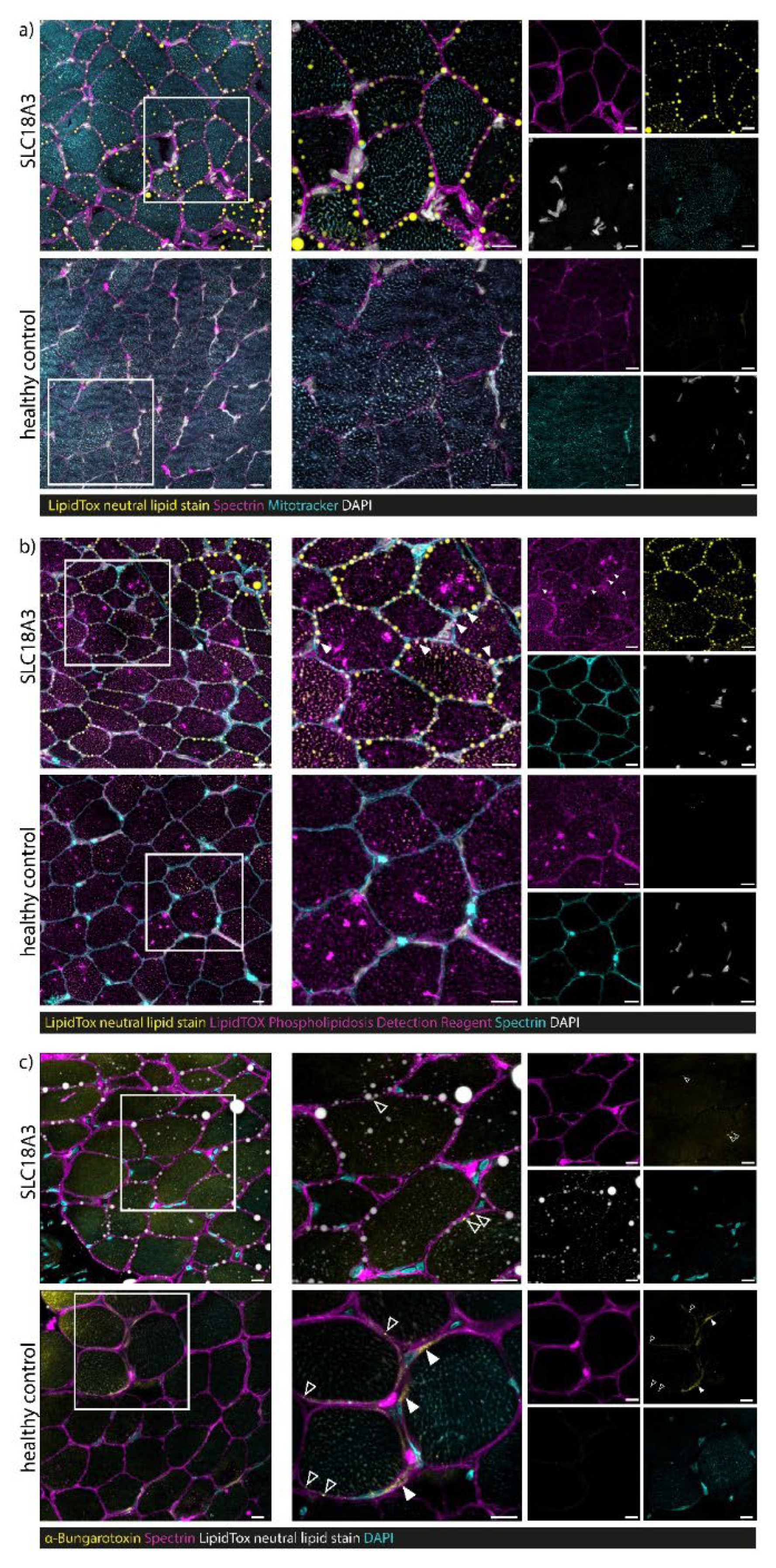
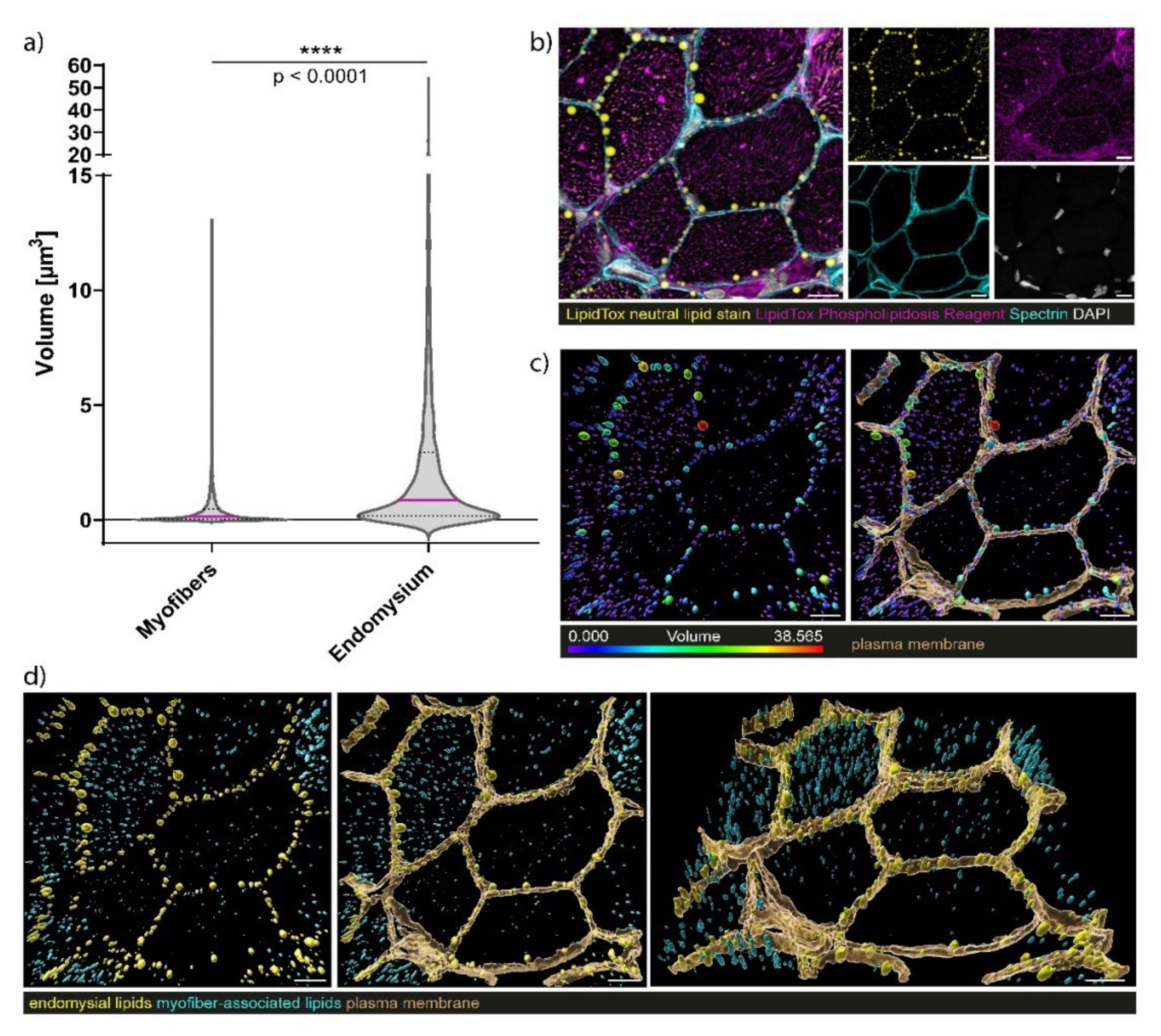
3.7. Confocal Laser Scanning Microscopy (CLSM) and Image Processing of Fluorescent-Labelled Muscle Sections
4. Discussion
4.1. Phenotype Associated with Bi-Allelic SLC18A3 Variants
4.2. Phenotype–Genotype Correlations
4.3. Myopathology
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McMacken, G.; Whittaker, R.G.; Evangelista, T.; Abicht, A.; Dusl, M.; Lochmüller, H. Congenital myasthenic syndrome with episodic apnoea: Clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J. Neurol. 2018, 265, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, J.C.; Milone, M.; Selcen, D.; Shen, X.M.; Engel, A.G.; Liewluck, T. Congenital myasthenic syndromes in adult neurology clinic: A long road to diagnosis and therapy. Neurology 2018, 91, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, G.L.; Verschuuren, C.; Yuen, M.; Webster, R.; Menezes, M.; Fock, J.M.; Pride, N.; Best, H.A.; Damm, T.B.; Turner, C.; et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology 2016, 87, 1442–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauché, S.; O’Regan, S.; Azuma, Y.; Laffargue, F.; McMacken, G.; Sternberg, D.; Brochier, G.; Buon, C.; Bouzidi, N.; Topf, A.; et al. Impaired Presynaptic High-Affinity Choline Transporter Causes a Congenital Myasthenic Syndrome with Episodic Apnea. Am. J. Hum. Genet. 2016, 99, 753–761. [Google Scholar] [CrossRef] [Green Version]
- De Castro, B.M.; De Jaeger, X.; Martins-Silva, C.; Lima, R.D.F.; Amaral, E.; Menezes, C.; Lima, P.; Neves, C.M.L.; Pires, R.G.; Gould, T.W.; et al. The Vesicular Acetylcholine Transporter Is Required for Neuromuscular Development and Function. Mol. Cell Biol. 2009, 29, 5238–5250. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebrouck, A.E.; Beeson, D. The congenital myasthenic syndromes: Expanding genetic and phenotypic spectrums and refining treatment strategies. Curr. Opin. Neurol. 2019, 32, 696–703. [Google Scholar] [CrossRef]
- Nicolau, S.; Milone, M. The Electrophysiology of Presynaptic Congenital Myasthenic Syndromes With and Without Facilitation: From Electrodiagnostic Findings to Molecular Mechanisms. Front. Neurol. 2019, 10, 257. [Google Scholar] [CrossRef]
- Hakonen, A.H.; Polvi, A.; Saloranta, C.; Paetau, A.; Heikkilä, P.; Almusa, H.; Ellonen, P.; Jakkula, E.; Saarela, J.; Aittomäki, K. SLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy. Am. J. Med. Genet. Part A 2019, 179, ajmg. [Google Scholar] [CrossRef]
- Mair, D.; Biskup, S.; Kress, W.; Abicht, A.; Brück, W.; Zechel, S.; Knop, K.C.; Koenig, F.B.; Tey, S.; Nikolin, S.; et al. Differential diagnosis of vacuolar myopathies in the NGS era. Brain Pathol. 2020, 30, 877–896. [Google Scholar] [CrossRef]
- Schwartz, M.; Sternberg, D.; Whalen, S.; Afenjar, A.; Isapof, A.; Chabrol, B.; Portnoï, M.-F.; Heide, S.; Keren, B.; Chantot-Bastaraud, S.; et al. How chromosomal deletions can unmask recessive mutations? Deletions in 10q11.2 associated with CHAT or SLC18A3 mutations lead to congenital myasthenic syndrome. Am. J. Med. Genet. Part A 2018, 176, 151–155. [Google Scholar] [CrossRef]
- Aran, A.; Segel, R.; Kaneshige, K.; Gulsuner, S.; Renbaum, P.; Oliphant, S.; Meirson, T.; Weinberg-Shukron, A.; Hershkovitz, Y.; Zeligson, S.; et al. Vesicular acetylcholine transporter defect underlies devastating congenital myasthenia syndrome. Neurology 2017, 88, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Lamond, A.; Buckley, D.; O’Dea, J.; Turner, L. Variants of SLC18A3 leading to congenital myasthenic syndrome in two children with varying presentations. BMJ Case Rep. 2021, 14, e237799. [Google Scholar] [CrossRef]
- Leite Schetino, L.P.; de Castro Fonseca, M.; Magalhães Gomes, M.P.S.; Costa Valadao, P.A.; de Camargo, W.L.; Rodrigues, H.A.; Andrade, J.N.; Arantes-Costa, F.M.; Naves, L.A.; Prado, C.M.; et al. Evaluation of the neuromuscular junction in a middle-aged mouse model of congenital myasthenic syndrome. Muscle Nerve 2019, 60, 790–800. [Google Scholar] [CrossRef]
- Hentschel, A.; Czech, A.; Münchberg, U.; Freier, E.; Schara-Schmidt, U.; Sickmann, A.; Reimann, J.; Roos, A. Protein signature of human skin fibroblasts allows the study of the molecular etiology of rare neurological diseases. Orphanet. J. Rare Dis. 2021, 16, 1–18. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Ajeti, V.; Nadiarnykh, O.; Ponik, S.M.; Keely, P.J.; Eliceiri, K.W.; Campagnola, P.J. Structural changes in mixed Col I/Col V collagen gels probed by SHG microscopy: Implications for probing stromal alterations in human breast cancer. Biomed. Opt. Express 2011, 2, 2307. [Google Scholar] [CrossRef] [Green Version]
- Rinia, H.A.; Burger, K.N.J.; Bonn, M.; Müller, M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophys. J. 2008, 95, 4908–4914. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.X.; Jia, Y.K.; Zheng, G.; Xie, X.S. Laser-scanning coherent anti-Stokes Raman scattering microscopy and applications to cell biology. Biophys. J. 2002, 83, 502–509. [Google Scholar] [CrossRef] [Green Version]
- González Coraspe, J.A.; Weis, J.; Anderson, M.E.; Münchberg, U.; Lorenz, K.; Buchkremer, S.; Carr, S.; Zahedi, R.; Brauers, E.; Michels, H.; et al. Biochemical and pathological changes result from mutated Caveolin-3 in muscle. Skelet Muscle 2018, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Saar, B.G.; Freudiger, C.W.; Reichman, J.; Stanley, C.M.; Holtom, G.R.; Xie, X.S. Video-rate molecular imaging in vivo with stimulated Raman scattering. Science 2010, 330, 1368–1370. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ramachandran, P.V.; Wang, M.C. Shedding new light on lipid functions with CARS and SRS microscopy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1120–1129. [Google Scholar] [CrossRef] [Green Version]
- Freudiger, C.W.; Yang, W.; Holtom, G.R.; Peyghambarian, N.; Xie, X.S.; Kieu, K.Q. Stimulated Raman scattering microscopy with a robust fibre laser source. Nat. Photonics 2014, 8, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Salter, C.G.; Refai, O.; Hardy, H.; Barwick, K.E.S.; Akpulat, U.; Kvarnung, M.; Chioza, B.A.; Harlalka, G.; Taylan, F.; et al. Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization. Brain 2017, 140, 2838–2850. [Google Scholar] [CrossRef]
- Schara, U.; Christen, H.-J.; Durmus, H.; Hietala, M.; Krabetz, K.; Rodolico, C.; Schreiber, G.; Topaloglu, H.; Talim, B.; Voss, W.; et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur. J. Paediatr. Neurol. 2010, 14, 326–333. [Google Scholar] [CrossRef]
- De Castro, B.M.; Pereira, G.S.; Magalhães, V.; Rossato, J.I.; De Jaeger, X.; Martins-Silva, C.; Leles, B.; Lima, P.; Gomez, M.V.; Gainetdinov, R.; et al. Reduced expression of the vesicular acetylcholine transporter causes learning deficits in mice. Genes Brain Behav. 2009, 8, 23–35. [Google Scholar] [CrossRef]
- Pascual, J.M.; Liu, P.; Mao, D.; Kelly, D.I.; Hernandez, A.; Sheng, M.; Good, L.B.; Ma, Q.; Marin-Valencia, I.; Zhang, X.; et al. Triheptanoin for glucose transporter type I deficiency (G1D): Modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014, 71, 1255–1265. [Google Scholar] [CrossRef]
- Liu, X.; Shi, Y.; Niu, B.; Shi, Z.; Li, J.; Ma, Z.; Wang, J.; Gong, P.; Zheng, A.; Zhang, F.; et al. Polymorphic variation in CHAT gene modulates general cognitive ability: An association study with random student cohort. Neurosci. Lett. 2016, 617, 122–126. [Google Scholar] [CrossRef]
- Prado, V.F.; Martins-Silva, C.; de Castro, B.M.; Lima, R.F.; Barros, D.M.; Amaral, E.; Ramsey, A.; Sotnikova, T.D.; Ramirez, M.R.; Kim, H.-G.; et al. Mice Deficient for the Vesicular Acetylcholine Transporter Are Myasthenic and Have Deficits in Object and Social Recognition. Neuron 2006, 51, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Joviano-Santos, J.V.; Kljakic, O.; Magalhães-Gomes, M.P.S.; Valadão, P.A.C.; de Oliveira, L.R.; Prado, M.A.M.; Prado, V.F.; Guatimosim, C. Motoneuron-specific loss of VAChT mimics neuromuscular defects seen in congenital myasthenic syndrome. FEBS J. 2021, 288, 5331–5349. [Google Scholar] [CrossRef]
- Jesse, C.M.; Bushuven, E.; Tripathi, P.; Chandrasekar, A.; Simon, C.M.; Drepper, C.; Yamoah, A.; Dreser, A.; Katona, I.; Johann, S.; et al. ALS-Associated Endoplasmic Reticulum Proteins in Denervated Skeletal Muscle: Implications for Motor Neuron Disease Pathology. Brain Pathol. 2017, 27, 781–794. [Google Scholar] [CrossRef]
- Freeman, S.S.; Engel, A.G.; Drachman, D.B. Experimental acetylcholine blockade of the neuromuscular junction. Effects on end plate and muscle fiber ultrastructure. Ann. N. Y. Acad. Sci. 1976, 274, 46–59. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| This Study | O’Grady et al., 2016 | Schwartz et al., 2017 | Aran et al., 2017 | Hakonen et al., 2019 | Lamon et al., 2021 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 (sibling 1) | Patient 6 (sibling 2) | Patient 7/8 (siblings) | Patient 9 | Patient 10 | |
| Causative variant | c.315G>A (p.Trp105*) & c.1192G>C (p.Asp398His); (NM_003055.2) compound heterozygous missense and nonsense | c.557G>C; (p.Gly186Ala); (NM_003055.2) hemizygous missense based on an additional 4.83MB deletion in 10q11.22-q11.23 | c.1192G>C, (p.Asp398His); (NM_003055.2) homozygous missense | c.154G>T (p.Val52Phe); (NM_003055.2) hemizygous missense based on an additional 5.55MB deletion in 10q11.22-q11.23 | c.1078G>A (p.Gly360Arg); (NM_003055.2) homozygous missense +NECAB2 p.R307H | c.1078G>A (p.Gly360Arg); (NM_003055.2) homozygous missense +NECAB2 p.R307H | c.1116C>A, p(Cys372*); (NM_003055.2) homozygous nonsense | c.945G>A, (p.Trp315*) & c.599T>A (p.Ile200Asn) compound heterozygous missense and nonsense | c.945G>A, (p.Trp315*) & c.599T>A, (p.Ile200Asn) compound heterozygous missense and nonsense |
| Age of onset | Neonatal | Infancy | Neonatal | Neonatal | Neonatal | Neonatal | Intrauterine death | Neonatal | Neonatal |
| Gender | Male | Male | Female | Female | Male | Male | n.n. | Male | Male |
| Descent | Poland/Turkey | Filipinos | Turkey | n.n. | Yemenite Jewish | Finnish | Caucasian | Caucasian | |
| Delivery (intrauterine symptoms) | 36 + 2 (reduced foetal movements) | n.n. | 36 + 5 | 34 + 0 (reduced foetal movements) | 37 weeks APGAR 4/5 | 40 weeks APGAR 3/3 | - | 38 + 1, APGAR 1/6/6 | 34 + 1, APGAR 7/8 |
| Initial symptoms | Apnoeas, feeding problems, ptosis, facial hypomimia, joint contractures, muscular hypotonia | Episodes of cyanosis 2–18 months, ptosis at 1 year fatigability | Apnoeas, feeding problems, muscular hypotonia, ptosis, horizontal nystagmus, ophthalmoplegia, knee flexure contractures | Cardiorespiratory arrest, poor sucking and swallowing, amimia, ptosis | Retrognathia, axial and peripheral hypotonia, distal arthrogryposis, dislocated hips, respiratory insufficiency | Retrognathia, axial and peripheral hypotonia, distal arthrogryposis, dislocated hips, respiratory insufficiency | Fetal akinesia at 11+6/, termination of pregnancy at 15+3/12+1 weeks: arthrogryposis, partial cleft palate, dysmorphic facial features, finger abnormalities, generalised body oedema | Respiratory problems, desaturations, excessive secretion, feeding problems | Respiratory problems, apnoeas, bulbar symptoms |
| Additional symptoms | Undescended testis | Meconium ileus | Bilateral undescended testis, micropenis, hirsutism | Necrotising enterocolitis at the age of 2 months | - | - | - | ||
| Motor development | Delayed | Normal | Delayed | - | Severe hypotonia | - | Muscular hypotonia | Delayed | |
| Mental development | Not tested | Partial learning deficits | Regular school | Regular school | - | Mental disability | - | Speech delay | n.n. |
| Follow up symptoms | Ptosis, ophthalmoplegia, facial hypomimia, swallowing problems, respiratory worsening during infections, joint contractures; can sit independently at the age of five for few minutes | Endurance problems, fatigability, ptosis, ophthalmoplegia, mild facial weakness | Walking possible at age of 4, lost of ambulance at 5, can stand, no walking | Ptosis, ophthalmoplegia, apnoeas, endurance problems, reduced walking distance | Died at the age of 5 days due to respiratory insufficiency | Ventilatory support, extreme muscular hypotonia, microcephaly, horizontal nystagmus, minimal voluntary movements in the upper extremities | - | Ptosis, “head drop”, truncal hypotonia; Speech delay, can walk with support | Muscular hypotonia, poor head control, nystagmus; no ptosis |
| Brain imagines | MRI: brain atrophy | MRI: mild hyperintensity of the white matter and small punctate haemorrhages in the frontal and parietal lobes | n.n. | CT: delayed myelinisation | CT: brain atrophy | - | MRI: normal | MRI: normal | |
| Echocardiography | normal | Mild left ventricular function insufficiency | normal | n.n. | n.d. | n.d. | n.d. | n.d. | normal |
| Repetitive stimulation | abnormal | First normal, abnormal after exercise | abnormal | EMG normal | n.d. | n.d. | - | abnormal | n.n. |
| Response to therapy (start) | PS+, 3,4 DAP-, Ephedrine - | PS (14) + | PS (3 mo)+ 3,4 DAP + Distigmine + Ephedrine + | PS (neonatal)+ | - | No therapy | - | PS (12 mo)+, 3,4 DAP+ | PS (2 mo)+ |
| Ratios | 2932/2921 | 2868/2847 | 2932/2868 | 2932/2847 | 2921/2868 | 2921/2947 |
|---|---|---|---|---|---|---|
| Inside fibres Between fibres Inside connective tissue | 0.92 0.93 1.01 | 0.70 0.56 0.57 | 0.88 0.73 0.86 | 0.62 0.41 0.49 | 0.95 0.78 0.85 | 0.67 0.44 0.49 |
| Subsarcolemmal protein | 1.02 | 0.99 | 1.21 | 1.20 | 1.19 | 1.18 |
| Reference | 0.86 | 1.14 | 0.88 | 1.00 | 1.02 | 1.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Della Marina, A.; Arlt, A.; Schara-Schmidt, U.; Depienne, C.; Gangfuß, A.; Kölbel, H.; Sickmann, A.; Freier, E.; Kohlschmidt, N.; Hentschel, A.; et al. Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3. Cells 2021, 10, 3481. https://doi.org/10.3390/cells10123481
Della Marina A, Arlt A, Schara-Schmidt U, Depienne C, Gangfuß A, Kölbel H, Sickmann A, Freier E, Kohlschmidt N, Hentschel A, et al. Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3. Cells. 2021; 10(12):3481. https://doi.org/10.3390/cells10123481
Chicago/Turabian StyleDella Marina, Adela, Annabelle Arlt, Ulrike Schara-Schmidt, Christel Depienne, Andrea Gangfuß, Heike Kölbel, Albert Sickmann, Erik Freier, Nicolai Kohlschmidt, Andreas Hentschel, and et al. 2021. "Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3" Cells 10, no. 12: 3481. https://doi.org/10.3390/cells10123481
APA StyleDella Marina, A., Arlt, A., Schara-Schmidt, U., Depienne, C., Gangfuß, A., Kölbel, H., Sickmann, A., Freier, E., Kohlschmidt, N., Hentschel, A., Weis, J., Czech, A., Grüneboom, A., & Roos, A. (2021). Phenotypical and Myopathological Consequences of Compound Heterozygous Missense and Nonsense Variants in SLC18A3. Cells, 10(12), 3481. https://doi.org/10.3390/cells10123481