
A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma

 ,
,  , , , , , , ,
, , , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. UAMS Clinical Cohort
2.2. RNA Extraction and Library Construction
2.3. Gene Expression Outlier Analysis
2.4. TumorMap Analysis
2.5. DNA Mutation Analysis
2.6. Cell Culture
2.7. RNA Extraction and RNAseq from Cell Cultures
2.8. Clonogenic Assay
2.9. Mini-Ring Organoid Viability Assay
2.10. 3D-PREDICT
3. Results
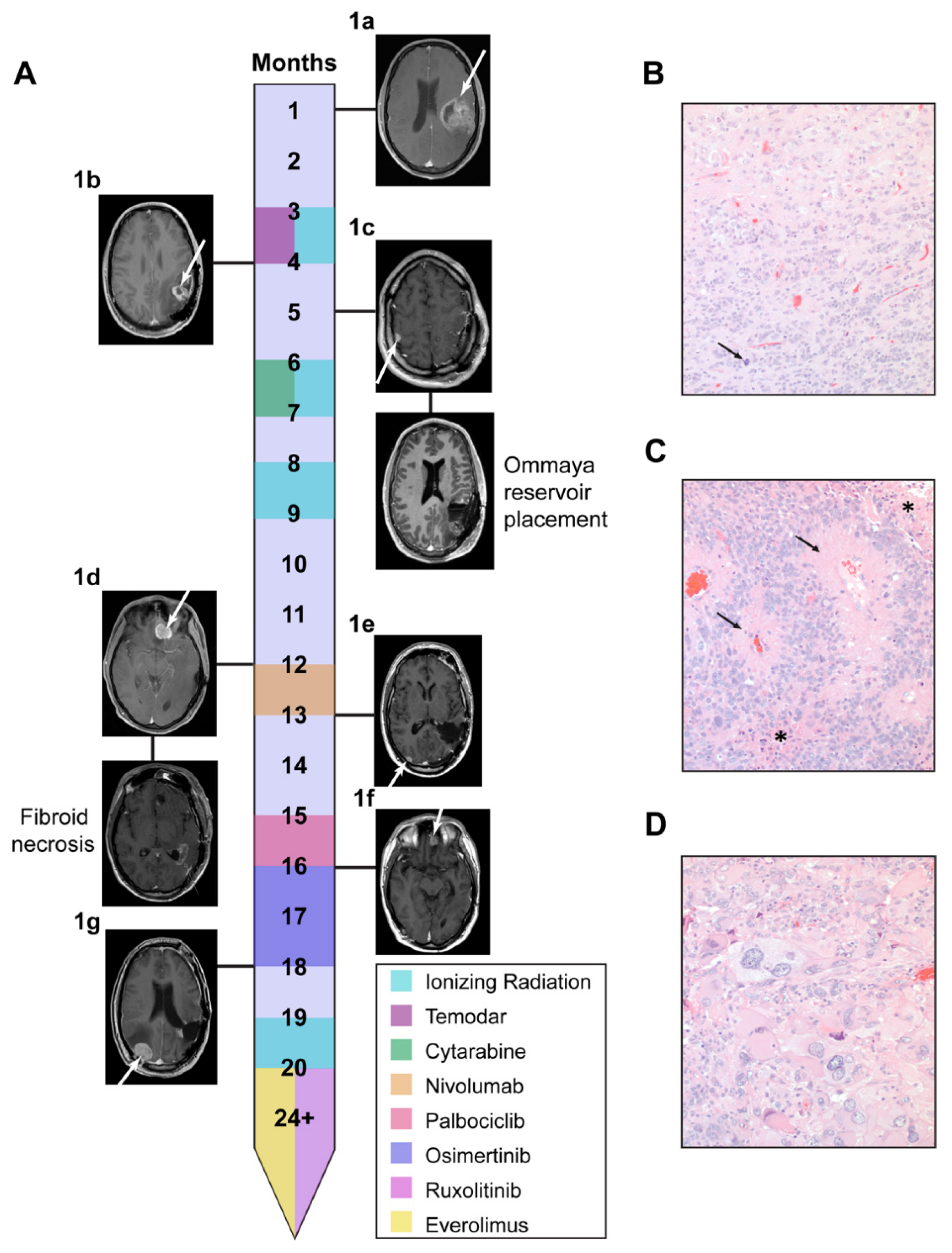
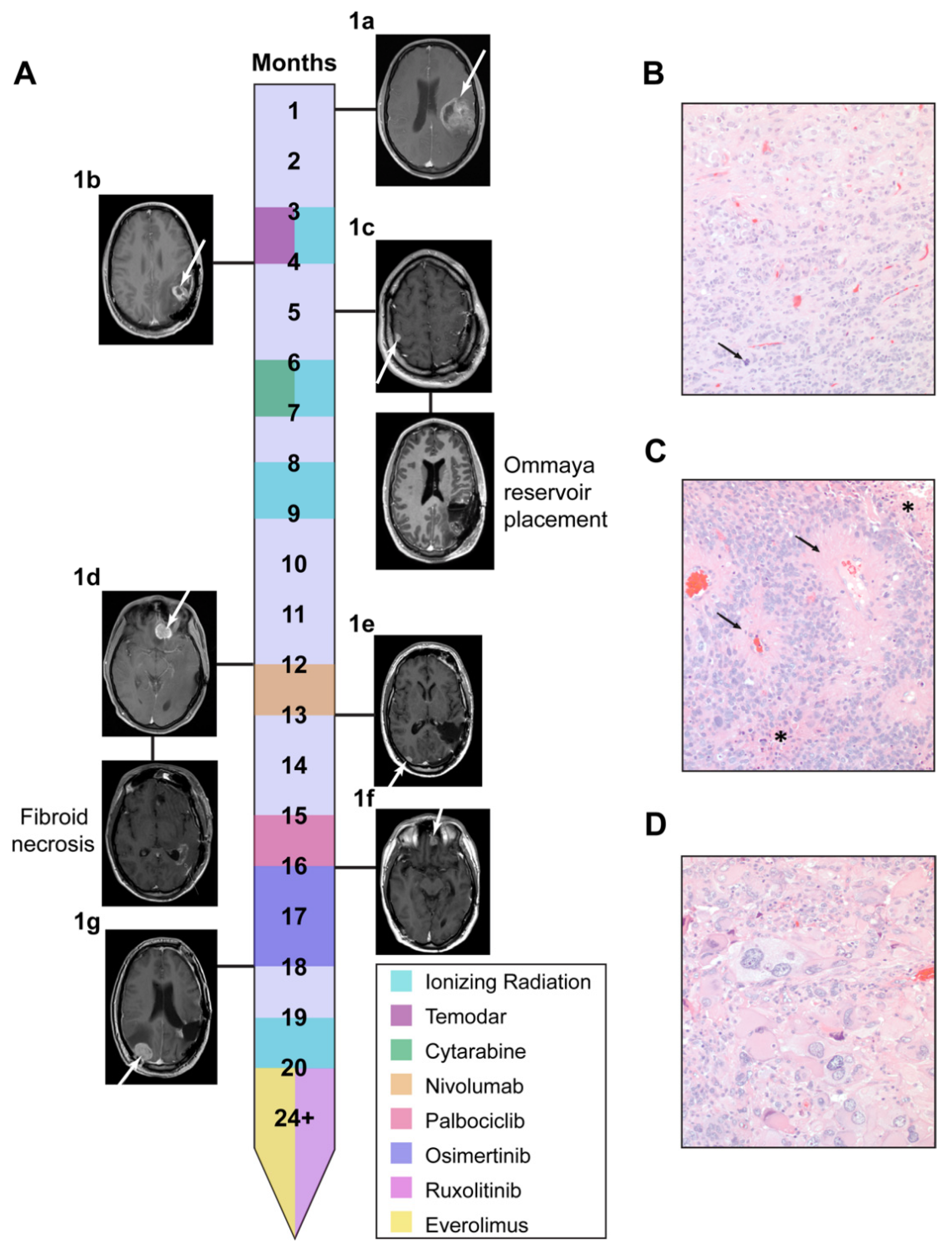
3.1. Li Fraumeni Patient Was Identified as a Candidate for Comparative Transcriptomics Pipeline
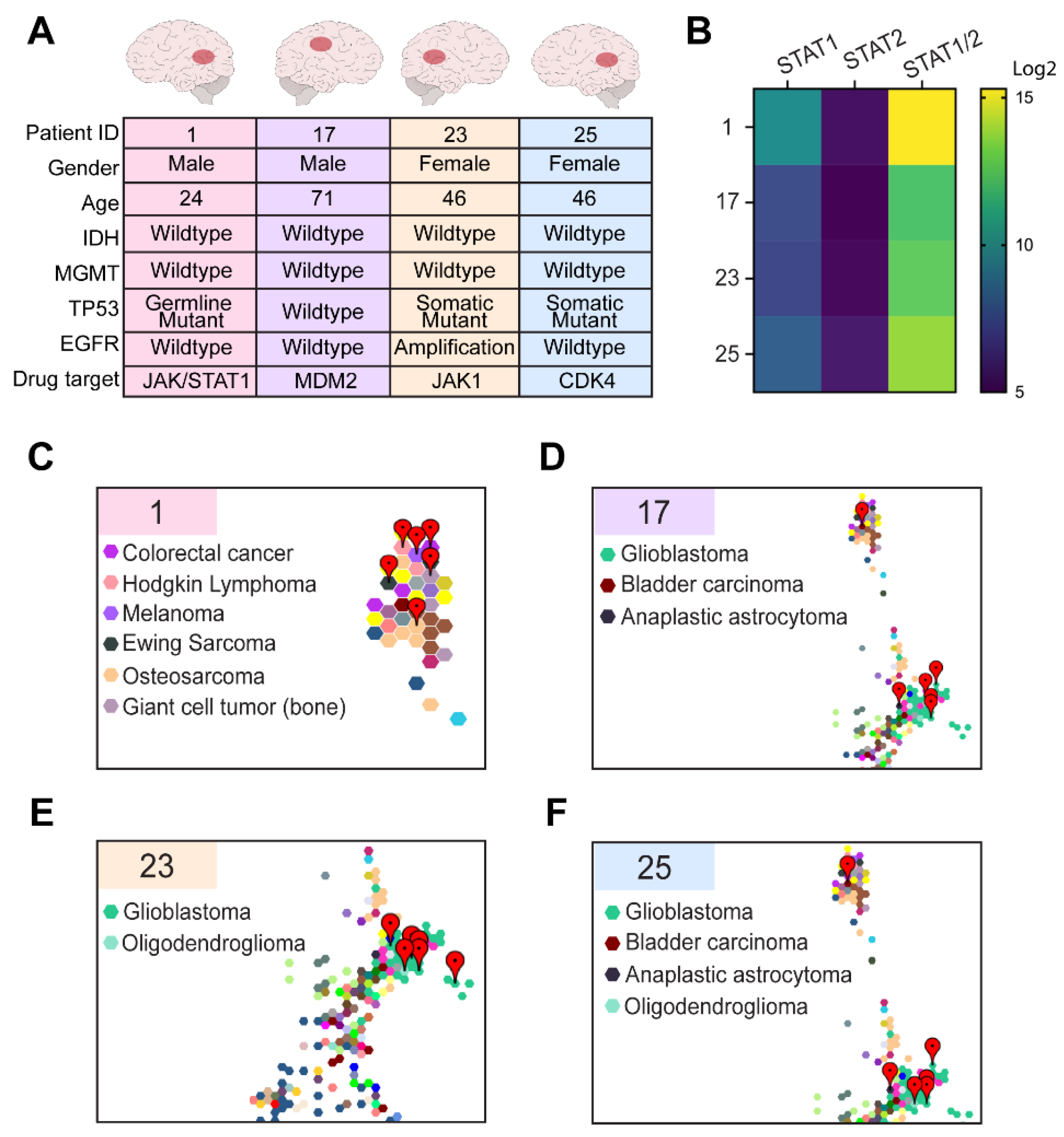
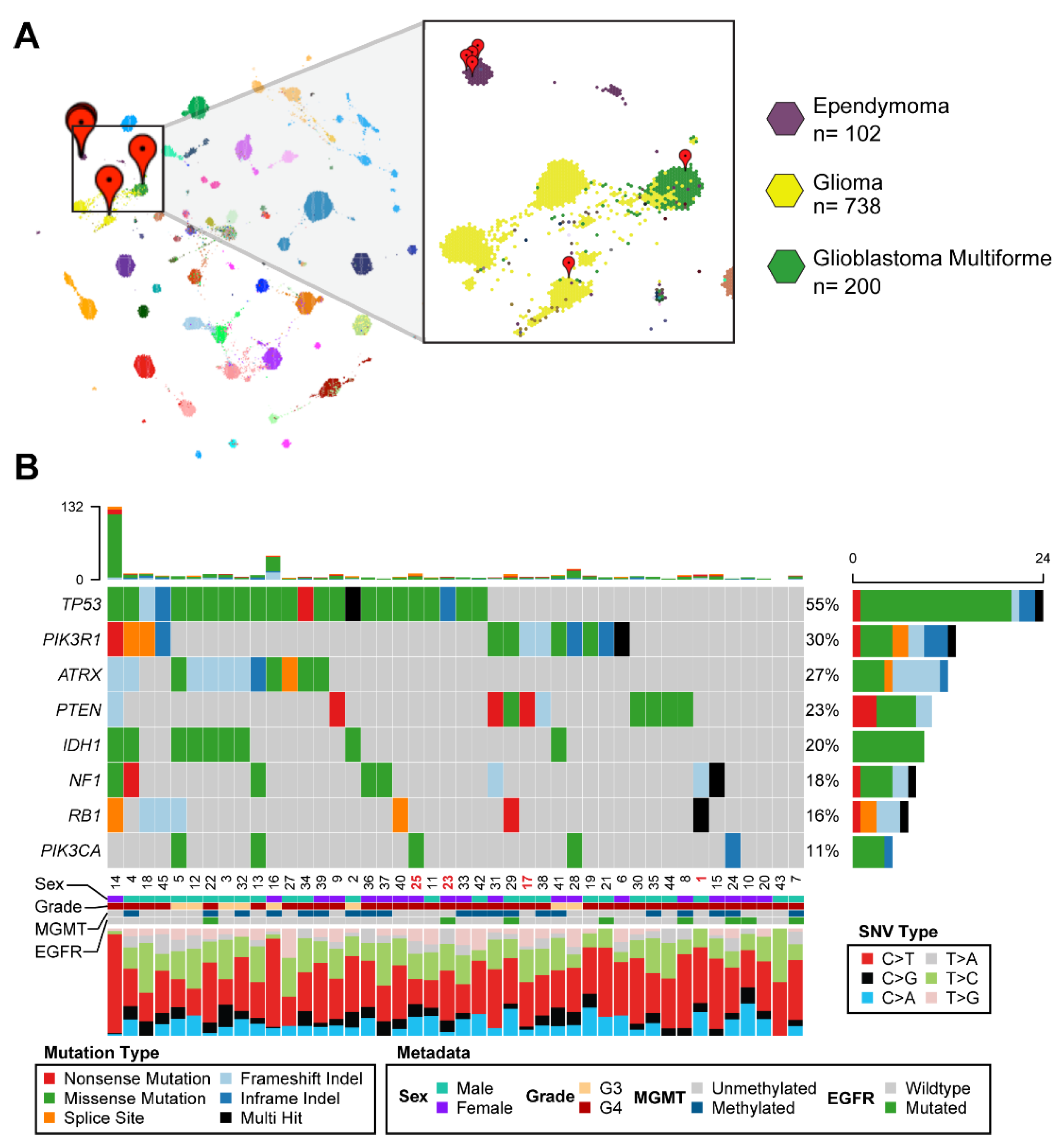
3.2. Comparative Transcriptomic Analysis Demonstrates Unique Features of the Tumor
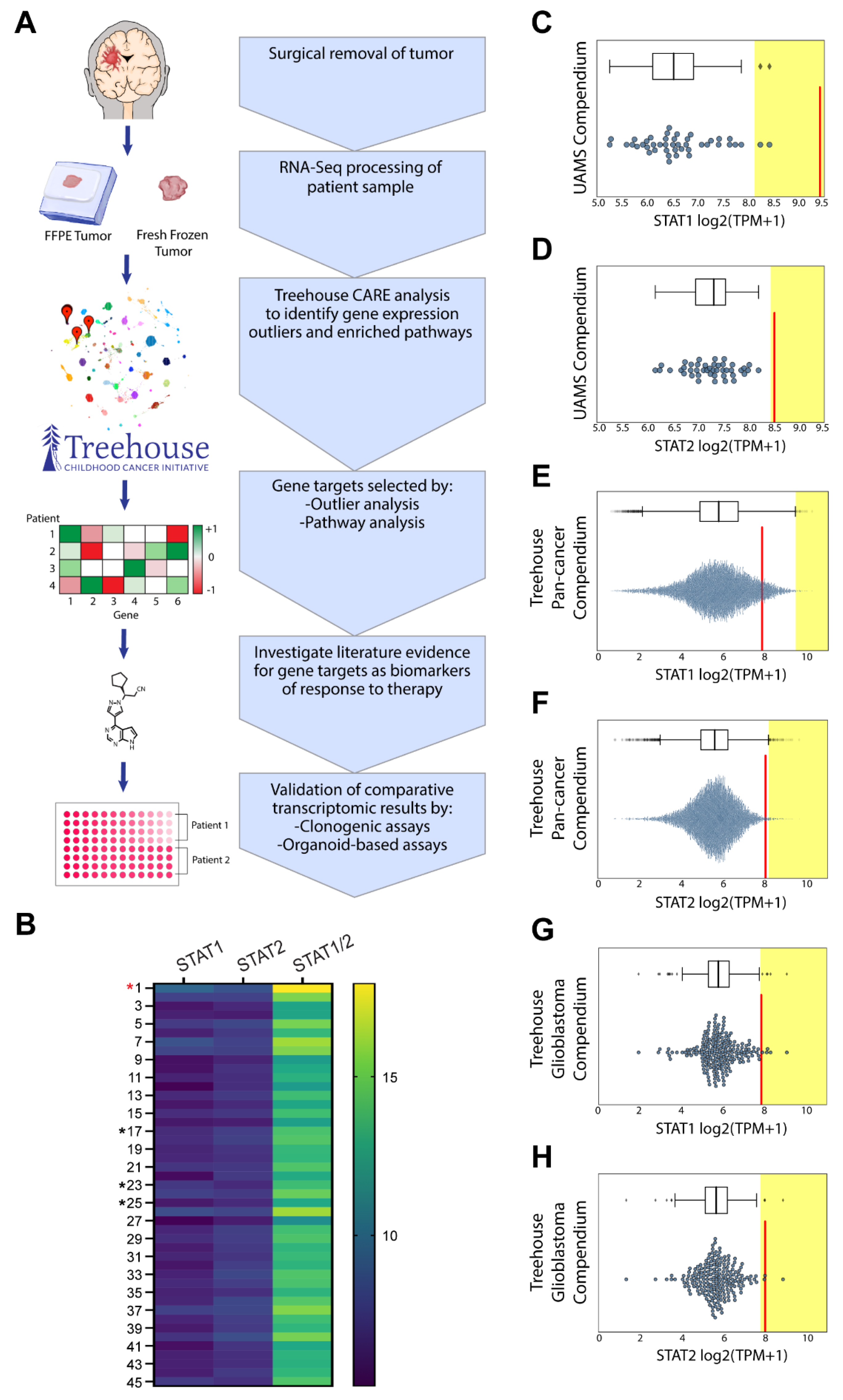
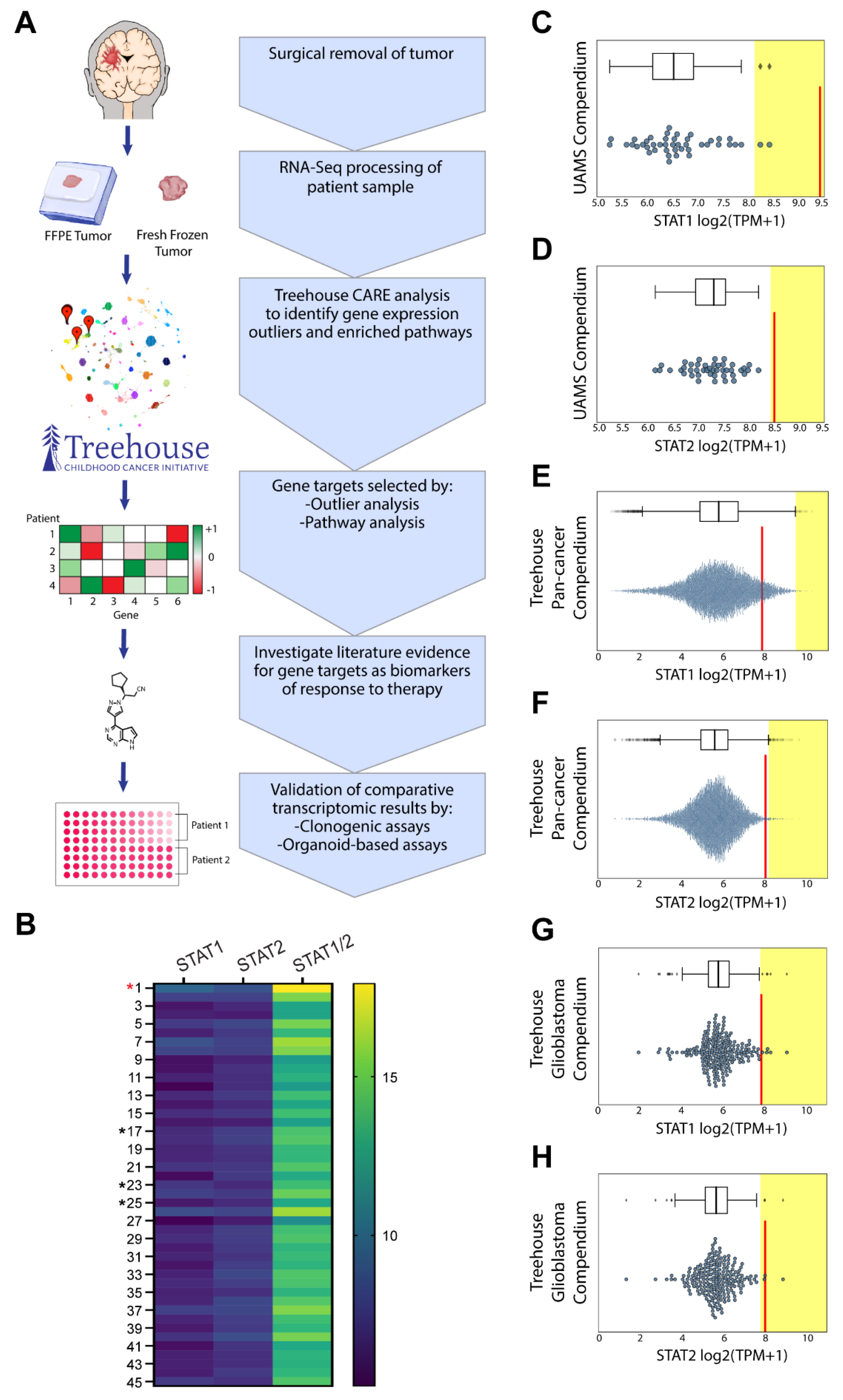
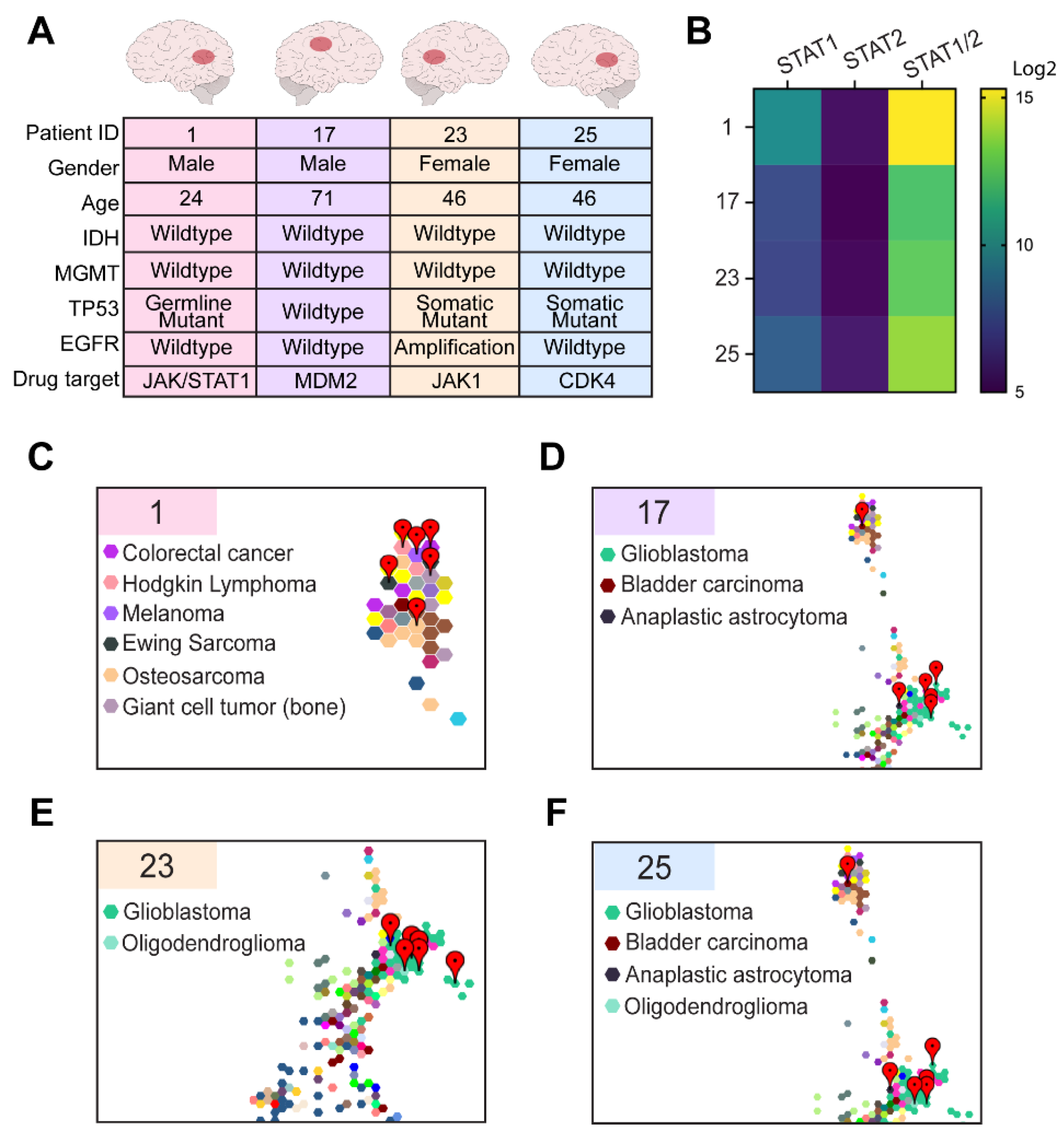
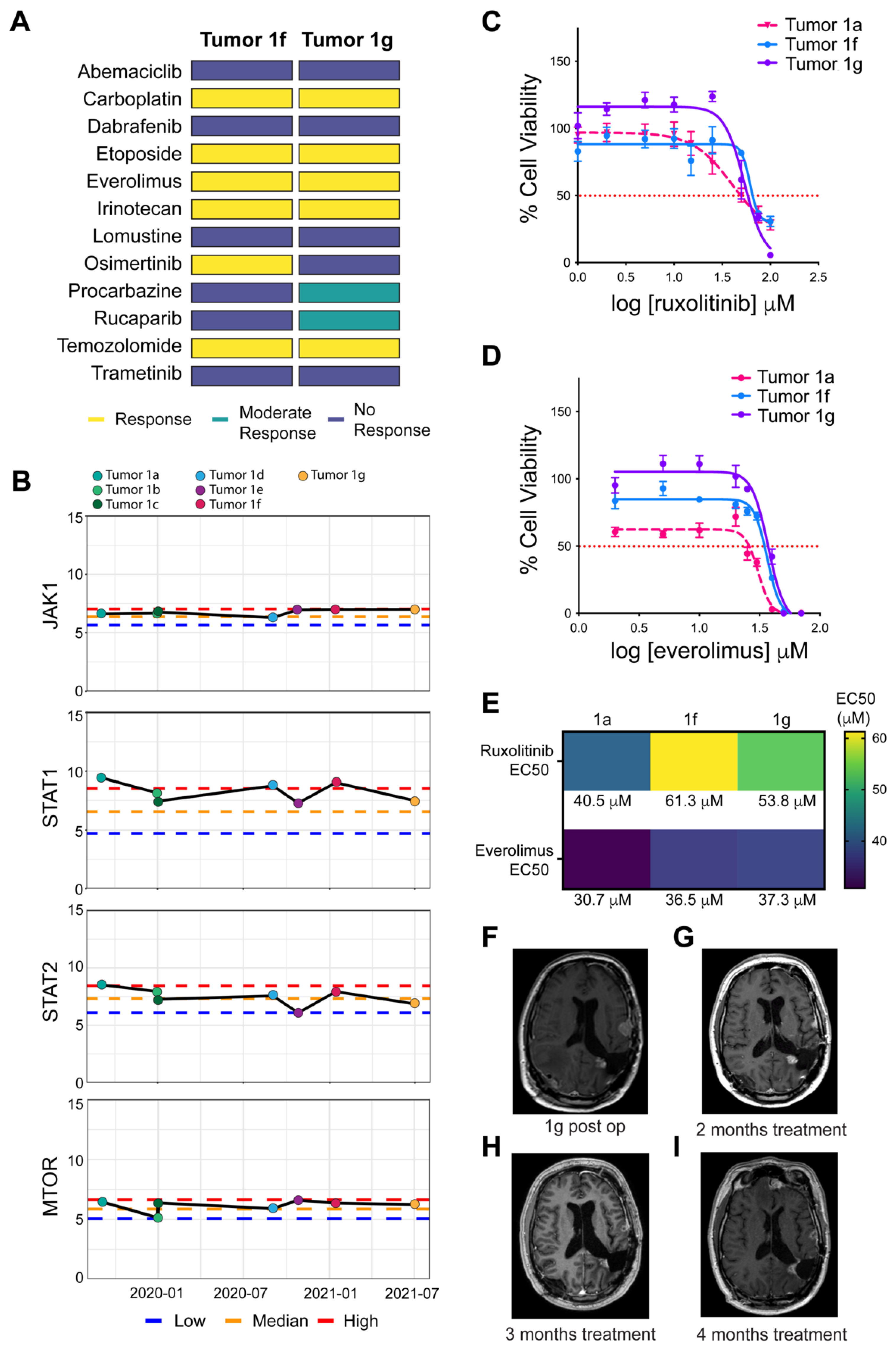
3.3. Comparative Transcriptomic Analysis Demonstrates Outlier Gene Expression in STAT1 and STAT2
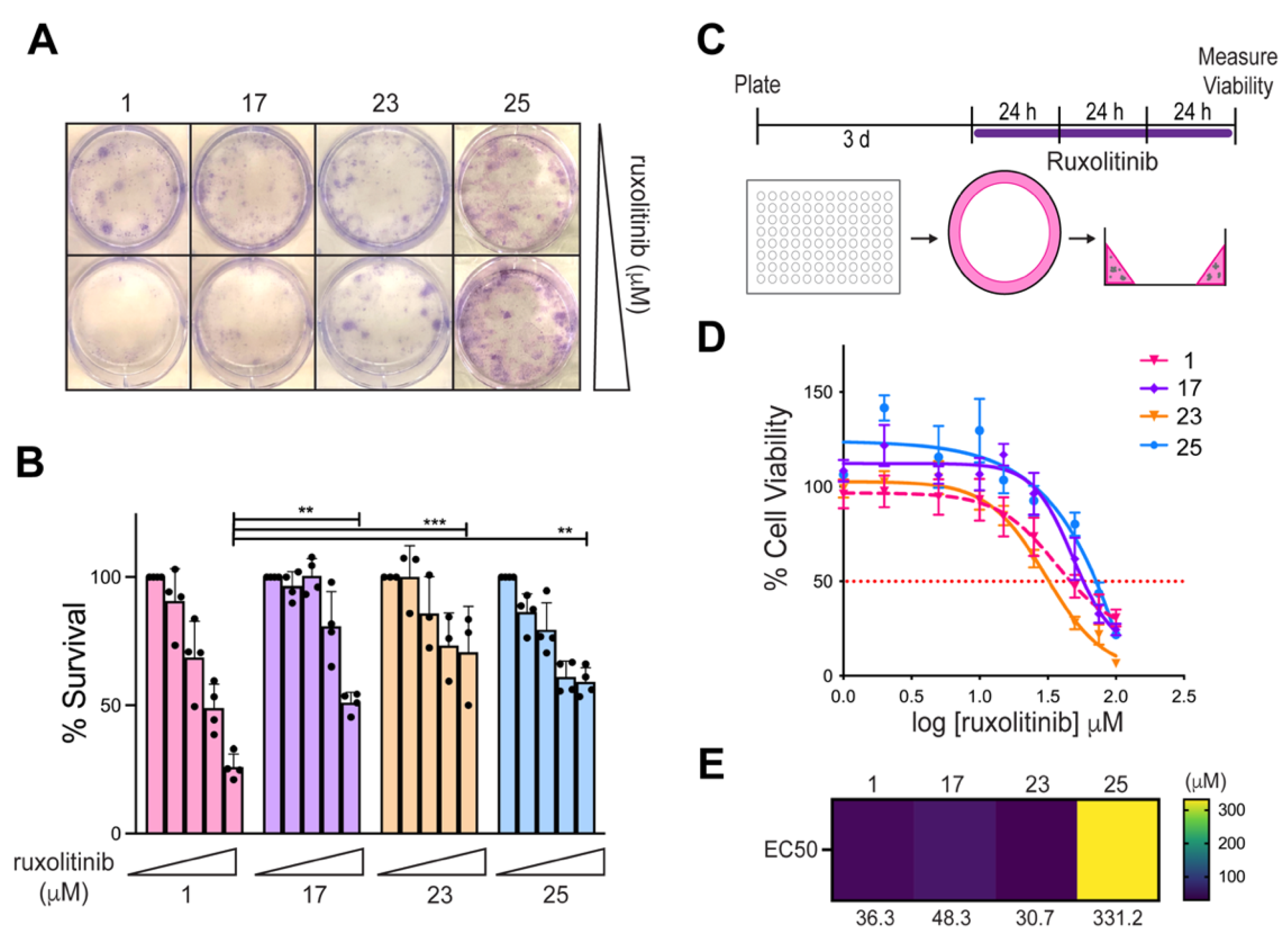
3.4. Comparative Transcriptomic Analysis Was Functionally Validated Using Patient-Derived Cell Lines and Organoids
3.5. 3D-PREDICT Trial Identifies mTOR Inhibition as an Additional Therapeutic Target
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Vecchione-Koval, T.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro. Oncol. 2017, 19, v1–v88. [Google Scholar] [CrossRef] [Green Version]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro. Oncol. 2019, 21, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Correa, H. Li-Fraumeni Syndrome. J. Pediatr. Genet. 2016, 5, 84–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.-X.; Liu, J.-P.; You, C.; Liu, Y.-H.; Mao, Q. Gain of function of mutant TP53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Park, C.; Na, D.; Han, J.Y.; Lee, J.; Park, O.-K.; Zhang, C.; Sung, C.O.; Moon, H.E.; Kim, Y.; et al. High prevalence of TP53 mutations is associated with poor survival and an EMT signature in gliosarcoma patients. Exp. Mol. Med. 2017, 49, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Xiao, W.; Song, T.; Feng, G.; Dai, Z. Expression and Prognostic Significance of p53 in Glioma Patients: A Meta-analysis. Neurochem. Res. 2016, 41, 1723–1731. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Gray, R.; Chen, A.; Li, S.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Mitchell, E.P.; Iafrate, A.J.; Sklar, J.; et al. The Molecular Analysis for Therapy Choice (NCI-MATCH) Trial: Lessons for Genomic Trial Design. J. Natl. Cancer Inst. 2020, 112, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaubier, N.; Bontrager, M.; Huether, R.; Igartua, C.; Lau, D.; Tell, R.; Bobe, A.M.; Bush, S.; Chang, A.L.; Hoskinson, D.C.; et al. Integrated genomic profiling expands clinical options for patients with cancer. Nat. Biotechnol. 2019, 37, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Vaske, O.M.; Bjork, I.; Salama, S.R.; Beale, H.; Tayi Shah, A.; Sanders, L.; Pfeil, J.; Lam, D.L.; Learned, K.; Durbin, A.; et al. Comparative Tumor RNA Sequencing Analysis for Difficult-to-Treat Pediatric and Young Adult Patients with Cancer. JAMA Netw. Open 2019, 2, e1913968. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Parekh, S.; Ziegenhain, C.; Vieth, B.; Enard, W.; Hellmann, I. The impact of amplification on differential expression analyses by RNA-seq. Sci. Rep. 2016, 6, 25533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, Y.; Novak, A.M.; Swatloski, T.; McColl, D.C.; Chopra, S.; Graim, K.; Weinstein, A.S.; Baertsch, R.; Salama, S.R.; Ellrott, K.; et al. TumorMap: Exploring the Molecular Similarities of Cancer Samples in an Interactive Portal. Cancer Res. 2017, 77, e111–e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Wardell, C.P.; Ashby, C.; Bauer, M.A. FiNGS: High quality somatic mutations using filters for next generation sequencing. BMC Bioinform. 2021, 22, 77. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef]
- Phan, N.; Hong, J.J.; Tofig, B.; Mapua, M.; Elashoff, D.; Moatamed, N.A.; Huang, J.; Memarzadeh, S.; Damoiseaux, R.; Soragni, A. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2019, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shuford, S.; Wilhelm, C.; Rayner, M.; Elrod, A.; Millard, M.; Mattingly, C.; Lotstein, A.; Smith, A.M.; Guo, Q.J.; O’Donnell, L.; et al. Prospective Validation of an Ex Vivo, Patient-Derived 3D Spheroid Model for Response Predictions in Newly Diagnosed Ovarian Cancer. Sci. Rep. 2019, 9, 11153. [Google Scholar] [CrossRef] [Green Version]
- Shuford, S.; Lipinski, L.; Abad, A.; Smith, A.M.; Rayner, M.; O’Donnell, L.; Stuart, J.; Mechtler, L.L.; Fabiano, A.J.; Edenfield, J.; et al. Prospective prediction of clinical drug response in high-grade gliomas using an ex vivo 3D cell culture assay. Neurooncol. Adv. 2021, 3, vdab065. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.-H.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro. Oncol. 2018, 20, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Lobbous, M.; Bernstock, J.D.; Coffee, E.; Friedman, G.K.; Metrock, L.K.; Chagoya, G.; Elsayed, G.; Nakano, I.; Hackney, J.R.; Korf, B.R.; et al. An Update on Neurofibromatosis Type 1-Associated Gliomas. Cancers 2020, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Wang, L.; Shao, Y.; Cao, W.; Xu, T.; Chen, S.; Wang, Z.; He, Q.; Yang, K. A novel dysfunctional germline P53 mutation identified in a family with Li-Fraumeni syndrome. Am. J. Cancer Res. 2018, 8, 165–169. [Google Scholar] [PubMed]
- Xu, J.; Qian, J.; Hu, Y.; Wang, J.; Zhou, X.; Chen, H.; Fang, J.-Y. Heterogeneity of Li-Fraumeni syndrome links to unequal gain-of-function effects of p53 mutations. Sci. Rep. 2014, 4, 4223. [Google Scholar] [CrossRef] [Green Version]
- Orr, B.A.; Clay, M.R.; Pinto, E.M.; Kesserwan, C. An update on the central nervous system manifestations of Li-Fraumeni syndrome. Acta Neuropathol. 2020, 139, 669–687. [Google Scholar] [CrossRef]
- Sloan, E.A.; Hilz, S.; Gupta, R.; Cadwell, C.; Ramani, B.; Hofmann, J.; Kline, C.N.; Banerjee, A.; Reddy, A.; Oberheim Bush, N.A.; et al. Gliomas arising in the setting of Li-Fraumeni syndrome stratify into two molecular subgroups with divergent clinicopathologic features. Acta Neuropathol. 2020, 139, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Gray, R.J.; Chen, A.P.; Li, S.; McShane, L.M.; Patton, D.; Hamilton, S.R.; Williams, P.M.; Iafrate, A.J.; Sklar, J.; et al. Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J. Clin. Oncol. 2020, 38, 3883–3894. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Hong, F.; McCourt, C.K.; Sachdev, J.C.; Mitchell, E.P.; Zwiebel, J.A.; Doyle, L.A.; McShane, L.M.; Li, S.; Gray, R.J.; et al. Effect of Capivasertib in Patients with an AKT1 E17K-Mutated Tumor: NCI-MATCH Subprotocol EAY131-Y Nonrandomized Trial. JAMA Oncol. 2021, 7, 271–278. [Google Scholar] [CrossRef]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.-M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.-S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef] [Green Version]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef] [PubMed]
- Meissl, K.; Macho-Maschler, S.; Müller, M.; Strobl, B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017, 89, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.H.; Wang, Y.; Sims, M.; Cai, C.; He, P.; Yue, J.; Cheng, J.; Boop, F.A.; Pfeffer, S.R.; Pfeffer, L.M. MiRNA203 suppresses the expression of protumorigenic STAT1 in glioblastoma to inhibit tumorigenesis. Oncotarget 2016, 7, 84017–84029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Imani, S.; Deng, Y.; Pathak, J.L.; Wen, Q.; Chen, Y.; Wu, J. Targeting IFN/STAT1 Pathway as a Promising Strategy to Overcome Radioresistance. Onco Targets Ther. 2020, 13, 6037–6050. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Cui, B.; Johnson, S.P.; Bullock, N.; Ali-Osman, F.; Bigner, D.D.; Friedman, H.S. Decoupling of DNA damage response signaling from DNA damages underlies temozolomide resistance in glioblastoma cells. J. Biomed. Res. 2010, 24, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, G.-Z.; Huang, G.; Guo, M.; Zhang, X.; Wang, H.; Deng, S.; Li, Y.; Xiang, W.; Chen, Z.; Pan, J.; et al. Acquired temozolomide resistance in MGMT-deficient glioblastoma cells is associated with regulation of DNA repair by DHC2. Brain 2019, 142, 2352–2366. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Spearman Rank Correlation | Disease |
|---|---|---|
| THR24_1804_S01 | 0.873 | Ependymoma |
| THR24_1821_S01 | 0.872 | Ependymoma |
| THR13_0969_S01 | 0.866 | Glioma |
| THR24_1839_S02 | 0.864 | Ependymoma |
| THR24_1786_S01 | 0.861 | Glioblastoma multiforme |
| Tumor ID | Sample ID | Spearman Rank Correlation | Disease |
|---|---|---|---|
| G28816 | 0.872 | Ewing sarcoma | |
| G28810 | 0.872 | Cutaneous melanoma | |
| Patient 1 | G28809 | 0.869 | Colorectal carcinoma |
| G28895 | 0.868 | Osteosarcoma | |
| G28837 | 0.868 | Malignant giant cell tumor of bone | |
| G28818 | 0.867 | Hodgkin lymphoma | |
| G27543 | 0.837 | Glioblastoma | |
| G26232 | 0.834 | Glioblastoma | |
| Patient 17 | G26195 | 0.833 | Glioblastoma |
| G26217 | 0.833 | Glioblastoma | |
| G28902 | 0.832 | Bladder carcinoma | |
| G27255 | 0.831 | Anaplastic astrocytoma | |
| G26217 | 0.873 | Glioblastoma | |
| G26232 | 0.869 | Glioblastoma | |
| Patient 23 | THR13_0970_S02 | 0.861 | Glioblastoma |
| G27205 | 0.860 | Glioblastoma | |
| G27462 | 0.858 | Glioblastoma | |
| G26228 | 0.858 | Oligodendroglioma | |
| G28902 | 0.846 | Bladder carcinoma | |
| G26217 | 0.834 | Glioblastoma | |
| G26228 | 0.833 | Oligodendroglioma | |
| Patient 25 | G26232 | 0.831 | Glioblastoma |
| G27255 | 0.826 | Anaplastic astrocytoma | |
| G26195 | 0.826 | Glioblastoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reed, M.R.; Lyle, A.G.; De Loose, A.; Maddukuri, L.; Learned, K.; Beale, H.C.; Kephart, E.T.; Cheney, A.; van den Bout, A.; Lee, M.P.; et al. A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma. Cells 2021, 10, 3400. https://doi.org/10.3390/cells10123400
Reed MR, Lyle AG, De Loose A, Maddukuri L, Learned K, Beale HC, Kephart ET, Cheney A, van den Bout A, Lee MP, et al. A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma. Cells. 2021; 10(12):3400. https://doi.org/10.3390/cells10123400
Chicago/Turabian StyleReed, Megan R., A. Geoffrey Lyle, Annick De Loose, Leena Maddukuri, Katrina Learned, Holly C. Beale, Ellen T. Kephart, Allison Cheney, Anouk van den Bout, Madison P. Lee, and et al. 2021. "A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma" Cells 10, no. 12: 3400. https://doi.org/10.3390/cells10123400
APA StyleReed, M. R., Lyle, A. G., De Loose, A., Maddukuri, L., Learned, K., Beale, H. C., Kephart, E. T., Cheney, A., van den Bout, A., Lee, M. P., Hundley, K. N., Smith, A. M., DesRochers, T. M., Vibat, C. R. T., Gokden, M., Salama, S., Wardell, C. P., Eoff, R. L., Vaske, O. M., & Rodriguez, A. (2021). A Functional Precision Medicine Pipeline Combines Comparative Transcriptomics and Tumor Organoid Modeling to Identify Bespoke Treatment Strategies for Glioblastoma. Cells, 10(12), 3400. https://doi.org/10.3390/cells10123400