
GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation

 , and
, and 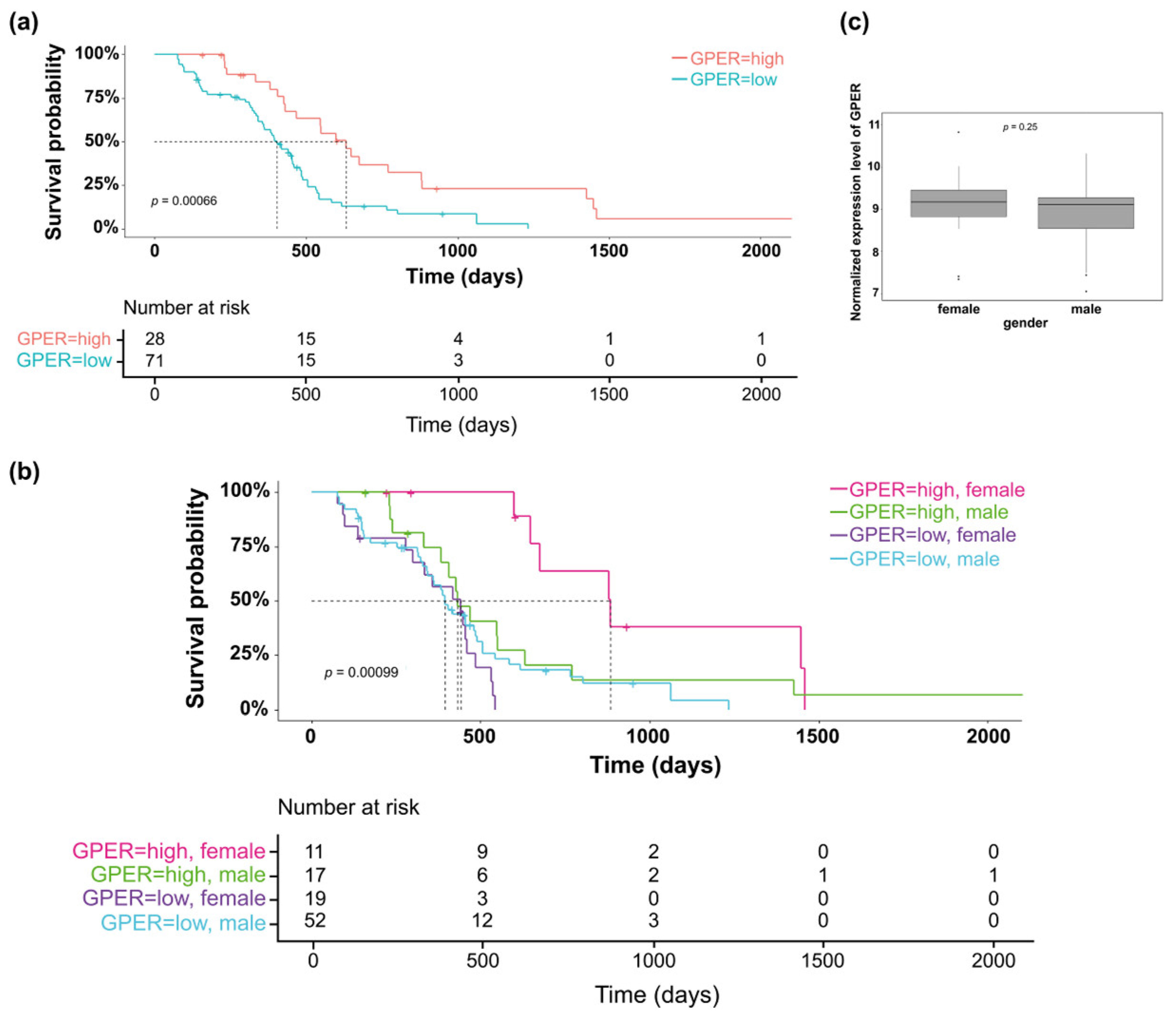{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. MTT(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide) Assay
2.3. Colony Formation Assay
2.4. Flow Cytometry
2.5. Immunofluorescence
2.6. Western Blot Analysis
2.7. Detection of Monomeric and Polymeric Tubulin
2.8. Time-Lapse Video Microscopy Imaging
2.9. GBM Patient Cohort and Survival Analysis
2.10. Statistical Analysis
3. Results
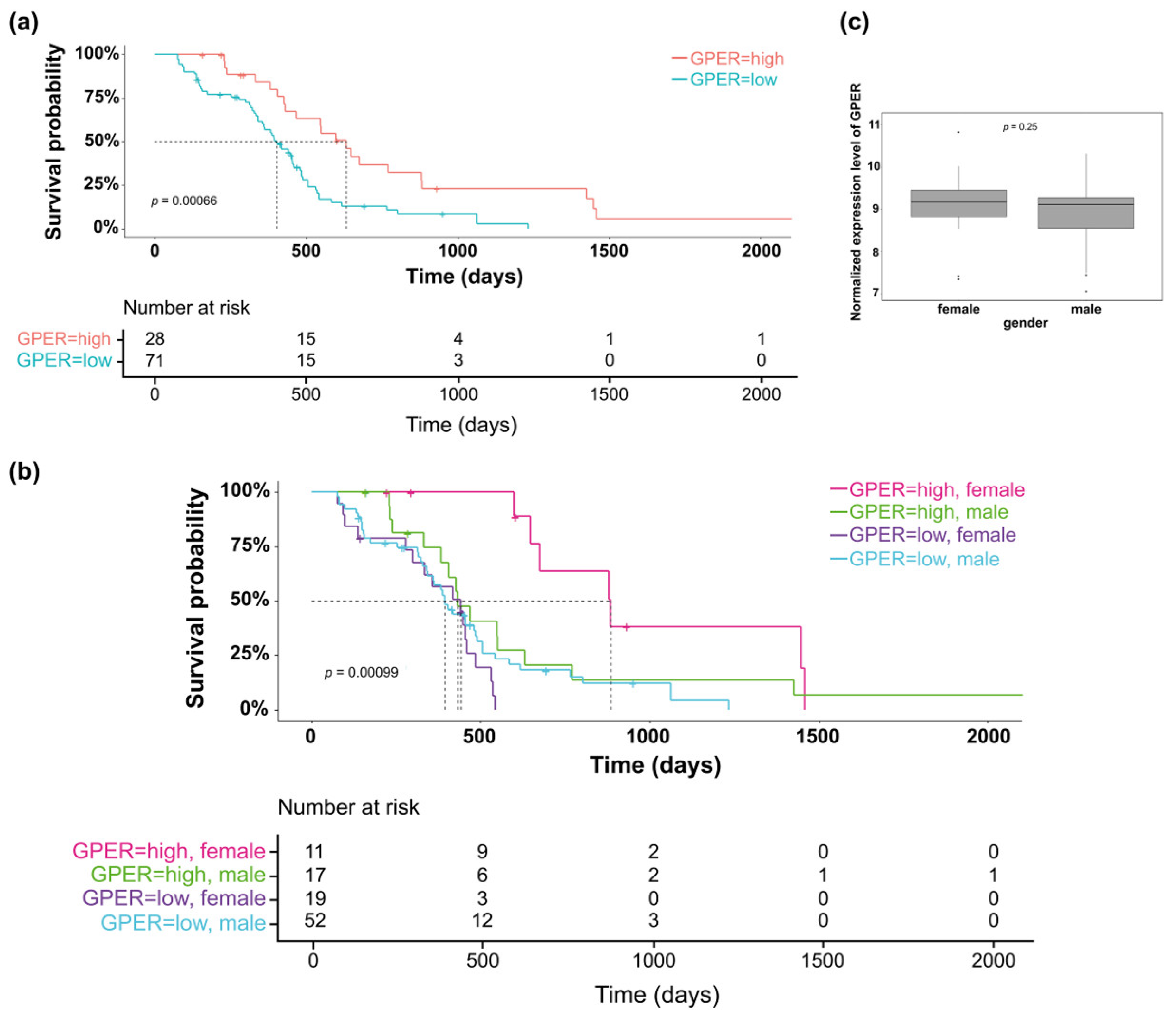
3.1. Clinical Relevance of GPER Expression in GBM Tumors
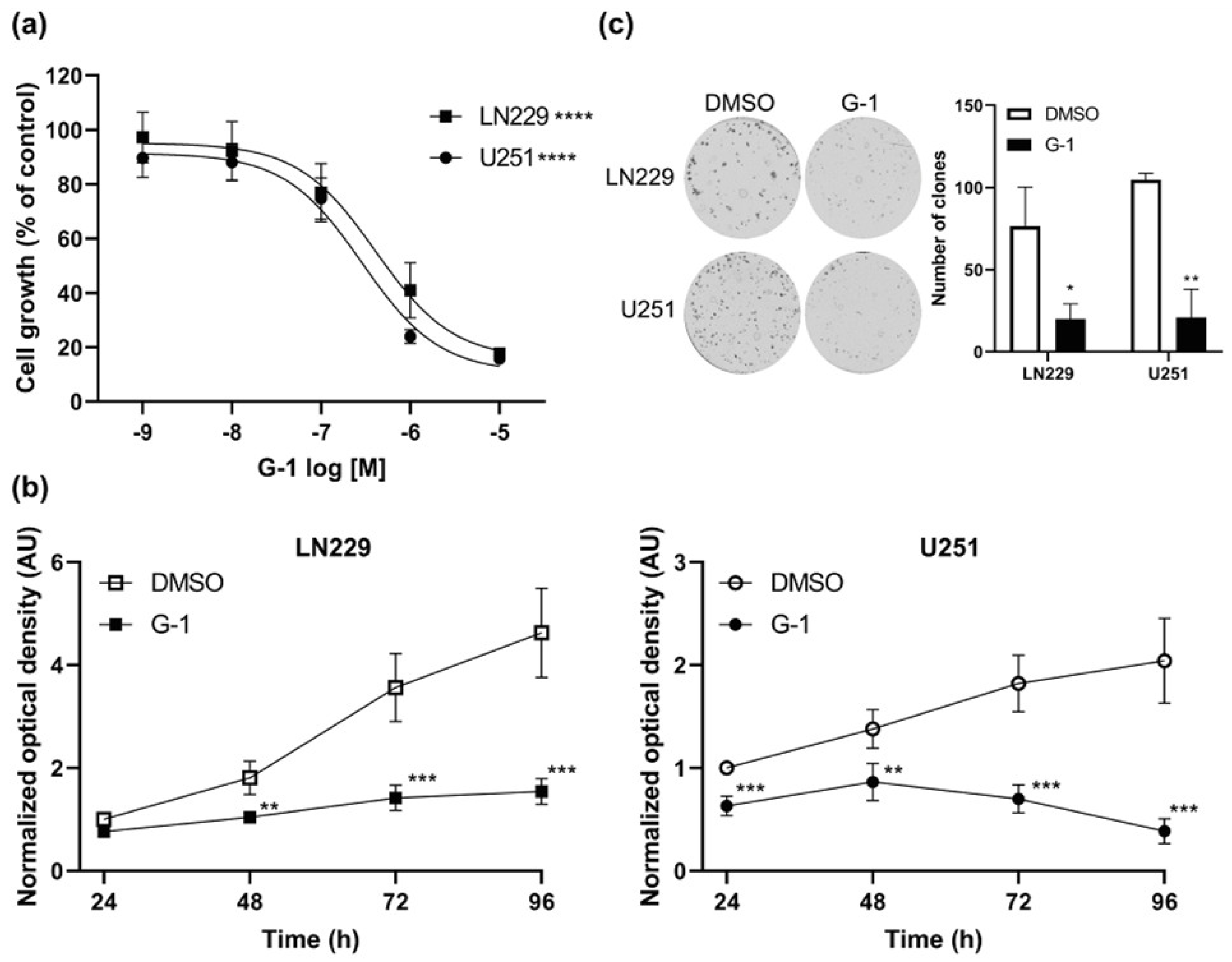
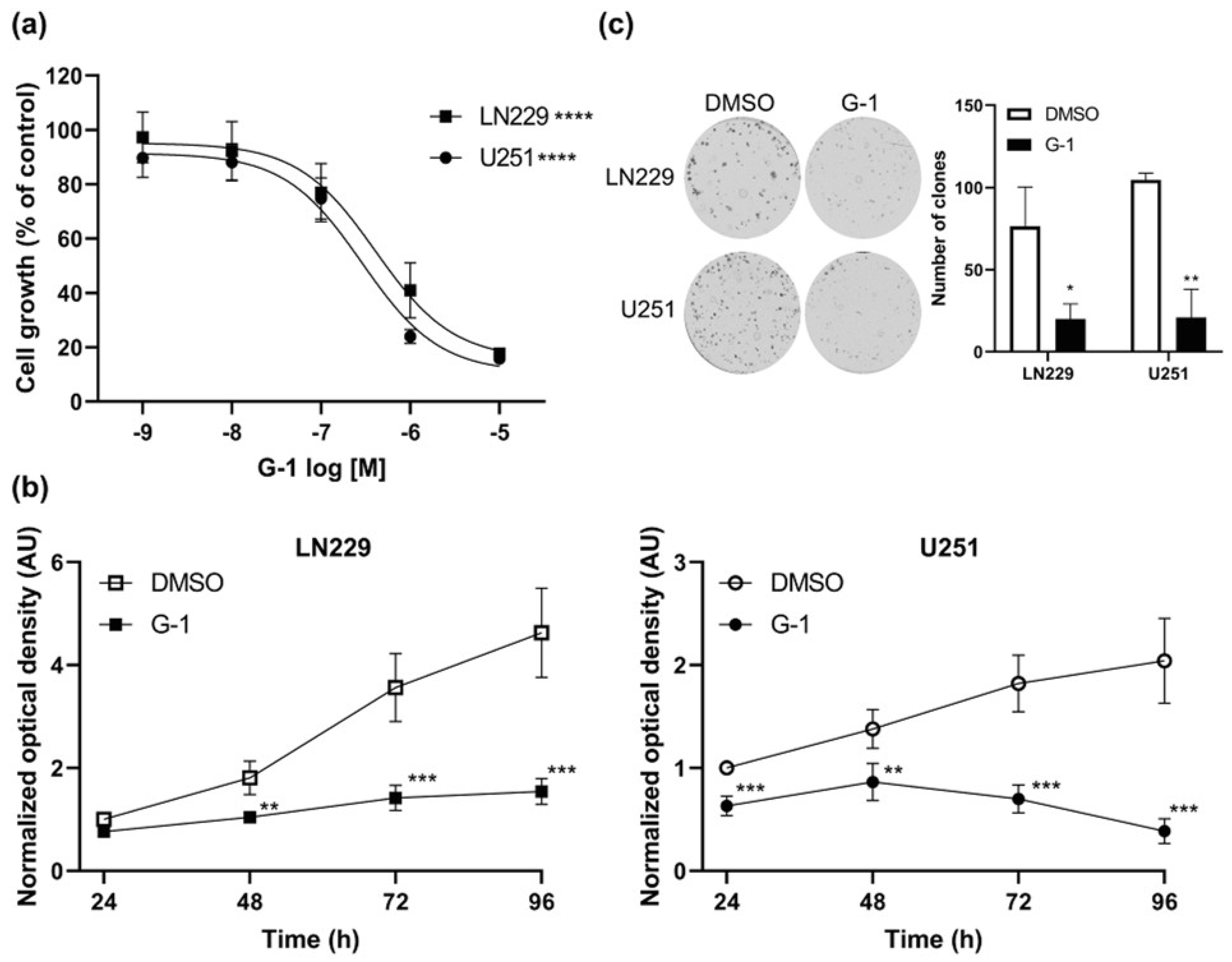
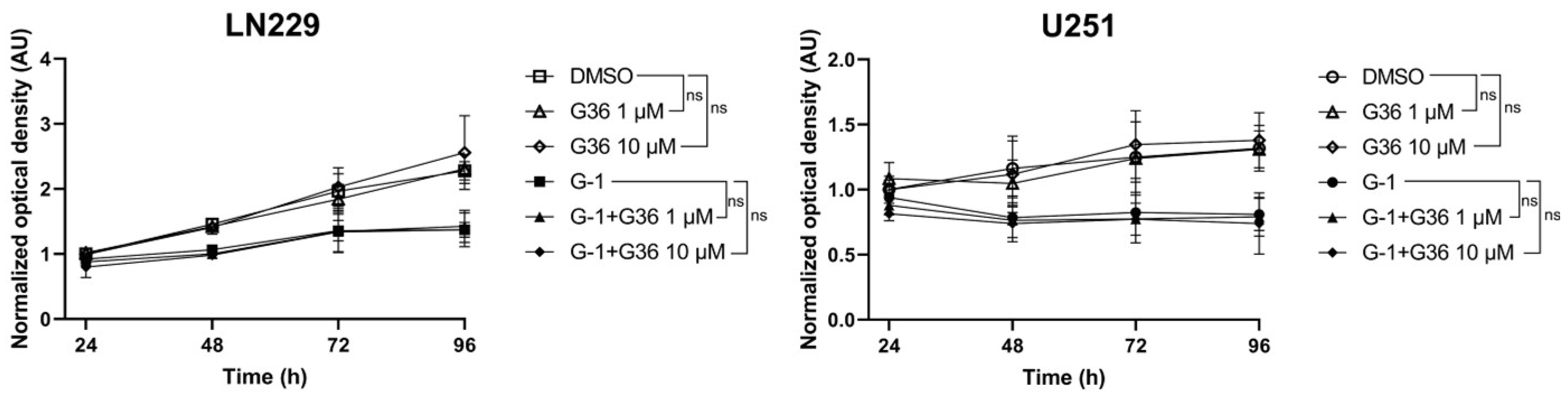
3.2. GPER Agonist G-1 Prevented GBM Cell Proliferation
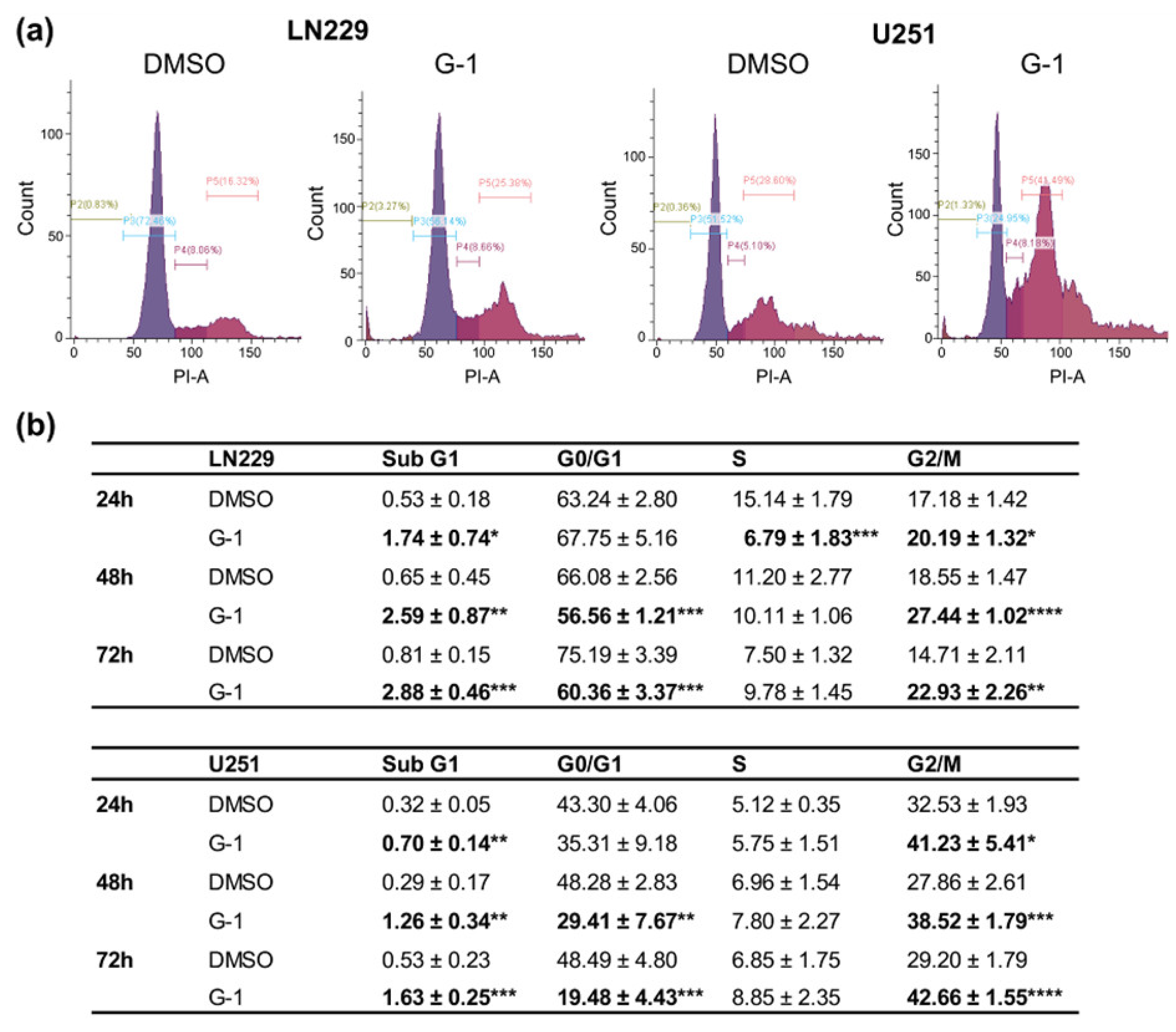
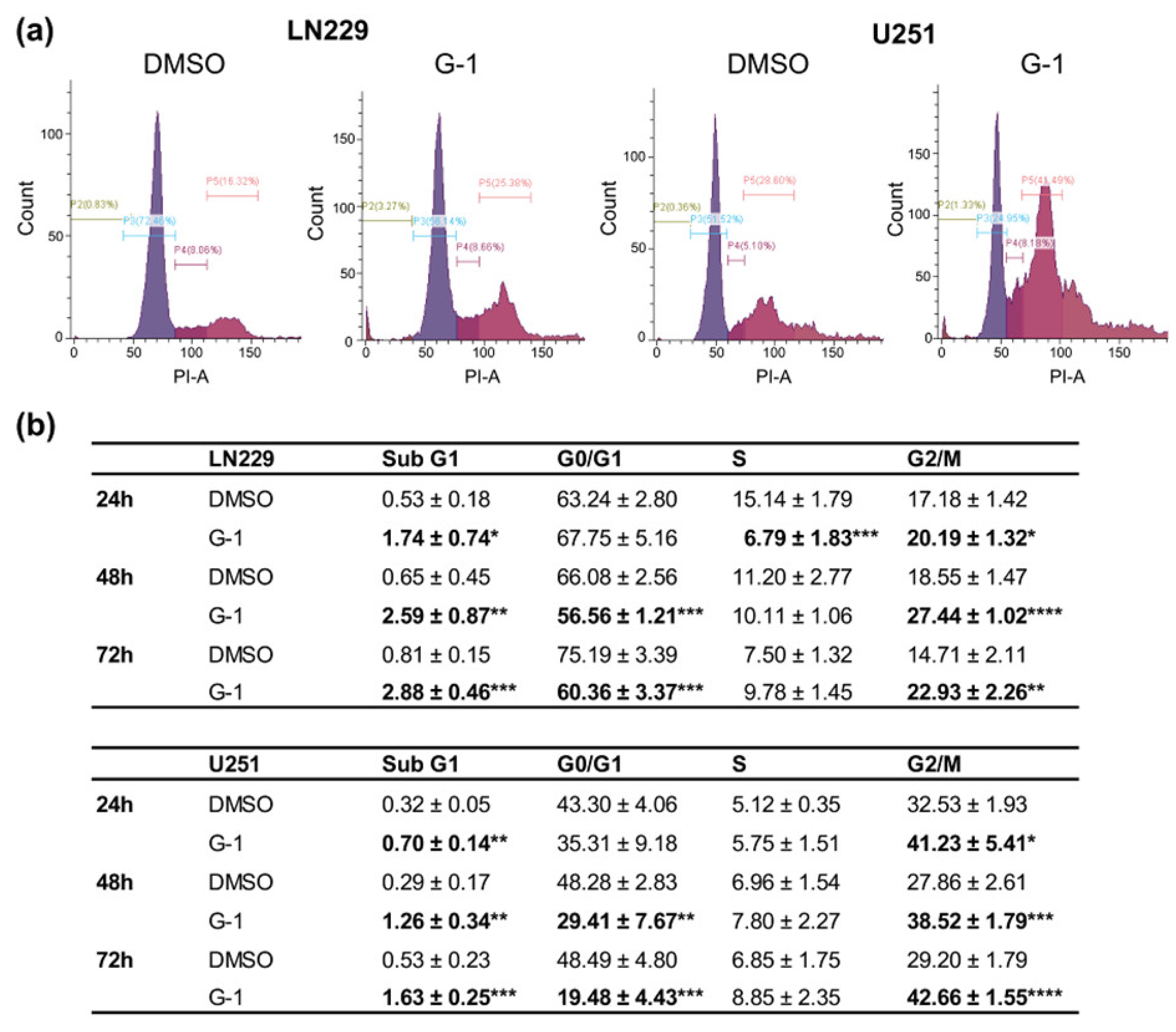
3.3. G-1 Exposure Triggered G2/M Cell Cycle Arrest in GBM Cells
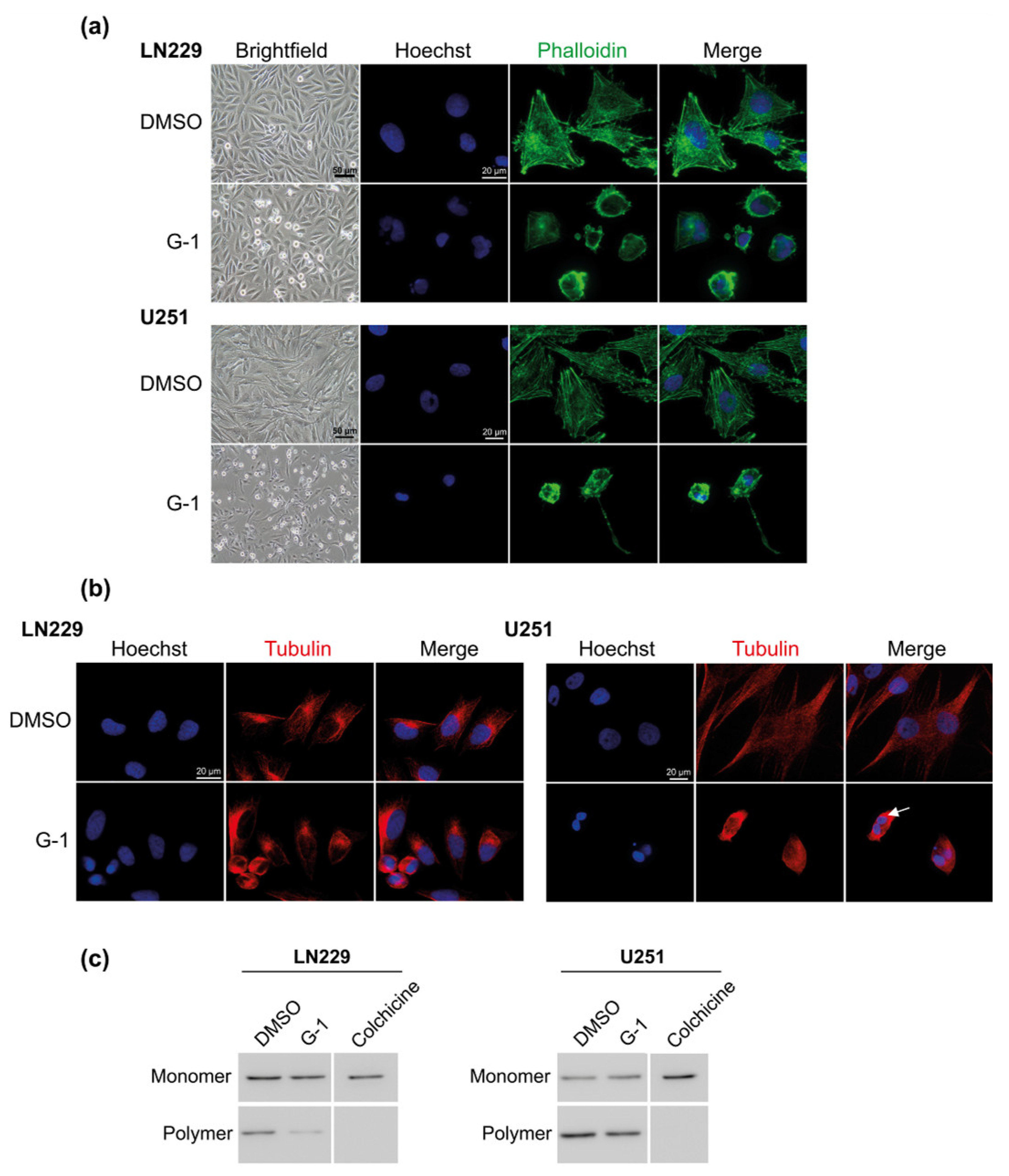
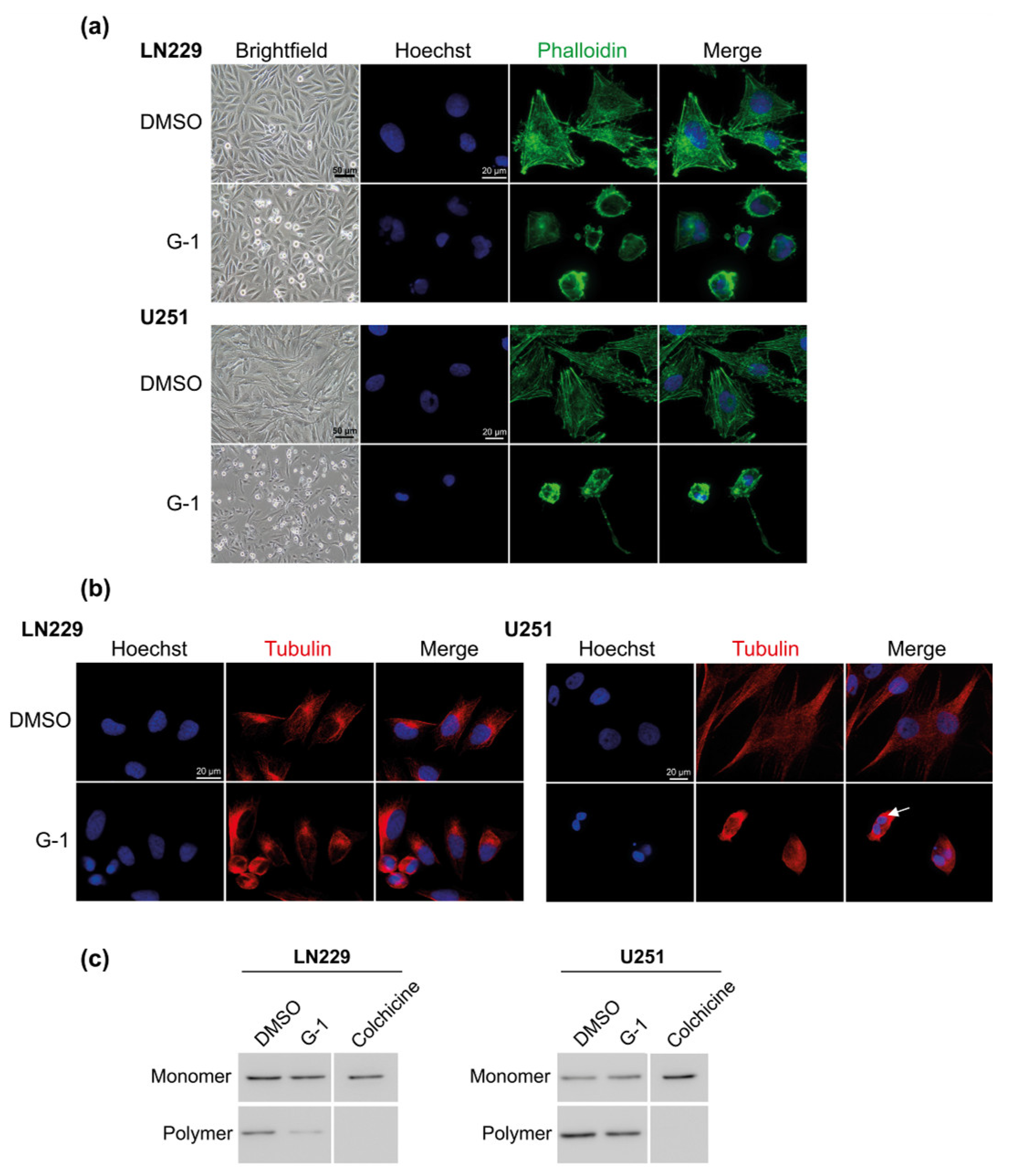
3.4. G-1 Induced a Major Reorganization of Cell Cytoskeleton
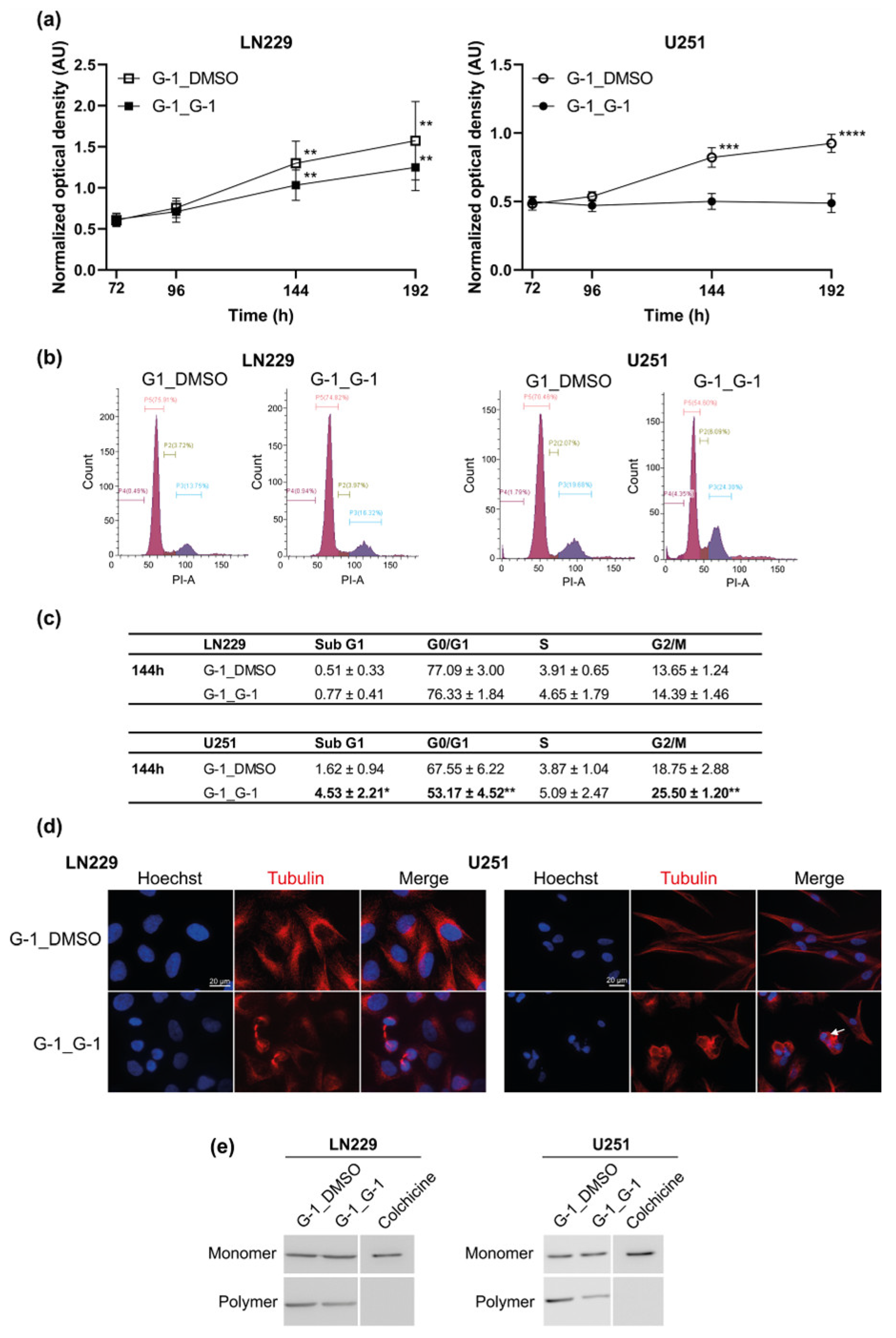
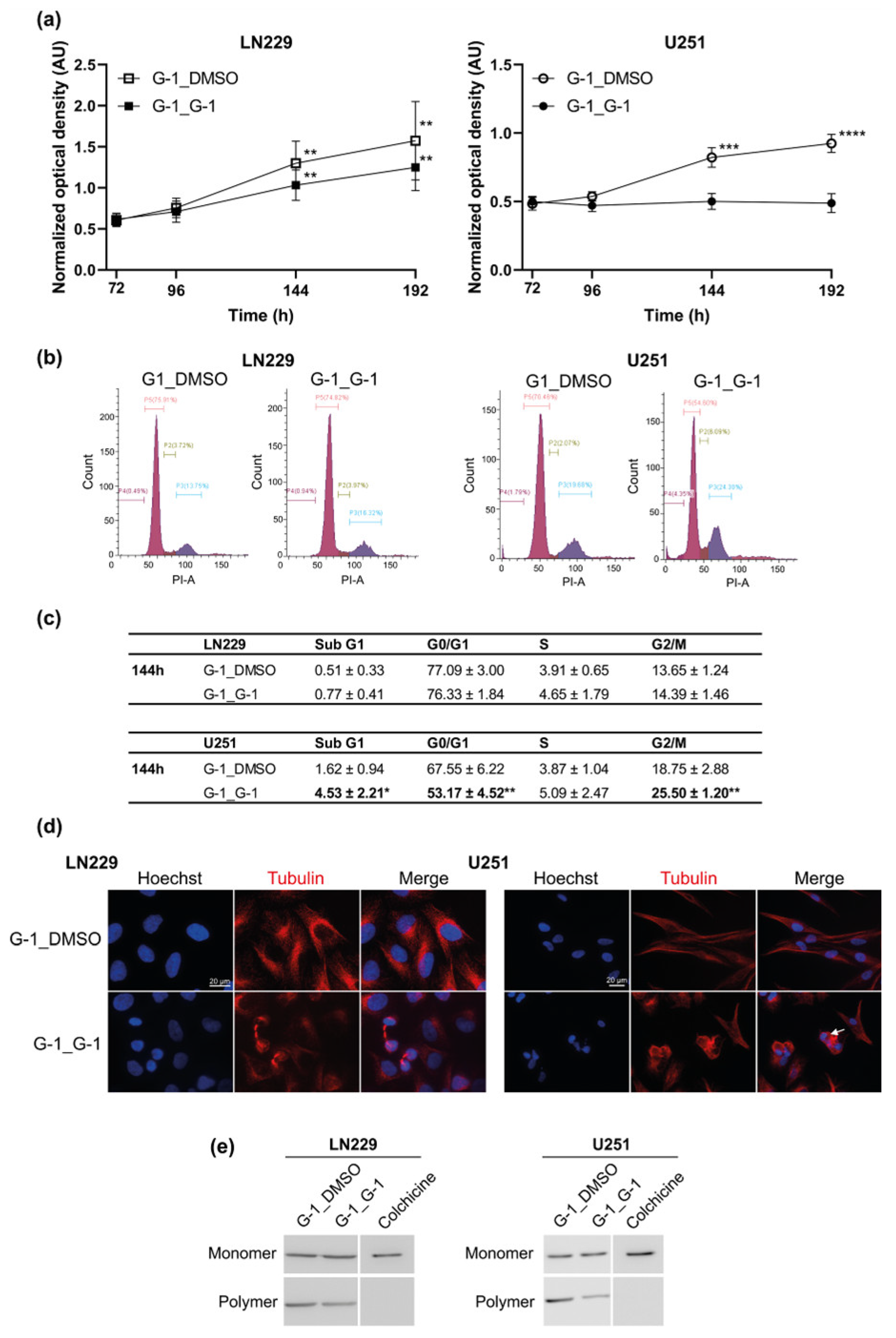
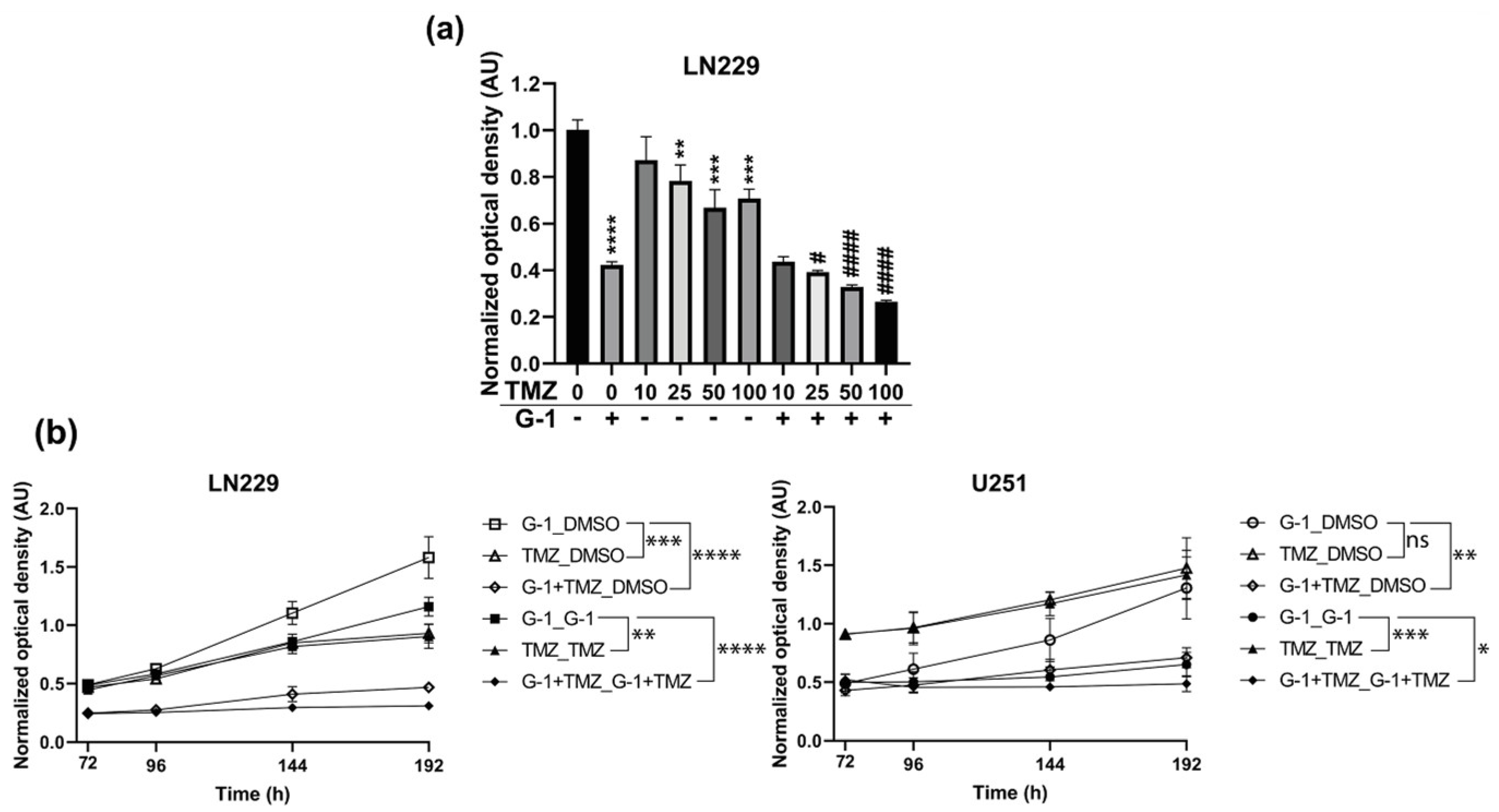
3.5. Additive Effect of G-1 and TMZ Suppressed Cell Proliferation Resumption
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Department of Neurosurgery, University Hospitals Leuven, Codon Publications: Leuven, Belgium, 2017; pp. 197–241. ISBN 978-0-9944381-2-6. [Google Scholar]
- Bauchet, L.; Mathieu-Daudé, H.; Fabbro-Peray, P.; Rigau, V.; Fabbro, M.; Chinot, O.; Pallusseau, L.; Carnin, C.; Lainé, K.; Schlama, A.; et al. Oncological Patterns of Care and Outcome for 952 Patients with Newly Diagnosed Glioblastoma in 2004. Neuro-oncology 2010, 12, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Baldi, I.; Gruber, A.; Alioum, A.; Berteaud, E.; Lebailly, P.; Huchet, A.; Tourdias, T.; Kantor, G.; Maire, J.P.; Vital, A.; et al. Descriptive Epidemiology of CNS Tumors in France: Results from the Gironde Registry for the Period 2000–2007. Neuro-oncology 2011, 13, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, N.; Sun, T.; Yu, K.; Rockwell, N.; Tinkum, K.L.; Qi, Z.; Warrington, N.M.; McDonald, P.; Roy, A.; Weir, S.J.; et al. Cooperative P16 and P21 Action Protects Female Astrocytes from Transformation. Acta Neuropathol. Commun. 2018, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirtz, A.; Rech, F.; Dubois-Pot-Schneider, H.; Dumond, H. Astrocytoma: A Hormone-Sensitive Tumor? Int. J. Mol. Sci. 2020, 21, 9114. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-oncology 2015, 17, iv1–iv62. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D.; Guan, X.; MacLusky, N.J.; Horvath, T.L.; Diano, S.; Singh, M.; Connolly, E.S.; Nethrapalli, I.S.; Tinnikov, A.A. ER-X: A Novel, Plasma Membrane-Associated, Putative Estrogen Receptor That Is Regulated during Development and after Ischemic Brain Injury. J. Neurosci. 2002, 22, 8391–8401. [Google Scholar] [CrossRef] [PubMed]
- Toran-Allerand, C.D.; Tinnikov, A.A.; Singh, R.J.; Nethrapalli, I.S. 17α-Estradiol: A Brain-Active Estrogen? Endocrinology 2005, 146, 3843–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brailoiu, E.; Dun, S.L.; Brailoiu, G.C.; Mizuo, K.; Sklar, L.A.; Oprea, T.I.; Prossnitz, E.R.; Dun, N.J. Distribution and Characterization of Estrogen Receptor G Protein-Coupled Receptor 30 in the Rat Central Nervous System. J. Endocrinol. 2007, 193, 311–321. [Google Scholar] [CrossRef]
- Almey, A.; Milner, T.A.; Brake, W.G. Estrogen Receptors in the Central Nervous System and Their Implication for Dopamine-Dependent Cognition in Females. Horm. Behav. 2015, 74, 125–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Szinte, J.S.; Boulware, M.I.; Frick, K.M. 17 -Estradiol and Agonism of G-Protein-Coupled Estrogen Receptor Enhance Hippocampal Memory via Different Cell-Signaling Mechanisms. J. Neurosci. 2016, 36, 3309–3321. [Google Scholar] [CrossRef] [Green Version]
- Roque, C.; Mendes-Oliveira, J.; Baltazar, G. G Protein-coupled Estrogen Receptor Activates Cell Type-specific Signaling Pathways in Cortical Cultures: Relevance to the Selective Loss of Astrocytes. J. Neurochem. 2019, 149, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, J.; Hu, L.; Tian, Z.; Zhang, K.; Yang, L.; Zhang, H.; Guo, Y.; Feng, B.; Liu, H.; et al. Activation of G Protein-coupled Receptor 30 Protects Neurons by Regulating Autophagy in Astrocytes. Glia 2020, 68, 27–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Ding, Q.; Hu, J.; He, S.; Shi, F.; Ma, L. GPER Expressed on Microglia Mediates the Anti-inflammatory Effect of Estradiol in Ischemic Stroke. Brain Behav. 2016, 6, e00449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Qin, P.; Deng, Y.; Ma, Z.; Guo, H.; Guo, H.; Hou, Y.; Wang, S.; Zou, W.; Sun, Y.; et al. The Novel Estrogenic Receptor GPR30 Alleviates Ischemic Injury by Inhibiting TLR4-Mediated Microglial Inflammation. J. Neuroinflamm. 2018, 15, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty Years of the G Protein-Coupled Estrogen Receptor GPER: Historical and Personal Perspectives. J. Steroid. Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez de Valdivia, E.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. G Protein-Coupled Estrogen Receptor 1 (GPER1)/GPR30 Increases ERK1/2 Activity through PDZ Motif-Dependent and -Independent Mechanisms. J. Biol. Chem. 2017, 292, 9932–9943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The Many Faces of Estrogen Signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, M.; Maggiolini, M.; Musti, A.M. GPER Mediates Non-Genomic Effects of Estrogen. In Estrogen Receptors; Eyster, K.M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1366, pp. 471–488. ISBN 978-1-4939-3126-2. [Google Scholar]
- Samartzis, E.P.; Noske, A.; Meisel, A.; Varga, Z.; Fink, D.; Imesch, P. The G Protein-Coupled Estrogen Receptor (GPER) Is Expressed in Two Different Subcellular Localizations Reflecting Distinct Tumor Properties in Breast Cancer. PLoS ONE 2014, 9, e83296. [Google Scholar] [CrossRef] [Green Version]
- Pupo, M.; Bodmer, A.; Berto, M.; Maggiolini, M.; Dietrich, P.-Y.; Picard, D. A Genetic Polymorphism Repurposes the G-Protein Coupled and Membrane-Associated Estrogen Receptor GPER to a Transcription Factor-like Molecule Promoting Paracrine Signaling between Stroma and Breast Carcinoma Cells. Oncotarget 2017, 8, 46728–46744. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shen, Y. Estrogen and Brain: Synthesis, Function and Diseases. Front Biosci. 2005, 10, 257–267. [Google Scholar] [CrossRef]
- Singh, M.; Dykens, J.A.; Simpkins, J.W. Novel Mechanisms for Estrogen-Induced Neuroprotection. Exp. Biol. Med. 2006, 231, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Barone, T.A.; Gorski, J.W.; Greenberg, S.J.; Plunkett, R.J. Estrogen Increases Survival in an Orthotopic Model of Glioblastoma. J. Neurooncol. 2009, 95, 37–48. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Nair, B.C.; Gonugunta, V.K.; Zhang, Q.; Brenner, A.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Therapeutic Significance of Estrogen Receptor β Agonists in Gliomas. Mol. Cancer 2012, 11, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Koganti, S.; Snyder, R.; Gumaste, U.; Karamyan, V.T.; Thekkumkara, T. 2-Methoxyestradiol Binding of GPR30 down-Regulates Angiotensin AT1 Receptor. Eur. J. Pharmacol. 2014, 723, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, L.; Kaza, N.; Coric, T.; Byer, S.J.; Brossier, N.M.; Klocke, B.J.; Bjornsti, M.-A.; Carroll, S.L.; Roth, K.A. 4-Hydroxytamoxifen Induces Autophagic Death through K-Ras Degradation. Cancer Res. 2013, 73, 4395–4405. [Google Scholar] [CrossRef] [Green Version]
- Harmalkar, M.; Upraity, S.; Kazi, S.; Shirsat, N.V. Tamoxifen-Induced Cell Death of Malignant Glioma Cells Is Brought About by Oxidative-Stress-Mediated Alterations in the Expression of BCL2 Family Members and Is Enhanced on MiR-21 Inhibition. J. Mol. Neurosci. 2015, 57, 197–202. [Google Scholar] [CrossRef]
- Spence, A.M.; Peterson, R.A.; Scharnhorst, J.D.; Silbergeld, D.L.; Rostomily, R.C. Phase II Study of Concurrent Continuous Temozolomide (TMZ) and Tamoxifen (TMX) for Recurrent Malignant Astrocytic Gliomas. J. Neurooncol. 2004, 70, 91–95. [Google Scholar] [CrossRef]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and Biomolecular Screening Converge on a Selective Agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In Vivo Effects of a GPR30 Antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.-I.; Sklar, L.A.; et al. Identification of a GPER/GPR30 Antagonist with Improved Estrogen Receptor Counterselectivity. J. Steroid. Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiebaut, C.; Chamard-Jovenin, C.; Chesnel, A.; Morel, C.; Djermoune, E.-H.; Boukhobza, T.; Dumond, H. Mammary Epithelial Cell Phenotype Disruption in Vitro and in Vivo through ERalpha36 Overexpression. PLoS ONE 2017, 12, e0173931. [Google Scholar] [CrossRef]
- Gui, Y.; Shi, Z.; Wang, Z.; Li, J.-J.; Xu, C.; Tian, R.; Song, X.; Walsh, M.P.; Li, D.; Gao, J.; et al. The GPER Agonist G-1 Induces Mitotic Arrest and Apoptosis in Human Vascular Smooth Muscle Cells Independent of GPER: G-1 and Smooth Muscle Cell Apoptosis. J. Cell. Physiol. 2015, 230, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.G.; Bland, J.M. Standard Deviations and Standard Errors. BMJ 2005, 331, 903. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Lv, X.; He, C.; Hua, G.; Tsai, M.-Y.; Davis, J.S. The G-Protein-Coupled Estrogen Receptor Agonist G-1 Suppresses Proliferation of Ovarian Cancer Cells by Blocking Tubulin Polymerization. Cell Death. Dis. 2013, 4, e869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.-J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 Predicts Poor Survival for Ovarian Cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Heublein, S.; Jeschke, U.; Kuhn, C.; Hester, A.; Czogalla, B.; Mahner, S.; Rottmann, M.; Mayr, D.; Schmoeckel, E.; et al. The G-Protein-Coupled Estrogen Receptor (GPER) Regulates Trimethylation of Histone H3 at Lysine 4 and Represses Migration and Proliferation of Ovarian Cancer Cells In Vitro. Cells 2021, 10, 619. [Google Scholar] [CrossRef]
- Filigheddu, N.; Sampietro, S.; Chianale, F.; Porporato, P.E.; Gaggianesi, M.; Gregnanin, I.; Rainero, E.; Ferrara, M.; Perego, B.; Riboni, F.; et al. Diacylglycerol Kinase α Mediates 17-β-Estradiol-Induced Proliferation, Motility, and Anchorage-Independent Growth of Hec-1A Endometrial Cancer Cell Line through the G Protein-Coupled Estrogen Receptor GPR30. Cell. Signal. 2011, 23, 1988–1996. [Google Scholar] [CrossRef]
- Smith, H.O.; Leslie, K.K.; Singh, M.; Qualls, C.R.; Revankar, C.M.; Joste, N.E.; Prossnitz, E.R. GPR30: A Novel Indicator of Poor Survival for Endometrial Carcinoma. Am. J. Obstet. Gynecol. 2007, 196, 386.e1–386.e11. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Brailoiu, E.; Yerrum, S.; Shupp, H.A.; Slifker, M.J.; Cunliffe, H.E.; Black, M.A.; Donato, A.L.; Arterburn, J.B.; Oprea, T.I.; et al. The G Protein–Coupled Receptor GPR30 Inhibits Proliferation of Estrogen Receptor–Positive Breast Cancer Cells. Cancer Res. 2010, 70, 1184–1194. [Google Scholar] [CrossRef] [Green Version]
- Ignatov, T.; Claus, M.; Nass, N.; Haybaeck, J.; Seifert, B.; Kalinski, T.; Ortmann, O.; Ignatov, A. G-Protein-Coupled Estrogen Receptor GPER-1 Expression in Hormone Receptor-Positive Breast Cancer Is Associated with Poor Benefit of Tamoxifen. Breast Cancer Res. Treat. 2019, 174, 121–127. [Google Scholar] [CrossRef]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front. Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef] [PubMed]
- Schüler-Toprak, S.; Skrzypczak, M.; Ignatov, T.; Ignatov, A.; Ortmann, O.; Treeck, O. G Protein-Coupled Estrogen Receptor 1 (GPER-1) and Agonist G-1 Inhibit Growth of Ovarian Cancer Cells by Activation of Anti-Tumoral Transcriptome Responses: Impact of GPER-1 MRNA on Survival. J. Cancer. Res. Clin. Oncol. 2020, 146, 3175–3188. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Valdivia, E.; Sandén, C.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. Human G Protein-Coupled Receptor 30 Is N-Glycosylated and N-Terminal Domain Asparagine 44 Is Required for Receptor Structure and Activity. Biosci. Rep. 2019, 39, BSR20182436. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Natale, C.A.; Li, J.; Pitarresi, J.R.; Norgard, R.J.; Dentchev, T.; Capell, B.C.; Seykora, J.T.; Stanger, B.Z.; Ridky, T.W. Pharmacologic Activation of the G Protein–Coupled Estrogen Receptor Inhibits Pancreatic Ductal Adenocarcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 868–880.e1. [Google Scholar] [CrossRef]
- Chimento, A. GPER Signaling in Spermatogenesis and Testicular Tumors. Front. Endocrin. 2014, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimento, A.; Sirianni, R.; Casaburi, I.; Zolea, F.; Rizza, P.; Avena, P.; Malivindi, R.; De Luca, A.; Campana, C.; Martire, E.; et al. GPER Agonist G-1 Decreases Adrenocortical Carcinoma (ACC) Cell Growth in vitro and in vivo. Oncotarget 2015, 6, 19190–19203. [Google Scholar] [CrossRef] [Green Version]
- Hsu, L.-H.; Chu, N.-M.; Lin, Y.-F.; Kao, S.-H. G-Protein Coupled Estrogen Receptor in Breast Cancer. IJMS 2019, 20, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajj, H.; Chesnel, A.; Pinel, S.; Plenat, F.; Flament, S.; Dumond, H. An Alkylphenol Mix Promotes Seminoma Derived Cell Proliferation through an ERalpha36-Mediated Mechanism. PLoS ONE 2013, 8, e61758. [Google Scholar] [CrossRef] [Green Version]
- Chimento, A.; De Luca, A.; Nocito, M.C.; Avena, P.; La Padula, D.; Zavaglia, L.; Pezzi, V. Role of GPER-Mediated Signaling in Testicular Functions and Tumorigenesis. Cells 2020, 9, 2115. [Google Scholar] [CrossRef] [PubMed]
- Tutzauer, J.; Gonzalez de Valdivia, E.; Swärd, K.; Alexandrakis Eilard, I.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. Ligand-Independent G Protein–Coupled Estrogen Receptor/G Protein–Coupled Receptor 30 Activity: Lack of Receptor-Dependent Effects of G-1 and 17 β-Estradiol. Mol. Pharm. 2021, 100, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Sixto-López, Y.; Marhuenda, E.; García-Vazquez, J.B.; Fragoso-Vazquez, J.; Rosales-Hernández, M.C.; Zacarías-Lara, O.; Méndez-Luna, D.; Gómez-Vidal, J.A.; Cornu, D.; Bakalara, N.; et al. Targeting Several Biologically Reported Targets of Glioblastoma Multiforme by Assaying 2D and 3D Cultured Cells; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; He, C.; Huang, C.; Hua, G.; Wang, Z.; Remmenga, S.W.; Rodabough, K.J.; Karpf, A.R.; Dong, J.; Davis, J.S.; et al. G-1 Inhibits Breast Cancer Cell Growth via Targeting Colchicine-Binding Site of Tubulin to Interfere with Microtubule Assembly. Mol. Cancer. 2017, 16, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.-W.; Chang, Y.-C.; Zhou, Y.; Zhang, H.; Chen, L.; Zhang, Y.; Si, J.-Q.; Li, L. GPER Agonist G1 Suppresses Neuronal Apoptosis Mediated by Endoplasmic Reticulum Stress after Cerebral Ischemia/Reperfusion Injury. Neural. Regen. Res. 2019, 14, 1221. [Google Scholar] [CrossRef]
- Horne, E.A.; Diaz, P.; Cimino, P.J.; Jung, E.; Xu, C.; Hamel, E.; Wagenbach, M.; Kumasaka, D.; Wageling, N.B.; Azorín, D.D.; et al. A Brain-Penetrant Microtubule-Targeting Agent That Disrupts Hallmarks of Glioma Tumorigenesis. Neuro-Oncol. Adv. 2021, 3, vdaa165. [Google Scholar] [CrossRef]
- Wang, H.; Feng, W.; Lu, Y.; Li, H.; Xiang, W.; Chen, Z.; He, M.; Zhao, L.; Sun, X.; Lei, B.; et al. Expression of Dynein, Cytoplasmic 2, Heavy Chain 1 (DHC2) Associated with Glioblastoma Cell Resistance to Temozolomide. Sci. Rep. 2016, 6, 28948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Shen, M.; Li, Y.; Sun, Y.; Teng, Y.; Wang, Y.; Duan, Y. The Synergic Antitumor Effects of Paclitaxel and Temozolomide Co-Loaded in MPEG-PLGA Nanoparticles on Glioblastoma Cells. Oncotarget 2016, 7, 20890–20901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellner, S.; Bauer, B.; Miller, D.S.; Schaffrik, M.; Fankhänel, M.; Spruß, T.; Bernhardt, G.; Graeff, C.; Färber, L.; Gschaidmeier, H.; et al. Transport of Paclitaxel (Taxol) across the Blood-Brain Barrier in Vitro and in Vivo. J. Clin. Investig. 2002, 110, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Hu, C.; Staquicini, D.I.; Brigman, J.L.; Liu, M.; Mauvais-Jarvis, F.; Pasqualini, R.; Arap, W.; Arterburn, J.B.; Hathaway, H.J.; et al. Preclinical Efficacy of the GPER-Selective Agonist G-1 in Mouse Models of Obesity and Diabetes. Sci. Transl. Med. 2020, 12, eaau5956. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirtz, A.; Lebourdais, N.; Rech, F.; Bailly, Y.; Vaginay, A.; Smaïl-Tabbone, M.; Dubois-Pot-Schneider, H.; Dumond, H. GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation. Cells 2021, 10, 3438. https://doi.org/10.3390/cells10123438
Hirtz A, Lebourdais N, Rech F, Bailly Y, Vaginay A, Smaïl-Tabbone M, Dubois-Pot-Schneider H, Dumond H. GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation. Cells. 2021; 10(12):3438. https://doi.org/10.3390/cells10123438
Chicago/Turabian StyleHirtz, Alex, Nolwenn Lebourdais, Fabien Rech, Yann Bailly, Athénaïs Vaginay, Malika Smaïl-Tabbone, Hélène Dubois-Pot-Schneider, and Hélène Dumond. 2021. "GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation" Cells 10, no. 12: 3438. https://doi.org/10.3390/cells10123438
APA StyleHirtz, A., Lebourdais, N., Rech, F., Bailly, Y., Vaginay, A., Smaïl-Tabbone, M., Dubois-Pot-Schneider, H., & Dumond, H. (2021). GPER Agonist G-1 Disrupts Tubulin Dynamics and Potentiates Temozolomide to Impair Glioblastoma Cell Proliferation. Cells, 10(12), 3438. https://doi.org/10.3390/cells10123438