
Role of ADGRG1/GPR56 in Tumor Progression


Abstract
1. Introduction
2. Overview of the ADGRG1/GPR56 Receptor
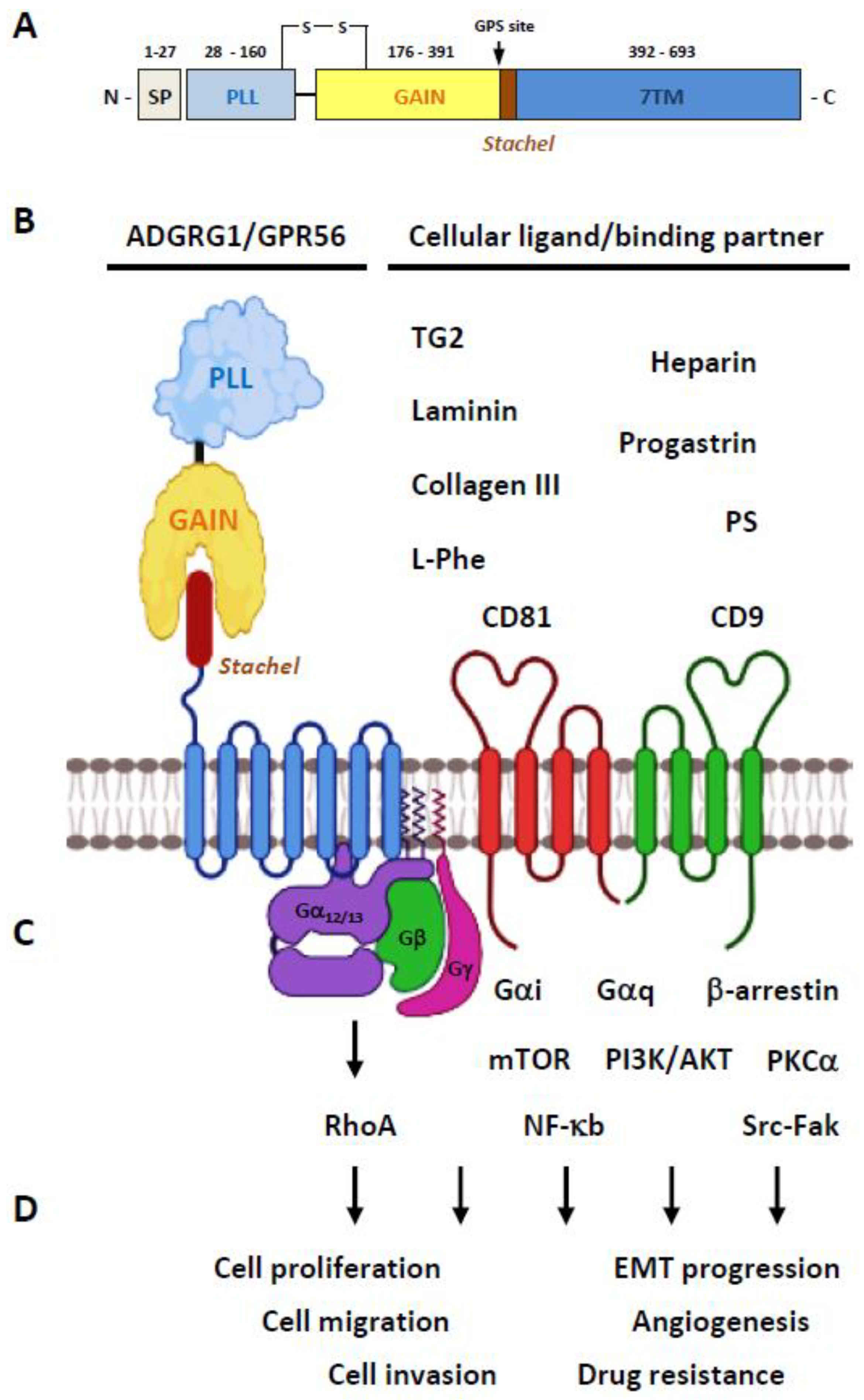
2.1. Structural Characteristics of the ADGRG1/GPR56 Protein
2.2. Ligands/Binding Partners of the ADGRG1/GPR56 Protein
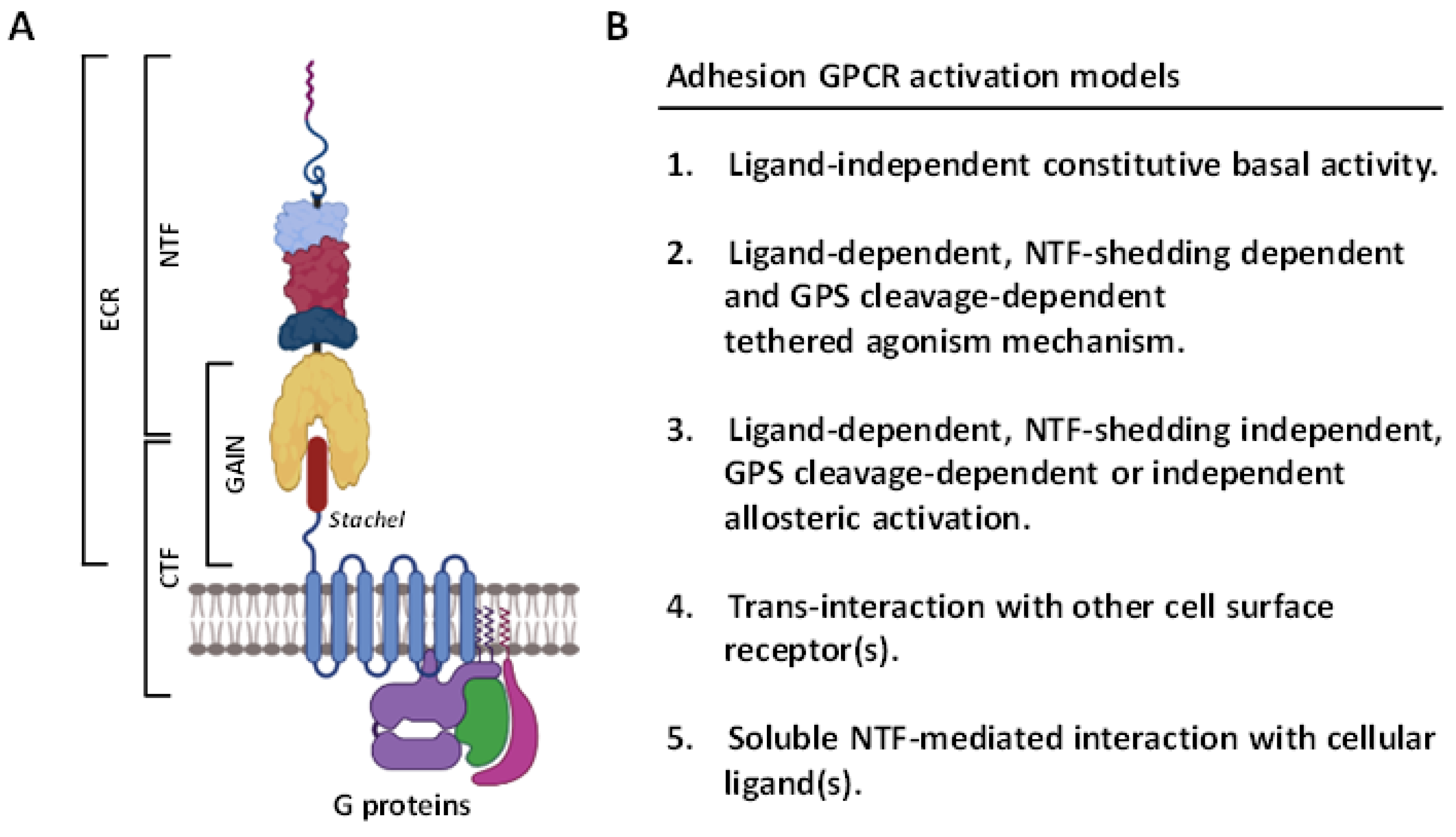
2.3. Activation and Signaling Mechanisms of ADGRG1/GPR56
2.4. The Biological Functions of GPR56
3. ADGRG1/GPR56 as a Cancer Marker and/or Prognostic Factor
4. The Tumor-Suppressive Role of ADGRG1/GPR56
5. The Tumor-Promoting Role of ADGRG1/GPR56
6. Unmet Challenges
7. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. 2013, 52, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Sriram, K.; Wiley, S.Z.; Wilderman, A.; Katakia, T.; McCann, T.; Yokouchi, H.; Zhang, L.; Corriden, R.; Liu, D.; et al. GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front. Pharmacol. 2018, 9, 431. [Google Scholar] [CrossRef]
- Gavi, S.; Shumay, E.; Wang, H.Y.; Malbon, C.C. G-protein-coupled receptors and tyrosine kinases: Crossroads in cell signaling and regulation. Trends Endocrinol. Metab. 2006, 17, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Receptor tyrosine kinase-G-protein-coupled receptor signalling platforms: Out of the shadow? Trends Pharmacol. Sci. 2011, 32, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Hamann, J.; Aust, G.; Arac, D.; Engel, F.B.; Formstone, C.; Fredriksson, R.; Hall, R.A.; Harty, B.L.; Kirchhoff, C.; Knapp, B.; et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol. Rev. 2015, 67, 338–367. [Google Scholar] [CrossRef]
- Yona, S.; Lin, H.H.; Siu, W.O.; Gordon, S.; Stacey, M. Adhesion-GPCRs: Emerging roles for novel receptors. Trends Biochem. Sci. 2008, 33, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Arac, D.; Aust, G.; Calebiro, D.; Engel, F.B.; Formstone, C.; Goffinet, A.; Hamann, J.; Kittel, R.J.; Liebscher, I.; Lin, H.H.; et al. Dissecting signaling and functions of adhesion G protein-coupled receptors. Ann. N. Y. Acad. Sci. 2012, 1276, 1–25. [Google Scholar] [CrossRef]
- Arac, D.; Boucard, A.A.; Bolliger, M.F.; Nguyen, J.; Soltis, S.M.; Sudhof, T.C.; Brunger, A.T. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 2012, 31, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Chang, G.W.; Davies, J.Q.; Stacey, M.; Harris, J.; Gordon, S. Autocatalytic cleavage of the EMR2 receptor occurs at a conserved G protein-coupled receptor proteolytic site motif. J. Biol. Chem. 2004, 279, 31823–31832. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Hall, R.A. Adhesion G protein-coupled receptors: Signaling, pharmacology, and mechanisms of activation. Mol. Pharmacol. 2012, 82, 777–783. [Google Scholar] [CrossRef]
- Kishore, A.; Hall, R.A. Versatile signaling activity of adhesion GPCRs. Handb. Exp. Pharmacol. 2016, 234, 127–146. [Google Scholar]
- Purcell, R.H.; Hall, R.A. Adhesion G protein-coupled receptors as drug targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 429–449. [Google Scholar] [CrossRef] [PubMed]
- Stoveken, H.M.; Hajduczok, A.G.; Xu, L.; Tall, G.G. Adhesion G protein-coupled receptors are activated by exposure of a cryptic tethered agonist. Proc. Natl. Acad. Sci. USA 2015, 112, 6194–6199. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, I.; Schon, J.; Petersen, S.C.; Fischer, L.; Auerbach, N.; Demberg, L.M.; Mogha, A.; Coster, M.; Simon, K.U.; Rothemund, S.; et al. A Tethered Agonist within the Ectodomain Activates the Adhesion G Protein-Coupled Receptors GPR126 and GPR133. Cell Rep. 2015, 10, 1021. [Google Scholar] [CrossRef] [PubMed]
- Monk, K.R.; Hamann, J.; Langenhan, T.; Nijmeijer, S.; Schoneberg, T.; Liebscher, I. Adhesion G protein-coupled receptors: From in vitro pharmacology to in vivo mechanisms. Mol. Pharmacol. 2015, 88, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Aust, G.; Zhu, D.; Van Meir, E.G.; Xu, L. Adhesion GPCRs in Tumorigenesis. Handb. Exp. Pharmacol. 2016, 234, 369–396. [Google Scholar]
- Gad, A.A.; Balenga, N. The emerging role of adhesion GPCRs in cancer. ACS Pharmacol. Transl. Sci. 2020, 3, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H. Adhesion family of G protein-coupled receptors and cancer. Chang. Gung Med. J. 2012, 35, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Piao, X.; Hill, R.S.; Bodell, A.; Chang, B.S.; Basel-Vanagaite, L.; Straussberg, R.; Dobyns, W.B.; Qasrawi, B.; Winter, R.M.; Innes, A.M.; et al. G protein-coupled receptor-dependent development of human frontal cortex. Science 2004, 303, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Han, J.M.; Park, C.R.; Shin, K.J.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Splicing variants of the orphan G-protein-coupled receptor GPR56 regulate the activity of transcription factors associated with tumorigenesis. J. Cancer Res. Clin. Oncol. 2010, 136, 47–53. [Google Scholar] [CrossRef]
- Salzman, G.S.; Ackerman, S.D.; Ding, C.; Koide, A.; Leon, K.; Luo, R.; Stoveken, H.M.; Fernandez, C.G.; Tall, G.G.; Piao, X.; et al. Structural basis for regulation of GPR56/ADGRG1 by its alternatively spliced extracellular domains. Neuron 2016, 91, 1292–1304. [Google Scholar] [CrossRef]
- Bae, B.I.; Tietjen, I.; Atabay, K.D.; Evrony, G.D.; Johnson, M.B.; Asare, E.; Wang, P.P.; Murayama, A.Y.; Im, K.; Lisgo, S.N.; et al. Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 2014, 343, 764–768. [Google Scholar] [CrossRef]
- Knierim, A.B.; Rothe, J.; Cakir, M.V.; Lede, V.; Wilde, C.; Liebscher, I.; Thor, D.; Schoneberg, T. Genetic basis of functional variability in adhesion G protein-coupled receptors. Sci. Rep. 2019, 9, 11036. [Google Scholar] [CrossRef] [PubMed]
- Promel, S.; Langenhan, T.; Arac, D. Matching structure with function: The GAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol. Sci. 2013, 34, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Little, K.D.; Hemler, M.E.; Stipp, C.S. Dynamic regulation of a GPCR-tetraspanin-G protein complex on intact cells: Central role of CD81 in facilitating GPR56-Galpha q/11 association. Mol. Biol. Cell 2004, 15, 2375–2387. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Begum, S.; Hearn, J.D.; Hynes, R.O. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 9023–9028. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, G.; Mohanty, S.; Scott, G.; Fazal, F.; Rahman, A.; Begum, S.; Hynes, R.O.; Xu, L. GPR56 Regulates VEGF production and angiogenesis during melanoma progression. Cancer Res. 2011, 71, 5558–5568. [Google Scholar] [CrossRef]
- Yang, L.; Friedland, S.; Corson, N.; Xu, L. GPR56 inhibits melanoma growth by internalizing and degrading its ligand TG2. Cancer Res. 2014, 74, 1022–1031. [Google Scholar] [CrossRef]
- Yang, L.; Xu, L. GPR56 in cancer progression: Current status and future perspective. Future Oncol. 2012, 8, 431–440. [Google Scholar] [CrossRef]
- Luo, R.; Jeong, S.J.; Jin, Z.; Strokes, N.; Li, S.; Piao, X. G protein-coupled receptor 56 and collagen III, a receptor-ligand pair, regulates cortical development and lamination. Proc. Natl. Acad. Sci. USA 2011, 108, 12925–12930. [Google Scholar] [CrossRef]
- Giera, S.; Luo, R.; Ying, Y.; Ackerman, S.D.; Jeong, S.J.; Stoveken, H.M.; Folts, C.J.; Welsh, C.A.; Tall, G.G.; Stevens, B.; et al. Microglial transglutaminase-2 drives myelination and myelin repair via GPR56/ADGRG1 in oligodendrocyte precursor cells. eLife 2018, 7, e33385. [Google Scholar] [CrossRef]
- Luo, R.; Jin, Z.; Deng, Y.; Strokes, N.; Piao, X. Disease-associated mutations prevent GPR56-collagen III interaction. PLoS ONE 2012, 7, e29818. [Google Scholar]
- Salzman, G.S.; Zhang, S.; Fernandez, C.G.; Arac, D.; Koide, S. Specific and direct modulation of the interaction between adhesion GPCR GPR56/ADGRG1 and tissue transglutaminase 2 using synthetic ligands. Sci. Rep. 2020, 10, 16912. [Google Scholar] [CrossRef]
- Chiang, N.Y.; Chang, G.W.; Huang, Y.S.; Peng, Y.M.; Hsiao, C.C.; Kuo, M.L.; Lin, H.H. Heparin interacts with the adhesion GPCR GPR56, reduces receptor shedding, and promotes cell adhesion and motility. J. Cell Sci. 2016, 129, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Sakitani, K.; Wang, H.; Jin, Y.; Dubeykovskiy, A.; Worthley, D.L.; Tailor, Y.; Wang, T.C. The G-protein coupled receptor 56, expressed in colonic stem and cancer cells, binds progastrin to promote proliferation and carcinogenesis. Oncotarget 2017, 8, 40606–40619. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Nwe, P.K.; Yang, Y.; Rosen, C.E.; Bielecka, A.A.; Kuchroo, M.; Cline, G.W.; Kruse, A.C.; Ring, A.M.; Crawford, J.M.; et al. A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 2019, 177, 1217–1231. [Google Scholar] [CrossRef]
- Li, T.; Chiou, B.; Gilman, C.K.; Luo, R.; Koshi, T.; Yu, D.; Oak, H.C.; Giera, S.; Johnson-Venkatesh, E.; Muthukumar, A.K.; et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 2020, 39, e104136. [Google Scholar] [CrossRef]
- Chang, G.W.; Hsiao, C.C.; Peng, Y.M.; Vieira Braga, F.A.; Kragten, N.A.; Remmerswaal, E.B.; van de Garde, M.D.; Straussberg, R.; Konig, G.M.; Kostenis, E.; et al. The adhesion g protein-coupled receptor GPR56/ADGRG1 is an inhibitory receptor on human NK cells. Cell Rep. 2016, 15, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.Y.; Peng, Y.M.; Juang, H.H.; Chen, T.C.; Pan, H.L.; Chang, G.W.; Lin, H.H. GPR56/ADGRG1 activation promotes melanoma cell migration via NTF dissociation and CTF-mediated galpha12/13/RhoA signaling. J. Investig. Dermatol. 2017, 137, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Sakaguchi, S.; Kobayashi, Y.; Mizuno, N.; Tago, K.; Itoh, H. Agonistic antibodies reveal the function of GPR56 in human glioma U87-MG cells. Biol. Pharm. Bull. 2015, 38, 594–600. [Google Scholar] [CrossRef]
- Iguchi, T.; Sakata, K.; Yoshizaki, K.; Tago, K.; Mizuno, N.; Itoh, H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a G alpha 12/13 and Rho pathway. J. Biol. Chem. 2008, 283, 14469–14478. [Google Scholar] [CrossRef] [PubMed]
- Salzman, G.S.; Zhang, S.; Gupta, A.; Koide, A.; Koide, S.; Arac, D. Stachel-independent modulation of GPR56/ADGRG1 signaling by synthetic ligands directed to its extracellular region. Proc. Natl. Acad. Sci. USA 2017, 114, 10095–10100. [Google Scholar] [CrossRef]
- Zhu, B.; Luo, R.; Jin, P.; Li, T.; Oak, H.C.; Giera, S.; Monk, K.R.; Lak, P.; Shoichet, B.K.; Piao, X. GAIN domain-mediated cleavage is required for activation of G protein-coupled receptor 56 (GPR56) by its natural ligands and a small-molecule agonist. J. Biol. Chem. 2019, 294, 19246–19254. [Google Scholar] [CrossRef] [PubMed]
- Stoveken, H.M.; Bahr, L.L.; Anders, M.W.; Wojtovich, A.P.; Smrcka, A.V.; Tall, G.G. Dihydromunduletone Is a small-molecule selective adhesion G protein-coupled receptor antagonist. Mol. Pharmacol. 2016, 90, 214–224. [Google Scholar] [CrossRef]
- Stoveken, H.M.; Larsen, S.D.; Smrcka, A.V.; Tall, G.G. Gedunin- and Khivorin-Derivatives are small-molecule partial agonists for adhesion G protein-coupled receptors GPR56/ADGRG1 and GPR114/ADGRG5. Mol. Pharmacol. 2018, 93, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Kishore, A.; Purcell, R.H.; Nassiri-Toosi, Z.; Hall, R.A. Stalk-dependent and stalk-independent signaling by the adhesion G protein-coupled receptors GPR56 (ADGRG1) and BAI1 (ADGRB1). J. Biol. Chem. 2016, 291, 3385–3394. [Google Scholar] [CrossRef] [PubMed]
- Paavola, K.J.; Stephenson, J.R.; Ritter, S.L.; Alter, S.P.; Hall, R.A. The N terminus of the adhesion G protein-coupled receptor GPR56 controls receptor signaling activity. J. Biol. Chem. 2011, 286, 28914–28921. [Google Scholar] [CrossRef]
- Luo, R.; Jeong, S.J.; Yang, A.; Wen, M.; Saslowsky, D.E.; Lencer, W.I.; Arac, D.; Piao, X. Mechanism for adhesion G protein-coupled receptor GPR56-mediated RhoA activation induced by collagen III stimulation. PLoS ONE 2014, 9, e100043. [Google Scholar] [CrossRef]
- Kishore, A.; Hall, R.A. Disease-associated extracellular loop mutations in the adhesion G protein-coupled receptor G1 (ADGRG1; GPR56) differentially regulate downstream signaling. J. Biol. Chem. 2017, 292, 9711–9720. [Google Scholar] [CrossRef] [PubMed]
- Beliu, G.; Altrichter, S.; Guixa-Gonzalez, R.; Hemberger, M.; Brauer, I.; Dahse, A.K.; Scholz, N.; Wieduwild, R.; Kuhlemann, A.; Batebi, H.; et al. Tethered agonist exposure in intact adhesion/class B2 GPCRs through intrinsic structural flexibility of the GAIN domain. Mol. Cell 2021, 81, 905–921. [Google Scholar] [CrossRef]
- Huang, K.Y.; Lin, H.H. The activation and signaling mechanisms of GPR56/ADGRG1 in Melanoma Cell. Front. Oncol. 2018, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Pedrosa, L.; Pare, L.; Pineda, E.; Bejarano, L.; Martinez, J.; Balasubramaniyan, V.; Ezhilarasan, R.; Kallarackal, N.; Kim, S.H.; et al. GPR56/ADGRG1 inhibits mesenchymal differentiation and radioresistance in glioblastoma. Cell Rep. 2017, 21, 2183–2197. [Google Scholar] [CrossRef]
- Chatterjee, T.; Zhang, S.; Posey, T.A.; Jacob, J.; Wu, L.; Yu, W.; Francisco, L.E.; Liu, Q.J.; Carmon, K.S. Anti-GPR56 monoclonal antibody potentiates GPR56-mediated Src-Fak signaling to modulate cell adhesion. J. Biol. Chem. 2021, 296, 100261. [Google Scholar] [CrossRef]
- Kitakaze, T.; Yoshikawa, M.; Kobayashi, Y.; Kimura, N.; Goshima, N.; Ishikawa, T.; Ogata, Y.; Yamashita, Y.; Ashida, H.; Harada, N.; et al. Extracellular transglutaminase 2 induces myotube hypertrophy through G protein-coupled receptor 56. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118563. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Falco, M.; Parolini, S.; Bellora, F.; Petretto, A.; Romeo, E.; Balsamo, M.; Gambarotti, M.; Scordamaglia, F.; Tabellini, G.; et al. GPR56 as a novel marker identifying the CD56dull CD16+ NK cell subset both in blood stream and in inflamed peripheral tissues. Int. Immunol. 2010, 22, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.M.; van de Garde, M.D.; Cheng, K.F.; Baars, P.A.; Remmerswaal, E.B.; van Lier, R.A.; Mackay, C.R.; Lin, H.H.; Hamann, J. Specific expression of GPR56 by human cytotoxic lymphocytes. J. Leukoc. Biol. 2011, 90, 735–740. [Google Scholar] [CrossRef]
- Ackerman, S.D.; Garcia, C.; Piao, X.; Gutmann, D.H.; Monk, K.R. The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Galpha12/13 and RhoA. Nat. Commun. 2015, 6, 6122. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, S.D.; Luo, R.; Poitelon, Y.; Mogha, A.; Harty, B.L.; D’Rozario, M.; Sanchez, N.E.; Lakkaraju, A.K.K.; Gamble, P.; Li, J.; et al. GPR56/ADGRG1 regulates development and maintenance of peripheral myelin. J. Exp. Med. 2018, 215, 941–961. [Google Scholar] [CrossRef]
- Giera, S.; Deng, Y.; Luo, R.; Ackerman, S.D.; Mogha, A.; Monk, K.R.; Ying, Y.; Jeong, S.J.; Makinodan, M.; Bialas, A.R.; et al. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 2015, 6, 6121. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Luo, R.; Singer, K.; Giera, S.; Kreidberg, J.; Kiyozumi, D.; Shimono, C.; Sekiguchi, K.; Piao, X. GPR56 functions together with alpha3beta1 integrin in regulating cerebral cortical development. PLoS ONE 2013, 8, e68781. [Google Scholar]
- Duner, P.; Al-Amily, I.M.; Soni, A.; Asplund, O.; Safi, F.; Storm, P.; Groop, L.; Amisten, S.; Salehi, A. Adhesion G protein-coupled receptor G1 (ADGRG1/GPR56) and pancreatic beta-cell function. J. Clin. Endocrinol. Metab. 2016, 101, 4637–4645. [Google Scholar] [CrossRef]
- Chen, G.; Yang, L.; Begum, S.; Xu, L. GPR56 is essential for testis development and male fertility in mice. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2010, 239, 3358–3367. [Google Scholar] [CrossRef] [PubMed]
- Roly, Z.Y.; Major, A.T.; Fulcher, A.; Estermann, M.A.; Hirst, C.E.; Smith, C.A. Adhesion G-protein-coupled receptor, GPR56, is required for Mullerian duct development in the chick. J. Endocrinol. 2020, 244, 395–413. [Google Scholar] [CrossRef]
- Singh, A.K.; Lin, H.H. The role of GPR56/ADGRG1 in health and disease. Biomed. J. 2021, in press. [Google Scholar] [CrossRef]
- Koirala, S.; Jin, Z.; Piao, X.; Corfas, G. GPR56-regulated granule cell adhesion is essential for rostral cerebellar development. J. Neurosci. 2009, 29, 7439–7449. [Google Scholar] [CrossRef]
- Belzeaux, R.; Gorgievski, V.; Fiori, L.M.; Lopez, J.P.; Grenier, J.; Lin, R.; Nagy, C.; Ibrahim, E.C.; Gascon, E.; Courtet, P.; et al. GPR56/ADGRG1 is associated with response to antidepressant treatment. Nat. Commun. 2020, 11, 1635. [Google Scholar] [CrossRef]
- White, J.P.; Wrann, C.D.; Rao, R.R.; Nair, S.K.; Jedrychowski, M.P.; You, J.S.; Martinez-Redondo, V.; Gygi, S.P.; Ruas, J.L.; Hornberger, T.A.; et al. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 15756–15761. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.P.; Doyle, J.R.; Barry, B.; Beauvais, A.; Rozkalne, A.; Piao, X.; Lawlor, M.W.; Kopin, A.S.; Walsh, C.A.; Gussoni, E. G-protein coupled receptor 56 promotes myoblast fusion through serum response factor- and nuclear factor of activated T-cell-mediated signalling but is not essential for muscle development in vivo. FEBS J. 2013, 280, 6097–6113. [Google Scholar] [CrossRef]
- Yeung, J.; Adili, R.; Stringham, E.N.; Luo, R.; Vizurraga, A.; Rosselli-Murai, L.K.; Stoveken, H.M.; Yu, M.; Piao, X.; Holinstat, M.; et al. GPR56/ADGRG1 is a platelet collagen-responsive GPCR and hemostatic sensor of shear force. Proc. Natl. Acad. Sci. USA 2020, 117, 28275–28286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Si, Y.; Ma, N.; Mei, J. The RNA-binding protein PCBP2 inhibits Ang II-induced hypertrophy of cardiomyocytes though promoting GPR56 mRNA degeneration. Biochem. Biophys. Res. Commun. 2015, 464, 679–684. [Google Scholar] [CrossRef]
- Maglitto, A.; Mariani, S.A.; de Pater, E.; Rodriguez-Seoane, C.; Vink, C.S.; Piao, X.; Lukke, M.L.; Dzierzak, E. Unexpected redundancy of Gpr56 and Gpr97 during hematopoietic cell development and differentiation. Blood Adv. 2021, 5, 829–842. [Google Scholar] [CrossRef]
- Olaniru, O.E.; Pingitore, A.; Giera, S.; Piao, X.; Castanera Gonzalez, R.; Jones, P.M.; Persaud, S.J. The adhesion receptor GPR56 is activated by extracellular matrix collagen III to improve beta-cell function. Cell. Mol. Life Sci. 2018, 75, 4007–4019. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.N.; Marks-Bluth, J.; Sullivan, J.; Gupta, M.K.; Chandrakanthan, V.; Fitch, S.R.; Ottersbach, K.; Jang, Y.C.; Piao, X.; Kulkarni, R.N.; et al. High-level Gpr56 expression is dispensable for the maintenance and function of hematopoietic stem and progenitor cells in mice. Stem Cell Res. 2015, 14, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Kaneda, K.; Suekane, A.; Ichihara, E.; Nakahata, S.; Yamakawa, N.; Nagai, K.; Mizuno, N.; Kogawa, K.; Miura, I.; et al. Maintenance of the hematopoietic stem cell pool in bone marrow niches by EVI1-regulated GPR56. Leukemia 2013, 27, 1637–1649. [Google Scholar] [CrossRef]
- Sud, N.; Sharma, R.; Ray, R.; Chattopadhyay, T.K.; Ralhan, R. Differential expression of G-protein coupled receptor 56 in human esophageal squamous cell carcinoma. Cancer Lett. 2006, 233, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.; Sundaram, R.; Liu, G.; Chionis, J.; Fan, W.; Rogers, C.; Awad, T.; Grifman, M.; Yu, D.; Wong-Staal, F.; et al. Orphan G protein-coupled receptor GPR56 plays a role in cell transformation and tumorigenesis involving the cell adhesion pathway. Mol. Cancer Ther. 2007, 6, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, Z.; Yang, W.; Li, Z.; Xing, S.; Li, H.; Hu, B.; Li, P. Expression of orphan GPR56 correlates with tumor progression in human epithelial ovarian cancer. Neoplasma 2017, 64, 32–39. [Google Scholar] [CrossRef]
- Shashidhar, S.; Lorente, G.; Nagavarapu, U.; Nelson, A.; Kuo, J.; Cummins, J.; Nikolich, K.; Urfer, R.; Foehr, E.D. GPR56 is a GPCR that is overexpressed in gliomas and functions in tumor cell adhesion. Oncogene 2005, 24, 1673–1682. [Google Scholar] [CrossRef]
- Daria, D.; Kirsten, N.; Muranyi, A.; Mulaw, M.; Ihme, S.; Kechter, A.; Hollnagel, M.; Bullinger, L.; Dohner, K.; Dohner, H.; et al. GPR56 contributes to the development of acute myeloid leukemia in mice. Leukemia 2016, 30, 1734–1741. [Google Scholar] [CrossRef]
- Pabst, C.; Bergeron, A.; Lavallee, V.P.; Yeh, J.; Gendron, P.; Norddahl, G.L.; Krosl, J.; Boivin, I.; Deneault, E.; Simard, J.; et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood 2016, 127, 2018–2027. [Google Scholar] [CrossRef]
- Liu, M.; Parker, R.M.; Darby, K.; Eyre, H.J.; Copeland, N.G.; Crawford, J.; Gilbert, D.J.; Sutherland, G.R.; Jenkins, N.A.; Herzog, H. GPR56, a novel secretin-like human G-protein-coupled receptor gene. Genomics 1999, 55, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Zendman, A.J.; Cornelissen, I.M.; Weidle, U.H.; Ruiter, D.J.; van Muijen, G.N. TM7XN1, a novel human EGF-TM7-like cDNA, detected with mRNA differential display using human melanoma cell lines with different metastatic potential. FEBS Lett. 1999, 446, 292–298. [Google Scholar] [CrossRef]
- Daga, S.; Rosenberger, A.; Quehenberger, F.; Krisper, N.; Prietl, B.; Reinisch, A.; Zebisch, A.; Sill, H.; Wolfler, A. High GPR56 surface expression correlates with a leukemic stem cell gene signature in CD34-positive AML. Cancer Med. 2019, 8, 1771–1778. [Google Scholar] [CrossRef]
- Lim, D.R.; Kang, D.H.; Kuk, J.C.; Kim, T.H.; Shin, E.J.; Ahn, T.S.; Kim, H.J.; Jeong, D.J.; Baek, M.J.; Kim, N.K. Prognostic impact of GPR56 in patients with colorectal cancer. Neoplasma 2021, 68, 580–589. [Google Scholar] [CrossRef]
- Zhang, S.; Chatterjee, T.; Godoy, C.; Wu, L.; Liu, Q.J.; Carmon, K.S. GPR56 drives colorectal tumor growth and promotes drug resistance through upregulation of MDR1 expression via a RhoA-mediated mechanism. Mol. Cancer Res. 2019, 17, 2196–2207. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-kappaB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Feng, Y.; Sun, Y.; Ji, D.; Qian, W.; Zhang, Z.; Wang, Q.; Zhang, Y.; Zhang, C.; Sun, Y. GPR56 promotes proliferation of colorectal cancer cells and enhances metastasis via epithelialmesenchymal transition through PI3K/AKT signaling activation. Oncol. Rep. 2018, 40, 1885–1896. [Google Scholar] [PubMed]
- Kausar, T.; Sharma, R.; Hasan, M.R.; Tripathi, S.C.; Saraya, A.; Chattopadhyay, T.K.; Gupta, S.D.; Ralhan, R. Clinical significance of GPR56, transglutaminase 2, and NF-kappaB in esophageal squamous cell carcinoma. Cancer Investig. 2011, 29, 42–48. [Google Scholar] [CrossRef]
- Song, Y.; Li, A.; Zhang, L.; Duan, L. Expression of G protein-coupled receptor 56 is associated with tumor progression in non-small-cell lung carcinoma patients. OncoTargets Ther. 2016, 9, 4105–4112. [Google Scholar]
- Chen, Z.; Gao, P.; Li, Z. Expression of G protein-coupled receptor 56 is an unfavorable prognostic factor in osteosarcoma patients. Tohoku J. Exp. Med. 2016, 239, 203–211. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sasaki, S.I.; Zhang, D.; Iwabuchi, S.; Tanabe, Y.; Hashimoto, S.; Yamauchi, A.; Hayashi, K.; Tsuchiya, H.; Hayakawa, Y.; Baba, T.; et al. Crucial contribution of GPR56/ADGRG1, expressed by breast cancer cells, to bone metastasis formation. Cancer Sci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, M.; Bill, M.; Grimm, J.; Schulz, J.; Schuhmann, L.; Brauer, D.; Goldmann, K.; Wilke, F.; Franke, G.N.; Behre, G.; et al. High expression of the stem cell marker GPR56 at diagnosis identifies acute myeloid leukemia patients at higher relapse risk after allogeneic stem cell transplantation in context with the CD34+/CD38- population. Haematologica 2020, 105, e507. [Google Scholar] [CrossRef] [PubMed]
- Xu, L. GPR56 interacts with extracellular matrix and regulates cancer progression. Adv. Exp. Med. Biol. 2010, 706, 98–108. [Google Scholar] [PubMed]
- Xu, L.; Begum, S.; Barry, M.; Crowley, D.; Yang, L.; Bronson, R.T.; Hynes, R.O. GPR56 plays varying roles in endogenous cancer progression. Clin. Exp. Metastasis 2010, 27, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Saha, H.R.; Kaneda-Nakashima, K.; Shimosaki, S.; Suekane, A.; Sarkar, B.; Saito, Y.; Ogoh, H.; Nakahata, S.; Inoue, K.; Watanabe, T.; et al. Suppression of GPR56 expression by pyrrole-imidazole polyamide represents a novel therapeutic drug for AML with high EVI1 expression. Sci. Rep. 2018, 8, 13741. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating proteomics and transcriptomics for systematic combinatorial chimeric antigen receptor therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cancer Type | Function | References |
|---|---|---|
| Melanoma | Potential negative metastatic marker/factor | [31,32,33,86] |
| Acute myeloid leukemia | Leukemia stem cell marker Unfavorable prognostic factor | [79,84,85,88] |
| Epithelial ovarian cancer | Unfavorable prognostic indicator | [82] |
| Colorectal cancer | Unfavorable prognostic indicator Promote drug-resistant cancer stem cell | [89,90] |
| Cancer Type | Potential Mechanisms | References |
|---|---|---|
| Melanoma | Binding of the ECM TG2 ligand inhibited tumor growth, angiogenesis, and metastasis due to GPR56-mediated TG2 internalization and degradation, reduced deposition of fibronectin in ECM, and reduced production of VEGF via a PKCα-dependent pathway. | [31,32,33] |
| Glioblastoma | Inhibitory effects on cell adhesion and migration via Gαq-Rho signaling. A restrictive role in mesenchymal differentiation and radioresistance due in part to the inhibition of the NF-κB signaling pathway. | [45,57,81] |
| Cancer Type | Potential Mechanisms | References |
|---|---|---|
| Breast, pancreatic, cervical, ovarian, prostate, and colorectal cancers, non-small-cell lung carcinoma (NSCLC), and esophageal squamous cell carcinoma | Upregulated GPR56 expression in cancer cells promoted cell growth, adhesion, migration, and/or drug resistance of cancer cells. | [80,81,82,90,96,97,98,99,100] |
| Colorectal cancer (CRC) | GPR56 promoted the EMT process via the induction of PI3K/AKT signaling pathway. GPR56 upregulated MDR1 expression via a RhoA-dependent signaling pathway. | [90,96] |
| Melanoma | GPR56 activation upregulated IL-6 production, promoting cell migration | [44] |
| Acute myeloid leukemia (AML) | GPR56 enhanced cell adhesion and antiapoptotic functions a RhoA-dependent signaling pathway. Gpr56+ HPCs increased primary colony formation in vitro and accelerated myeloid leukemogenesis in vivo. | [79,84,85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, K.-F.; Chen, T.-C.; Stacey, M.; Lin, H.-H. Role of ADGRG1/GPR56 in Tumor Progression. Cells 2021, 10, 3352. https://doi.org/10.3390/cells10123352
Ng K-F, Chen T-C, Stacey M, Lin H-H. Role of ADGRG1/GPR56 in Tumor Progression. Cells. 2021; 10(12):3352. https://doi.org/10.3390/cells10123352
Chicago/Turabian StyleNg, Kwai-Fong, Tse-Ching Chen, Martin Stacey, and Hsi-Hsien Lin. 2021. "Role of ADGRG1/GPR56 in Tumor Progression" Cells 10, no. 12: 3352. https://doi.org/10.3390/cells10123352
APA StyleNg, K.-F., Chen, T.-C., Stacey, M., & Lin, H.-H. (2021). Role of ADGRG1/GPR56 in Tumor Progression. Cells, 10(12), 3352. https://doi.org/10.3390/cells10123352