
Metabolite G-Protein Coupled Receptors in Cardio-Metabolic Diseases


 , and
, and
Abstract
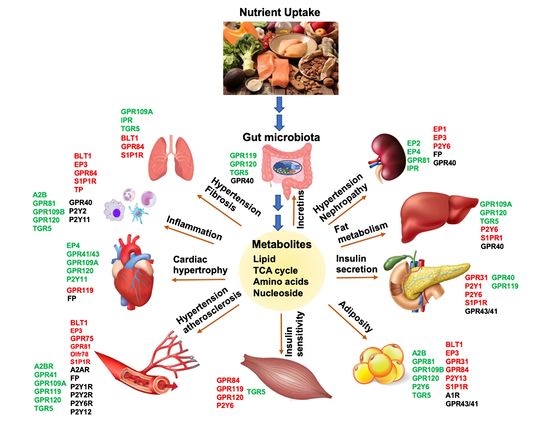
1. Introduction
2. Lipid Metabolites
2.1. Free Fatty Acid Receptors (FFAR)
2.1.1. Short-Chain Fatty Acid Receptors (SCFA)
2.1.2. Medium and Long-Chain Fatty Acid Receptors
2.2. Ketone Bodies
Hydroxycarboxylic Acid Receptors (HCA)
2.3. Bile Acids
2.4. Ceramide
2.5. Arachidonic Acid
2.5.1. Prostaglandins
2.5.2. Leukotriene
2.5.3. Hydroxy Eicosatetraenoic Acids
3. TCA Cycle Metaboltes
4. Amino Acid Metabolites
5. Nucleotide-Nucleoside Metabolites
5.1. P1 Receptors
5.2. P2 Purinergic Receptors
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoyas, I.; Leon-Sanz, M. Nutritional Challenges in Metabolic Syndrome. J. Clin. Med. 2019, 8, 1301. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef] [PubMed]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.D., Jr.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Backhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Han, H.; Yi, B.; Zhong, R.; Wang, M.; Zhang, S.; Ma, J.; Yin, Y.; Yin, J.; Chen, L.; Zhang, H. From gut microbiota to host appetite: Gut microbiota-derived metabolites as key regulators. Microbiome 2021, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.L.; Stephens, J.W.; Harris, D.A. A review on gut microbiota: A central factor in the pathophysiology of obesity. Lipids Health Dis. 2021, 20, 65. [Google Scholar] [CrossRef]
- Mongraw-Chaffin, M.; Foster, M.C.; Anderson, C.A.M.; Burke, G.L.; Haq, N.; Kalyani, R.R.; Ouyang, P.; Sibley, C.T.; Tracy, R.; Woodward, M.; et al. Metabolically Healthy Obesity, Transition to Metabolic Syndrome, and Cardiovascular Risk. J. Am. Coll. Cardiol. 2018, 71, 1857–1865. [Google Scholar] [CrossRef]
- Mendizabal, Y.; Llorens, S.; Nava, E. Hypertension in metabolic syndrome: Vascular pathophysiology. Int. J. Hypertens. 2013, 2013, 230868. [Google Scholar] [CrossRef]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [PubMed]
- Barella, L.F.; Jain, S.; Kimura, T.; Pydi, S.P. Metabolic roles of G protein-coupled receptor signaling in obesity and type 2 diabetes. FEBS J. 2021, 288, 2622–2644. [Google Scholar] [CrossRef]
- Guimaraes, R.C.; Goncalves, T.T.; Leiria, L.O. Exploiting oxidized lipids and the lipid-binding GPCRs against cardiometabolic diseases. Br. J. Pharmacol. 2021, 178, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Kihara, Y. Druggable Lipid GPCRs: Past, Present, and Prospects. Adv. Exp. Med. Biol. 2020, 1274, 223–258. [Google Scholar] [PubMed]
- Cosin-Roger, J.; Ortiz-Masia, D.; Barrachina, M.D.; Calatayud, S. Metabolite Sensing GPCRs: Promising Therapeutic Targets for Cancer Treatment? Cells 2020, 9, 2345. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Dragsted, L.O. Diet-derived microbial metabolites in health and disease. Nutr. Bull. 2019, 44, 216–227. [Google Scholar] [CrossRef]
- Katikaneni, A.; Jelcic, M.; Gerlach, G.F.; Ma, Y.; Overholtzer, M.; Niethammer, P. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat. Cell Biol. 2020, 22, 1049–1055. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef]
- Acosta-Montano, P.; Garcia-Gonzalez, V. Effects of Dietary Fatty Acids in Pancreatic Beta Cell Metabolism, Implications in Homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef]
- Schonfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Ichimura, A.; Hasegawa, S.; Kasubuchi, M.; Kimura, I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 2014, 5, 236. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Kasubuchi, M.; Hasegawa, S.; Hiramatsu, T.; Ichimura, A.; Kimura, I. Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients 2015, 7, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Overby, H.B.; Ferguson, J.F. Gut Microbiota-Derived Short-Chain Fatty Acids Facilitate Microbiota:Host Cross talk and Modulate Obesity and Hypertension. Curr. Hypertens. Rep. 2021, 23, 8. [Google Scholar] [CrossRef] [PubMed]
- Janssen, A.W.; Kersten, S. The role of the gut microbiota in metabolic health. FASEB J. 2015, 29, 3111–3123. [Google Scholar] [CrossRef]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef]
- Byrne, C.S.; Chambers, E.S.; Morrison, D.J.; Frost, G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int. J. Obes. 2015, 39, 1331–1338. [Google Scholar] [CrossRef]
- McNelis, J.C.; Lee, Y.S.; Mayoral, R.; van der Kant, R.; Johnson, A.M.; Wollam, J.; Olefsky, J.M. GPR43 Potentiates beta-Cell Function in Obesity. Diabetes 2015, 64, 3203–3217. [Google Scholar] [CrossRef]
- Tang, C.; Ahmed, K.; Gille, A.; Lu, S.; Gröne, H.-J.; Tunaru, S.; Offermanns, S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. [Google Scholar] [CrossRef]
- Hong, Y.-H.; Nishimura, Y.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Gotoh, C.; Choi, K.-C.; Feng, D.D.; Chen, C.; Lee, H.-G.; et al. Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 2005, 146, 5092–5099. [Google Scholar] [CrossRef]
- Bellahcene, M.; O’Dowd, J.F.; Wargent, E.T.; Zaibi, M.S.; Hislop, D.C.; Ngala, R.A.; Smith, D.M.; Cawthorne, M.A.; Stocker, C.J.; Arch, J.R. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br. J. Nutr. 2013, 109, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Al-Lahham, S.H.; Roelofsen, H.; Priebe, M.; Weening, D.; Dijkstra, M.; Hoek, A.; Rezaee, F.; Venema, K.; Vonk, R.J. Regulation of adipokine production in human adipose tissue by propionic acid. Eur. J. Clin. Investig. 2010, 40, 401–407. [Google Scholar] [CrossRef]
- Veprik, A.; Laufer, D.; Weiss, S.; Rubins, N.; Walker, M.D. GPR41 modulates insulin secretion and gene expression in pancreatic beta-cells and modifies metabolic homeostasis in fed and fasting states. FASEB J. 2016, 30, 3860–3869. [Google Scholar] [CrossRef] [PubMed]
- Yue, C.; Li, M.; Li, J.; Han, X.; Zhu, H.; Yu, G.; Cheng, J. Medium-, long- and medium-chain-type structured lipids ameliorate high-fat diet-induced atherosclerosis by regulating inflammation, adipogenesis, and gut microbiota in ApoE(−/−) mice. Food Funct. 2020, 11, 5142–5155. [Google Scholar] [CrossRef]
- Aguilar, E.C.; Santos, L.C.; Leonel, A.J.; de Oliveira, J.S.; Santos, E.A.; Navia-Pelaez, J.M.; da Silva, J.F.; Mendes, B.P.; Capettini, L.S.; Teixeira, L.G.; et al. Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J. Nutr. Biochem. 2016, 34, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism during Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Pluznick, J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes 2014, 5, 202–207. [Google Scholar] [CrossRef]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef]
- Pluznick, J.L. Microbial Short-Chain Fatty Acids and Blood Pressure Regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef]
- Kamp, M.E.; Shim, R.; Nicholls, A.J.; Oliveira, A.C.; Mason, L.J.; Binge, L.; Mackay, C.; Wong, C.H.Y. G Protein-Coupled Receptor 43 Modulates Neutrophil Recruitment during Acute Inflammation. PLoS ONE 2016, 11, e0163750. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed]
- Robles-Vera, I.; Toral, M.; De La Visitación, N.; Aguilera-Sánchez, N.; Redondo, J.M.; Duarte, J. Protective Effects of Short-Chain Fatty Acids on Endothelial Dysfunction Induced by Angiotensin II. Front. Physiol. 2020, 11, 277. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, G.; Peng, Y.; Zhong, W.; Wang, Y.; Zhang, B. Sodium butyrate alleviates adipocyte inflammation by inhibiting NLRP3 pathway. Sci. Rep. 2015, 5, 12676. [Google Scholar] [CrossRef]
- Sanna, S.; Van Zuydam, N.R.; Mahajan, A.; Kurilshikov, A.; Vila, A.V.; Võsa, U.; Mujagic, Z.; Masclee, A.A.M.; Jonkers, D.M.A.E.; Oosting, M.; et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 2019, 51, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation—Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Jones, P.J. Physiological effects of medium-chain triglycerides: Potential agents in the prevention of obesity. J. Nutr. 2002, 132, 329–332. [Google Scholar] [CrossRef]
- St-Onge, M.P.; Bosarge, A. Weight-loss diet that includes consumption of medium-chain triacylglycerol oil leads to a greater rate of weight and fat mass loss than does olive oil. Am. J. Clin. Nutr. 2008, 87, 621–626. [Google Scholar] [CrossRef]
- Wojciechowicz, M.L.; Ma’ayan, A. GPR84: An Immune Response Dial? Nat. Rev. Drug Discov. 2020, 19, 374. [Google Scholar] [CrossRef]
- Luscombe, V.B.; Lucy, D.; Bataille, C.J.; Russell, A.J.; Greaves, D.R. 20 Years an Orphan: Is GPR84 a Plausible Medium-Chain Fatty Acid-Sensing Receptor? DNA Cell Biol. 2020, 39, 1926–1937. [Google Scholar] [CrossRef]
- Puengel, T.; De Vos, S.; Hundertmark, J.; Kohlhepp, M.; Guldiken, N.; Pujuguet, P.; Auberval, M.; Marsais, F.; Shoji, K.F.; Saniere, L.; et al. The Medium-Chain Fatty Acid Receptor GPR84 Mediates Myeloid Cell Infiltration Promoting Steatohepatitis and Fibrosis. J. Clin. Med. 2020, 9, 1140. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, H.; Kondo, T.; Fuchigami, M.; Hashimoto, H.; Sugimura, Y.; Ozaki, N.; Arima, H.; Ota, A.; Oiso, Y.; Hamada, Y. Inflammatory changes in adipose tissue enhance expression of GPR84, a medium-chain fatty acid receptor: TNFalpha enhances GPR84 expression in adipocytes. FEBS Lett. 2012, 586, 368–372. [Google Scholar] [CrossRef]
- Simard, J.-C.; Thibodeau, J.-F.; LeDuc, M.; Tremblay, M.; Laverdure, A.; Sarra-Bournet, F.; Gagnon, W.; Ouboudinar, J.; Gervais, L.; Felton, A.; et al. Fatty acid mimetic PBI-4547 restores metabolic homeostasis via GPR84 in mice with non-alcoholic fatty liver disease. Sci. Rep. 2020, 10, 12778. [Google Scholar] [CrossRef]
- Zaibi, M.S.; Kępczyńska, M.A.; Harikumar, P.; AlOmar, S.Y.; Trayhurn, P. IL-33 stimulates expression of the GPR84 (EX33) fatty acid receptor gene and of cytokine and chemokine genes in human adipocytes. Cytokine 2018, 110, 189–193. [Google Scholar] [CrossRef]
- Marsango, S.; Barki, N.; Jenkins, L.; Tobin, A.B.; Milligan, G. Therapeutic validation of an orphan G protein-coupled receptor: The case of GPR84. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Tsalamandris, S.; Antonopoulos, A.S.; Oikonomou, E.; Papamikroulis, G.-A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur. Cardiol. 2019, 14, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, E.; Browne, L.; Irving-Rodgers, H.; Massa, H.M.; Fozzard, N.; Jennings, M.P.; Peak, I.R. Effect of GPR84 deletion on obesity and diabetes development in mice fed long chain or medium chain fatty acid rich diets. Eur. J. Nutr. 2018, 57, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [PubMed]
- Recio, C.; Lucy, D.; Purvis, G.; Iveson, P.; Zeboudj, L.; Iqbal, A.; Lin, D.; O’Callaghan, C.; Davison, L.; Griesbach, E.; et al. Activation of the Immune-Metabolic Receptor GPR84 Enhances Inflammation and Phagocytosis in Macrophages. Front. Immunol. 2018, 9, 1419. [Google Scholar] [CrossRef]
- Gaidarov, I.; Anthony, T.; Gatlin, J.; Chen, X.; Mills, D.; Solomon, M.; Han, S.; Semple, G.; Unett, D.J. Embelin and its derivatives unravel the signaling, proinflammatory and antiatherogenic properties of GPR84 receptor. Pharmacol. Res. 2018, 131, 185–198. [Google Scholar] [CrossRef]
- Recio, C.; Lucy, D.; Iveson, P.; Iqbal, A.; Valaris, S.; Wynne, G.; Russell, A.; Choudhury, R.P.; O’Callaghan, C.; Monaco, C.; et al. The Role of Metabolite-Sensing G Protein-Coupled Receptors in Inflammation and Metabolic Disease. Antioxid. Redox Signal. 2018, 29, 237–256. [Google Scholar] [CrossRef]
- Lim, S.L.; Tran, D.N.; Kieu, Z.; Chen, C.; Villanueva, E.; Ghiaar, S.; Gallup, V.; Zumkehr, J.; Cribbs, D.H.; Rodriguez-Ortiz, C.J.; et al. Genetic Ablation of Hematopoietic Cell Kinase Accelerates Alzheimer’s Disease-Like Neuropathology in Tg2576 Mice. Mol. Neurobiol. 2020, 57, 2447–2460. [Google Scholar] [CrossRef]
- Kiepura, A.; Stachyra, K.; Olszanecki, R. Anti-Atherosclerotic Potential of Free Fatty Acid Receptor 4 (FFAR4). Biomedicines. 2021, 9, 467. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. GPR40 Receptor Agonists for the Treatment of Type 2 Diabetes and Related Diseases. ACS Med. Chem. Lett. 2018, 9, 870–871. [Google Scholar] [CrossRef][Green Version]
- Tomita, T.; Hosoda, K.; Fujikura, J.; Inagaki, N.; Nakao, K. The G-Protein-Coupled Long-Chain Fatty Acid Receptor GPR40 and Glucose Metabolism. Front. Endocrinol. 2014, 5, 152. [Google Scholar] [CrossRef]
- Vettor, R.; Granzotto, M.; De Stefani, D.; Trevellin, E.; Rossato, M.; Farina, M.G.; Milan, G.; Pilon, C.; Nigro, A.; Federspil, G.; et al. Loss-of-function mutation of the GPR40 gene associates with abnormal stimulated insulin secretion by acting on intracellular calcium mobilization. J. Clin. Endocrinol. Metab. 2008, 93, 3541–3550. [Google Scholar] [CrossRef] [PubMed]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.-H.; Poitout, V. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Kebede, M.; Alquier, T.; Latour, M.G.; Semache, M.; Tremblay, C.; Poitout, V. The fatty acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes 2008, 57, 2432–2437. [Google Scholar] [CrossRef]
- Lan, H.; Hoos, L.M.; Liu, L.; Tetzloff, G.; Hu, W.; Abbondanzo, S.J.; Vassileva, G.; Gustafson, E.L.; Hedrick, J.A.; Davis, H.R. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes 2008, 57, 2999–3006. [Google Scholar] [CrossRef] [PubMed]
- Steneberg, P.; Rubins, N.; Bartoov-Shifman, R.; Walker, M.D.; Edlund, H. The FFA receptor GPR40 links hyperinsulinemia, hepatic steatosis, and impaired glucose homeostasis in mouse. Cell Metab. 2005, 1, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Meidute Abaraviciene, S.; Muhammed, S.J.; Amisten, S.; Lundquist, I.; Salehi, A. GPR40 protein levels are crucial to the regulation of stimulated hormone secretion in pancreatic islets. Lessons from spontaneous obesity-prone and non-obese type 2 diabetes in rats. Mol. Cell Endocrinol. 2013, 381, 150–159. [Google Scholar] [CrossRef][Green Version]
- Del Guerra, S.; Bugliani, M.; D’Aleo, V.; Del Prato, S.; Boggi, U.; Mosca, F.; Filipponi, F.; Lupi, R. G-protein-coupled receptor 40 (GPR40) expression and its regulation in human pancreatic islets: The role of type 2 diabetes and fatty acids. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 22–25. [Google Scholar] [CrossRef]
- Nagasumi, K.; Esaki, R.; Iwachidow, K.; Yasuhara, Y.; Ogi, K.; Tanaka, H.; Nakata, M.; Yano, T.; Shimakawa, K.; Taketomi, S.; et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 2009, 58, 1067–1076. [Google Scholar] [CrossRef]
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 is expressed in enteroendocrine cells and mediates free fatty acid stimulation of incretin secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef]
- Syed, I.; Lee, J.; Moraes-Vieira, P.M.; Donaldson, C.J.; Sontheimer, A.; Aryal, P.; Wellenstein, K.; Kolar, M.J.; Nelson, A.T.; Siegel, D.; et al. Palmitic Acid Hydroxystearic Acids Activate GPR40, Which Is Involved in Their Beneficial Effects on Glucose Homeostasis. Cell Metab. 2018, 27, 419–427. [Google Scholar] [CrossRef]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K.; et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef]
- Ichimura, A.; Hara, T.; Hirasawa, A. Regulation of Energy Homeostasis via GPR120. Front. Endocrinol. 2014, 5, 111. [Google Scholar] [CrossRef]
- Satapati, S.; Qian, Y.; Wu, M.S.; Petrov, A.; Dai, G.; Wang, S.-P.; Zhu, Y.; Shen, X.; Muise, E.; Chen, Y.; et al. GPR120 suppresses adipose tissue lipolysis and synergizes with GPR40 in antidiabetic efficacy. J. Lipid Res. 2017, 58, 1561–1578. [Google Scholar] [CrossRef] [PubMed]
- Du, M.-q.; Huang, Y.-q.; Lu, N.-S.; Shu, G.; Zhu, X.-t.; Wang, L.-n.; Gao, P.; Xi, Q.-y.; Zhang, Y.-l.; Wang, S.-b.; et al. Characterization and Differentiation into Adipocytes and Myocytes of Porcine Bone Marrow Mesenchymal Stem Cells. J. Integr. Agric. 2014, 13, 837–848. [Google Scholar] [CrossRef]
- Paschoal, V.A.; Walenta, E.; Talukdar, S.; Pessentheiner, A.R.; Osborn, O.; Hah, N.; Chi, T.J.; Tye, G.L.; Armando, A.M.; Evans, R.M.; et al. Positive Reinforcing Mechanisms between GPR120 and PPARgamma Modulate Insulin Sensitivity. Cell Metab. 2020, 31, 1173–1188. [Google Scholar] [CrossRef]
- Christian, M. Elucidation of the roles of brown and brite fat genes: GPR120 is a modulator of brown adipose tissue function. Exp. Physiol. 2020, 105, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.Y.; Jun, H.S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, T.; Zhang, D.; Leung, P.S. GPR120 protects lipotoxicity-induced pancreatic beta-cell dysfunction through regulation of PDX1 expression and inhibition of islet inflammation. Clin. Sci. 2019, 133, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Leung, P.S. Potential roles of GPR120 and its agonists in the management of diabetes. Drug Des. Devel. Ther. 2014, 8, 1013–1027. [Google Scholar]
- Murphy, K.A.; Harsch, B.A.; Healy, C.L.; Joshi, S.S.; Huang, S.; Walker, R.E.; Wagner, B.M.; Ernste, K.M.; Huang, W.; Block, R.; et al. Free fatty acid receptor 4 is a nutrient sensor that resolves inflammation to maintain cardiac homeostasis. bioRxiv 2019, 776294. [Google Scholar] [CrossRef]
- Murphy, K.A.; Harsch, B.A.; Healy, C.L.; Joshi, S.S.; Huang, S.; Walker, R.E.; Wagner, B.M.; Ernste, K.M.; Huang, W.; Block, R.C.; et al. Free fatty acid receptor 4 responds to endogenous fatty acids to protect the heart from pressure overload. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- An, T.; Zhang, X.; Li, H.; Dou, L.; Huang, X.; Man, Y.; Zhang, X.; Shen, T.; Li, G.; Li, J.; et al. GPR120 facilitates cholesterol efflux in macrophages through activation of AMPK signaling pathway. FEBS J. 2020, 287, 5080–5095. [Google Scholar] [CrossRef] [PubMed]
- Gettys, T.W.; Burrows, P.M.; Henricks, D.M. Variance weighting functions in radioimmunoassay calibration. Am. J. Physiol. 1986, 251, E357–E361. [Google Scholar] [CrossRef]
- Liu, R.; Cheng, F.; Zeng, K.; Li, W.; Lan, J. GPR120 Agonist GW9508 Ameliorated Cellular Senescence Induced by ox-LDL. ACS Omega 2020, 5, 32195–32202. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Olefsky, J.M.; Osborn, O. Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol. Sci. 2011, 32, 543–550. [Google Scholar] [CrossRef]
- Yamada, H.; Umemoto, T.; Kakei, M.; Momomura, S.-I.; Kawakami, M.; Ishikawa, S.-E.; Hara, K. Eicosapentaenoic acid shows anti-inflammatory effect via GPR120 in 3T3-L1 adipocytes and attenuates adipose tissue inflammation in diet-induced obese mice. Nutr. Metab. 2017, 14, 33. [Google Scholar] [CrossRef]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.Q.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, Y.; Liang, C.-P.; Thorp, E.B.; Han, S.; Jehle, A.W.; Saraswathi, V.; Pridgen, B.; Kanter, J.E.; Li, R.; et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ. Res. 2009, 105, 1072–1082. [Google Scholar] [CrossRef]
- Overton, H.A.; Fyfe, M.C.; Reynet, C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S76–S81. [Google Scholar] [CrossRef]
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 2009, 58, 1058–1066. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Langhans, C.D.; Hanson, J.; Michalski, C.W.; Kolker, S.; Jones, P.M.; Okun, J.G.; Offermanns, S. Deorphanization of GPR109B as a receptor for the beta-oxidation intermediate 3-OH-octanoic acid and its role in the regulation of lipolysis. J. Biol. Chem. 2009, 284, 21928–21933. [Google Scholar] [CrossRef]
- Adewale, Q.; Khan, A.F.; Carbonell, F.; Iturria-Medina, Y. Integrated transcriptomic and neuroimaging brain model decodes biological mechanisms in aging and Alzheimer’s disease. Elife 2021, 10, e62589. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, W.; Qian, H. GPR119 Agonists: A Novel Strategy for Type 2 Diabetes Treatment. In Diabetes Mellitus: Insights and Perspectives; Oguntibeju, O., Ed.; BoD–Books on Demand: Hamburg, Germany, 2013. [Google Scholar]
- Panaro, B.L.; Flock, G.B.; Campbell, J.E.; Beaudry, J.L.; Cao, X.; Drucker, D.J. beta-Cell Inactivation of Gpr119 Unmasks Incretin Dependence of GPR119-Mediated Glucoregulation. Diabetes 2017, 66, 1626–1635. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, Y.; Hu, Y.; Peng, J. Targeting the GPR119/incretin axis: A promising new therapy for metabolic-associated fatty liver disease. Cell Mol. Biol. Lett. 2021, 26, 32. [Google Scholar] [CrossRef]
- Li, H.; Fang, Y.; Guo, S.; Yang, Z. GPR119 agonists for the treatment of type 2 diabetes: An updated patent review (2014–present). Expert Opin. Ther. Pat. 2021, 31, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Cornall, L.M.; Hryciw, D.; Mathai, M.L.; McAinch, A.J. Direct activation of the proposed anti-diabetic receptor, GPR119 in cardiomyoblasts decreases markers of muscle metabolic activity. Mol. Cell Endocrinol. 2015, 402, 72–85. [Google Scholar] [CrossRef]
- Yamada, Y.; Terauchi, Y.; Watada, H.; Nakatsuka, Y.; Shiosakai, K.; Washio, T.; Taguchi, T. Efficacy and Safety of GPR119 Agonist DS-8500a in Japanese Patients with Type 2 Diabetes: A Randomized, Double-Blind, Placebo-Controlled, 12-Week Study. Adv. Ther. 2018, 35, 367–381. [Google Scholar] [CrossRef]
- Manaithiya, A.; Alam, O.; Sharma, V.; Naim, M.J.; Mittal, S.; Khan, I.A. GPR119 agonists: Novel therapeutic agents for type 2 diabetes mellitus. Bioorganic Chem. 2021, 113, 104998. [Google Scholar] [CrossRef] [PubMed]
- Lillich, F.F.; Imig, J.D.; Proschak, E. Multi-Target Approaches in Metabolic Syndrome. Front. Pharmacol. 2021, 11, 554961. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Kim, H.S.; Choi, Y.W.; Kim, Y.M.; Kang, K.W. Therapeutic application of GPR119 ligands in metabolic disorders. Diabetes Obes. Metab. 2018, 20, 257–269. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Dąbek, A.; Wojtala, M.; Pirola, L.; Balcerczyk, A. Modulation of Cellular Biochemistry, Epigenetics and Metabolomics by Ketone Bodies. Implications of the Ketogenic Diet in the Physiology of the Organism and Pathological States. Nutrients 2020, 12, 788. [Google Scholar] [CrossRef]
- Prattichizzo, F.; De Nigris, V.; Micheloni, S.; La Sala, L.; Ceriello, A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes Obes. Metab. 2018, 20, 2515–2522. [Google Scholar] [CrossRef]
- Offermanns, S. Hydroxy-Carboxylic Acid Receptor Actions in Metabolism. Trends Endocrinol. Metab. 2017, 28, 227–236. [Google Scholar] [CrossRef]
- Ahmed, K. Biological roles and therapeutic potential of hydroxy-carboxylic Acid receptors. Front. Endocrinol. 2011, 2, 51. [Google Scholar] [CrossRef] [PubMed]
- Blad, C.C.; Ahmed, K.; Ijzerman, A.; Offermanns, S. Biological and pharmacological roles of HCA receptors. Adv. Pharmacol. 2011, 62, 219–250. [Google Scholar] [PubMed]
- Blad, C.C.; Tang, C.; Offermanns, S. G protein-coupled receptors for energy metabolites as new therapeutic targets. Nat. Rev. Drug Discov. 2012, 11, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Stuttgen, G.M.; Sahoo, D. FFAR4: A New Player in Cardiometabolic Disease? Endocrinology 2021, 162, bqab111. [Google Scholar] [CrossRef]
- Kanikarla-Marie, P.; Jain, S.K. Hyperketonemia and ketosis increase the risk of complications in type 1 diabetes. Free Radic. Biol. Med. 2016, 95, 268–277. [Google Scholar] [CrossRef]
- Wallenius, K.; Thalén, P.; Björkman, J.-A.; Johannesson, P.; Wiseman, J.; Böttcher, G.; Fjellström, O.; Oakes, N.D. Involvement of the metabolic sensor GPR81 in cardiovascular control. JCI Insight 2017, 2, e92564. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef]
- Sun, S.; Li, H.; Chen, J.; Qian, Q. Lactic Acid: No Longer an Inert and End-Product of Glycolysis. Physiology 2017, 32, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. Inflammation inhibits GPR81 expression in adipose tissue. Inflamm. Res. 2011, 60, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Graff, E.C.; Judd, R.L. Effects of high fat diet on GPR109A and GPR81 gene expression. Biochem. Biophys. Res. Commun. 2012, 425, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Jeninga, E.H.; Bugge, A.; Nielsen, R.; Kersten, S.; Hamers, N.; Dani, C.; Wabitsch, M.; Berger, R.; Stunnenberg, H.G.; Mandrup, S.; et al. Peroxisome proliferator-activated receptor gamma regulates expression of the anti-lipolytic G-protein-coupled receptor 81 (GPR81/Gpr81). J. Biol. Chem. 2009, 284, 26385–26393. [Google Scholar] [CrossRef]
- Weiss, H.J.; Angiari, S. Metabolite Transporters as Regulators of Immunity. Metabolites 2020, 10, 418. [Google Scholar] [CrossRef]
- Rooney, K.; Trayhurn, P. Lactate and the GPR81 receptor in metabolic regulation: Implications for adipose tissue function and fatty acid utilisation by muscle during exercise. Br. J. Nutr. 2011, 106, 1310–1316. [Google Scholar] [CrossRef]
- Schulz, T.J.; Tseng, Y.H. Brown adipose tissue: Development, metabolism and beyond. Biochem. J. 2013, 453, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.; Yoo, T.; Joung, H.-Y.; Jo, Y.-H. Hydrocarboxylic acid receptor 1 in BAT regulates glucose uptake in mice fed a high-fat diet. PLoS ONE 2020, 15, e0228320. [Google Scholar] [CrossRef]
- Jones, N.K.; Stewart, K.; Czopek, A.; Menzies, R.I.; Thomson, A.; Moran, C.M.; Cairns, C.; Conway, B.R.; Denby, L.; Livingstone, D.E.; et al. Endothelin-1 Mediates the Systemic and Renal Hemodynamic Effects of GPR81 Activation. Hypertension 2020, 75, 1213–1222. [Google Scholar] [CrossRef]
- Manoharan, I.; Prasad, P.D.; Thangaraju, M.; Manicassamy, S. Lactate-Dependent Regulation of Immune Responses by Dendritic Cells and Macrophages. Front. Immunol. 2021, 12, 691134. [Google Scholar] [CrossRef]
- Yang, K.; Xu, J.; Fan, M.; Tu, F.; Wang, X.; Ha, T.; Williams, D.L.; Li, C. Lactate Suppresses Macrophage Pro-Inflammatory Response to LPS Stimulation by Inhibition of YAP and NF-kappaB Activation via GPR81-Mediated Signaling. Front. Immunol. 2020, 11, 587913. [Google Scholar] [CrossRef]
- Sun, Z.; Han, Y.; Song, S.; Chen, T.; Han, Y.; Liu, Y. Activation of GPR81 by lactate inhibits oscillatory shear stress-induced endothelial inflammation by activating the expression of KLF2. IUBMB Life 2019, 71, 2010–2019. [Google Scholar] [CrossRef]
- Hu, J.; Cai, M.; Liu, Y.; Liu, B.; Xue, X.; Ji, R.; Bian, X.; Lou, S. The roles of GRP81 as a metabolic sensor and inflammatory mediator. J. Cell Physiol. 2020, 235, 8938–8950. [Google Scholar] [CrossRef] [PubMed]
- Shantha, G.P.S.; Wasserman, B.; Astor, B.C.; Coresh, J.; Brancati, F.; Sharrett, A.R.; Young, J.H. Association of blood lactate with carotid atherosclerosis: The Atherosclerosis Risk in Communities (ARIC) Carotid MRI Study. Atherosclerosis 2013, 228, 249–255. [Google Scholar] [CrossRef]
- Lerch, M.M.; Conwell, D.L.; Mayerle, J. The anti-inflammasome effect of lactate and the lactate GPR81-receptor in pancreatic and liver inflammation. Gastroenterology 2014, 146, 1602–1605. [Google Scholar] [CrossRef] [PubMed]
- Errea, A.; Cayet, D.; Marchetti, P.; Tang, C.; Kluza, J.; Offermanns, S.; Sirard, J.-C.; Rumbo, M. Lactate Inhibits the Pro-Inflammatory Response and Metabolic Reprogramming in Murine Macrophages in a GPR81-Independent Manner. PLoS ONE 2016, 11, e0163694. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar]
- Riddy, D.; Delerive, P.; Summers, R.; Sexton, P.; Langmead, C.J. G Protein-Coupled Receptors Targeting Insulin Resistance, Obesity, and Type 2 Diabetes Mellitus. Pharmacol Rev. 2018, 70, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Jadeja, R.N.; Jones, M.A.; Fromal, O.; Powell, F.L.; Khurana, S.; Singh, N.; Martin, P.M. Loss of GPR109A/HCAR2 induces aging-associated hepatic steatosis. Aging 2019, 11, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Marcos, E.; Parpaleix, A.; Amsellem, V.; Breau, M.; Houssaini, A.; Vienney, N.; Lefevre, M.; Derumeaux, G.; Evans, S.; et al. CCR2/CCR5-mediated macrophage-smooth muscle cell crosstalk in pulmonary hypertension. Eur. Respir. J. 2019, 54, 1802308. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.T.; Digby, J.E.; Choudhury, R.P. GPR109A and vascular inflammation. Curr. Atheroscler. Rep. 2013, 15, 325. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; McKenzie, C.I.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef]
- Ratajczak, W.; Ryl, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczynska, M. Immunomodulatory potential of gut microbiome-derived short-chain fatty acids (SCFAs). Acta Biochim. Pol. 2019, 66, 1–12. [Google Scholar] [CrossRef]
- Zhou, E.; Li, Y.; Yao, M.; Wei, Z.; Fu, Y.; Yang, Z. Niacin attenuates the production of pro-inflammatory cytokines in LPS-induced mouse alveolar macrophages by HCA2 dependent mechanisms. Int. Immunopharmacol. 2014, 23, 121–126. [Google Scholar] [CrossRef]
- Feingold, K.R.; Moser, A.; Shigenaga, J.K.; Grunfeld, C. Inflammation stimulates niacin receptor (GPR109A/HCA2) expression in adipose tissue and macrophages. J. Lipid Res. 2014, 55, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Mandrika, I.; Tilgase, A.; Petrovska, R.; Klovins, J. Hydroxycarboxylic Acid Receptor Ligands Modulate Proinflammatory Cytokine Expression in Human Macrophages and Adipocytes without Affecting Adipose Differentiation. Biol. Pharm. Bull. 2018, 41, 1574–1580. [Google Scholar] [CrossRef]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 2016, 65, 102–113. [Google Scholar] [CrossRef]
- Montserrat-de la Paz, S.; Naranjo, M.C.; Lopez, S.; Abia, R.; Muriana, F.J.G.; Bermudez, B. Niacin and its metabolites as master regulators of macrophage activation. J. Nutr. Biochem. 2017, 39, 40–47. [Google Scholar] [CrossRef]
- Wanders, D.; Graff, E.C.; White, B.D.; Judd, R.L. Niacin increases adiponectin and decreases adipose tissue inflammation in high fat diet-fed mice. PLoS ONE 2013, 8, e71285. [Google Scholar] [CrossRef]
- Schaub, A.; Futterer, A.; Pfeffer, K. PUMA-G, an IFN-gamma-inducible gene in macrophages is a novel member of the seven transmembrane spanning receptor superfamily. Eur. J. Immunol. 2001, 31, 3714–3725. [Google Scholar] [CrossRef]
- El Kebir, D.; Filep, J.G. Modulation of Neutrophil Apoptosis and the Resolution of Inflammation through beta2 Integrins. Front. Immunol. 2013, 4, 60. [Google Scholar] [CrossRef]
- Mierziak, J.; Burgberger, M.; Wojtasik, W. 3-Hydroxybutyrate as a Metabolite and a Signal Molecule Regulating Processes of Living Organisms. Biomolecules 2021, 11, 402. [Google Scholar] [CrossRef] [PubMed]
- Gille, A.; Bodor, E.T.; Ahmed, K.; Offermanns, S. Nicotinic acid: Pharmacological effects and mechanisms of action. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 79–106. [Google Scholar] [CrossRef]
- Horimatsu, T.; Blomkalns, A.L.; Ogbi, M.; Moses, M.; Kim, D.; Patel, S.; Gilreath, N.; Reid, L.; Benson, T.W.; Pye, J.; et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: Role of NAD+/nicotinamide. Cardiovasc. Res. 2020, 116, 2226–2238. [Google Scholar] [CrossRef]
- Lukasova, M.; Malaval, C.; Gille, A.; Kero, J.; Offermanns, S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J. Clin. Investig. 2011, 121, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.J.; Sayanlar, J.J.; Dooley, D.J.; Srichai, M.B.; Taylor, A.J. A randomized pilot study on the effect of niacin on pulmonary arterial pressure. Trials 2015, 16, 530. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Bai, P.; Wan, N.; Liu, J.; Zhu, Q.; He, Y.; Chen, G.; Wang, J.; Chen, H.; Wang, C.; et al. Niacin Attenuates Pulmonary Hypertension Through H-PGDS in Macrophages. Circ. Res. 2020, 127, 1323–1336. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.C.; Naveed, M.; Gordon, A.; Majeed, F.; Saeed, M.; Ogbuke, M.I.; Atif, M.; Zubair, H.M.; Changxing, L. beta-Adrenergic receptor, an essential target in cardiovascular diseases. Heart Fail. Rev. 2020, 25, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.M.; Shihata, W.A.; Jama, H.A.; Tsyganov, K.; Ziemann, M.; Kiriazis, H.; Horlock, D.; Vijay, A.; Giam, B.; Vinh, A.; et al. Deficiency of Prebiotic Fiber and Insufficient Signaling Through Gut Metabolite-Sensing Receptors Leads to Cardiovascular Disease. Circulation 2020, 141, 1393–1403. [Google Scholar] [CrossRef]
- Tuteja, S.; Wang, L.; Dunbar, R.; Chen, J.; DerOhannessian, S.; Marcovina, S.M.; Elam, M.; Lader, E.; Rader, D.J. Genetic coding variants in the niacin receptor, hydroxyl-carboxylic acid receptor 2, and response to niacin therapy. Pharm. Genom. 2017, 27, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Dubrall, D.; Pflock, R.; Kosinska, J.; Schmid, M.; Bleich, M.; Himmerkus, N.; Offermanns, S.; Schwaninger, M.; Sachs, B. Do dimethyl fumarate and nicotinic acid elicit common, potentially HCA2 -mediated adverse reactions? A combined epidemiological-experimental approach. Br. J. Clin. Pharmacol. 2021, 87, 3813–3824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, G.; Deng, X.Y.; He, X.B.; Chen, L.J.; Wu, C.; Shi, Y.; Wu, K.P.; Mei, L.J.; Lu, J.X.; et al. Activated human hydroxy-carboxylic acid receptor-3 signals to MAP kinase cascades via the PLC-dependent PKC and MMP-mediated EGFR pathways. Br. J. Pharmacol. 2012, 166, 1756–1773. [Google Scholar] [CrossRef]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef]
- Kapolka, N.J.; Taghon, G.J.; Rowe, J.B.; Morgan, W.M.; Enten, J.F.; Lambert, N.A.; Isom, D.G. DCyFIR: A high-throughput CRISPR platform for multiplexed G protein-coupled receptor profiling and ligand discovery. Proc. Natl. Acad. Sci. USA 2020, 117, 13117–13126. [Google Scholar] [CrossRef]
- Cervenka, I.; Agudelo, L.Z.; Ruas, J.L. Kynurenines: Tryptophan’s metabolites in exercise, inflammation, and mental health. Science 2017, 357, eaaf9794. [Google Scholar] [CrossRef]
- Semple, G.; Skinner, P.J.; Cherrier, M.C.; Webb, P.J.; Sage, C.R.; Tamura, S.Y.; Chen, R.; Richman, J.G.; Connolly, D. 1-Alkyl-benzotriazole-5-carboxylic acids are highly selective agonists of the human orphan G-protein-coupled receptor GPR109b. J. Med. Chem. 2006, 49, 1227–1230. [Google Scholar] [CrossRef]
- Kapolka, N.J.; Isom, D.G. HCAR3: An underexplored metabolite sensor. Nat. Rev. Drug Discov. 2020, 19, 745. [Google Scholar] [CrossRef]
- Skinner, P.J.; Cherrier, M.C.; Webb, P.J.; Sage, C.R.; Dang, H.T.; Pride, C.C.; Chen, R.; Tamura, S.Y.; Richman, J.G.; Connolly, D.T.; et al. 3-Nitro-4-amino benzoic acids and 6-amino nicotinic acids are highly selective agonists of GPR109b. Bioorg. Med. Chem. Lett. 2007, 17, 6619–6622. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Guo, C.; Chen, W.D.; Wang, Y.D. TGR5, Not Only a Metabolic Regulator. Front. Physiol. 2016, 7, 646. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [PubMed]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Van Nierop, F.S.; Scheltema, M.J.; Eggink, H.M.; Pols, T.W.; Sonne, D.P.; Knop, F.K.; Soeters, M.R. Clinical relevance of the bile acid receptor TGR5 in metabolism. Lancet Diabetes Endocrinol. 2017, 5, 224–233. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef]
- Vassileva, G.; Golovko, A.; Markowitz, L.; Abbondanzo, S.J.; Zeng, M.; Yang, S.; Hoos, L.; Tetzloff, G.; Levitan, D.; Murgolo, N.J.; et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem. J. 2006, 398, 423–430. [Google Scholar] [CrossRef]
- Prinz, P.; Hofmann, T.; Ahnis, A.; Elbelt, U.; Goebel-Stengel, M.; Klapp, B.F.; Rose, M.; Stengel, A. Plasma bile acids show a positive correlation with body mass index and are negatively associated with cognitive restraint of eating in obese patients. Front. Neurosci. 2015, 9, 199. [Google Scholar] [CrossRef]
- Chen, X.; Lou, G.; Meng, Z.; Huang, W. TGR5: A novel target for weight maintenance and glucose metabolism. Exp. Diabetes Res. 2011, 2011, 853501. [Google Scholar] [CrossRef]
- Donepudi, A.C.; Boehme, S.; Li, F.; Chiang, J.Y. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology 2017, 65, 813–827. [Google Scholar] [CrossRef]
- Zarrinpar, A.; Loomba, R. Review article: The emerging interplay among the gastrointestinal tract, bile acids and incretins in the pathogenesis of diabetes and non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 2012, 36, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, W.; Wang, T.; You, P.; Zhao, Y.; Yang, Y.; Wang, X.; Luo, J.; Chen, Y.; Liu, M.; et al. A Novel TGR5 Activator WB403 Promotes GLP-1 Secretion and Preserves Pancreatic beta-Cells in Type 2 Diabetic Mice. PLoS ONE 2015, 10, e0134051. [Google Scholar]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef] [PubMed]
- Holter, M.M.; Chirikjian, M.K.; Govani, V.N.; Cummings, B.P. TGR5 Signaling in Hepatic Metabolic Health. Nutrients 2020, 12, 2598. [Google Scholar] [CrossRef]
- Ethanic, M.; Stanimirov, B.; Pavlovic, N.; Golocorbin-Kon, S.; Al-Salami, H.; Stankov, K.; Mikov, M. Pharmacological Applications of Bile Acids and Their Derivatives in the Treatment of Metabolic Syndrome. Front. Pharmacol. 2018, 9, 1382. [Google Scholar]
- Pols, T.W.; Nomura, M.; Harach, T.; Sasso, G.L.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef]
- Miyazaki-Anzai, S.; Masuda, M.; Levi, M.; Keenan, A.L.; Miyazaki, M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS ONE 2014, 9, e108270. [Google Scholar]
- Steiner, C.; Othman, A.; Saely, C.H.; Rein, P.; Drexel, H.; von Eckardstein, A.; Rentsch, K.M. Bile acid metabolites in serum: Intraindividual variation and associations with coronary heart disease, metabolic syndrome and diabetes mellitus. PLoS ONE 2011, 6, e25006. [Google Scholar] [CrossRef] [PubMed]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef]
- Kang, Y.M.; Jung, C.H. Cardiovascular Effects of Glucagon-Like Peptide-1 Receptor Agonists. Endocrinol. Metab. 2016, 31, 258–274. [Google Scholar] [CrossRef]
- Zhou, H.; Hylemon, P.B. Bile acids are nutrient signaling hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Kwong, E.; Li, Y.; Hylemon, P.B.; Zhou, H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm. Sin. B 2015, 5, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Cheng, K.; Zimniak, P. Activation of muscarinic receptor signaling by bile acids: Physiological and medical implications. Dig. Dis. Sci. 2003, 48, 1431–1444. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Cheng, K.; Saxena, N.; Chahdi, A.; Belo, A.; Khurana, S.; Xie, G. Muscarinic receptor agonists stimulate matrix metalloproteinase 1-dependent invasion of human colon cancer cells. Biochem. Biophys. Res. Commun. 2011, 415, 319–324. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Fyrst, H.; Saba, J.D. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat. Chem. Biol. 2010, 6, 489–497. [Google Scholar] [CrossRef]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell-Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R.; et al. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018, 3, e96805. [Google Scholar] [CrossRef] [PubMed]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Vestri, A.; Pierucci, F.; Frati, A.; Monaco, L.; Meacci, E. Sphingosine 1-Phosphate Receptors: Do They Have a Therapeutic Potential in Cardiac Fibrosis? Front. Pharmacol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015, 61, 1216–1226. [Google Scholar] [CrossRef]
- Chen, W.; Lu, H.; Yang, J.; Xiang, H.; Peng, H. Sphingosine 1-phosphate in metabolic syndrome (Review). Int. J. Mol. Med. 2016, 38, 1030–1038. [Google Scholar] [CrossRef]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar]
- Samad, F.; Hester, K.D.; Yang, G.; Hannun, Y.A.; Bielawski, J. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 2006, 55, 2579–2587. [Google Scholar] [CrossRef]
- Moon, M.H.; Jeong, J.K.; Lee, J.H.; Park, Y.G.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Antiobesity activity of a sphingosine 1-phosphate analogue FTY720 observed in adipocytes and obese mouse model. Exp. Mol. Med. 2012, 44, 603–614. [Google Scholar] [CrossRef]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Park, S.Y. Sphingosine-1-phosphate inhibits the adipogenic differentiation of 3T3-L1 preadipocytes. Int. J. Mol. Med. 2014, 34, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Wang, W.; Song, Z.; Aji, G.; Liu, X.T.; Xia, P. Role of Sphingosine Kinase in Type 2 Diabetes Mellitus. Front. Endocrinol. 2020, 11, 627076. [Google Scholar] [CrossRef]
- Wang, J.; Badeanlou, L.; Bielawski, J.; Ciaraldi, T.P.; Samad, F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E756–E768. [Google Scholar] [CrossRef]
- Cantrell Stanford, J.; Morris, A.J.; Sunkara, M.; Popa, G.J.; Larson, K.L.; Ozcan, S. Sphingosine 1-phosphate (S1P) regulates glucose-stimulated insulin secretion in pancreatic beta cells. J. Biol. Chem. 2012, 287, 13457–13464. [Google Scholar] [CrossRef]
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V. Obesity and dyslipidemia. Metabolism. 2019, 92, 71–81. [Google Scholar] [CrossRef]
- Yang, J.; Yang, L.; Tian, L.; Ji, X.; Yang, L.; Li, L. Sphingosine 1-Phosphate (S1P)/S1P Receptor2/3 Axis Promotes Inflammatory M1 Polarization of Bone Marrow-Derived Monocyte/Macrophage via G(alpha)i/o/PI3K/JNK Pathway. Cell Physiol. Biochem. 2018, 49, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Hajeyah, A.A.; Griffiths, W.J.; Wang, Y.; Finch, A.J.; O’Donnell, V.B. The Biosynthesis of Enzymatically Oxidized Lipids. Front. Endocrinol. 2020, 11, 591819. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance-A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef]
- Simopoulos, A.P. An Increase in the Omega-6/Omega-3 Fatty Acid Ratio Increases the Risk for Obesity. Nutrients 2016, 8, 128. [Google Scholar] [CrossRef]
- An, Y.A.; Chen, S.; Deng, Y.; Wang, Z.V.; Funcke, J.B.; Shah, M.; Shan, B.; Gordillo, R.; Yoshino, J.; Klein, S.; et al. The mitochondrial dicarboxylate carrier prevents hepatic lipotoxicity by inhibiting white adipocyte lipolysis. J. Hepatol. 2021, 75, 387–399. [Google Scholar] [CrossRef]
- Martin Arias, L.H.; Martin Gonzalez, A.; Sanz Fadrique, R.; Vazquez, E.S. Cardiovascular Risk of Nonsteroidal Anti-inflammatory Drugs and Classical and Selective Cyclooxygenase-2 Inhibitors: A Meta-analysis of Observational Studies. J. Clin. Pharmacol. 2019, 59, 55–73. [Google Scholar] [CrossRef]
- Woodward, D.F.; Jones, R.L.; Narumiya, S. International Union of Basic and Clinical Pharmacology. LXXXIII: Classification of prostanoid receptors, updating 15 years of progress. Pharmacol. Rev. 2011, 63, 471–538. [Google Scholar] [CrossRef]
- Hirata, T.; Narumiya, S. Prostanoid receptors. Chem. Rev. 2011, 111, 6209–6230. [Google Scholar] [CrossRef]
- Boie, Y.; Rushmore, T.H.; Darmon-Goodwin, A.; Grygorczyk, R.; Slipetz, D.M.; Metters, K.M.; Abramovitz, M. Cloning and expression of a cDNA for the human prostanoid IP receptor. J. Biol. Chem. 1994, 269, 12173–12178. [Google Scholar] [CrossRef]
- Coleman, R.A.; Smith, W.L.; Narumiya, S. International Union of Pharmacology classification of prostanoid receptors: Properties, distribution, and structure of the receptors and their subtypes. Pharmacol. Rev. 1994, 46, 205–229. [Google Scholar] [PubMed]
- Negrel, R. Prostacyclin as a critical prostanoid in adipogenesis. Prostaglandins Leukot. Essent. Fatty Acids 1999, 60, 383–386. [Google Scholar] [CrossRef]
- Sasaki, Y.; Kuwata, H.; Akatsu, M.; Yamakawa, Y.; Ochiai, T.; Yoda, E.; Nakatani, Y.; Yokoyama, C.; Hara, S. Involvement of prostacyclin synthase in high-fat-diet-induced obesity. Prostaglandins Other Lipid Mediat. 2021, 153, 106523. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Eckman, K.; Lee, Y.K.; Abdelmegeed, M.A.; Esterle, A.; Chilian, W.M.; Chiang, J.Y.; Song, B.J. Eicosanoids in metabolic syndrome. Adv. Pharmacol. 2013, 66, 157–266. [Google Scholar] [PubMed]
- Hodnett, B.L.; Dearman, J.A.; Carter, C.B.; Hester, R.L. Attenuated PGI2 synthesis in obese Zucker rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R715–R721. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Kaneko, M.; Tamura, M.; Kurumatani, H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes 2010, 59, 1092–1100. [Google Scholar] [CrossRef]
- Smyth, E.M.; Grosser, T.; Wang, M.; Yu, Y.; FitzGerald, G.A. Prostanoids in health and disease. J. Lipid Res. 2009, 50, S423–S428. [Google Scholar] [CrossRef] [PubMed]
- Arehart, E.; Stitham, J.; Asselbergs, F.W.; Douville, K.; MacKenzie, T.; Fetalvero, K.M.; Gleim, S.; Kasza, Z.; Rao, Y.; Martel, L.; et al. Acceleration of cardiovascular disease by a dysfunctional prostacyclin receptor mutation: Potential implications for cyclooxygenase-2 inhibition. Circ. Res. 2008, 102, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Geraci, M.W.; Gao, B.; Shepherd, D.C.; Moore, M.D.; Westcott, J.Y.; Fagan, K.A.; Alger, L.A.; Tuder, R.M.; Voelkel, N.F. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J. Clin. Investig. 1999, 103, 1509–1515. [Google Scholar] [CrossRef][Green Version]
- Montani, D.; Chaumais, M.C.; Guignabert, C.; Gunther, S.; Girerd, B.; Jais, X.; Algalarrondo, V.; Price, L.C.; Savale, L.; Sitbon, O.; et al. Targeted therapies in pulmonary arterial hypertension. Pharmacol. Ther. 2014, 141, 172–191. [Google Scholar] [CrossRef]
- Cheng, Y.; Austin, S.C.; Rocca, B.; Koller, B.H.; Coffman, T.M.; Grosser, T.; Lawson, J.A.; FitzGerald, G.A. Role of prostacyclin in the cardiovascular response to thromboxane A2. Science 2002, 296, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Ushikubi, F.; Matsuoka, T.; Hirata, M.; Yamasaki, A.; Sugimoto, Y.; Ichikawa, A.; Aze, Y.; Tanaka, T.; Yoshida, N.; et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature 1997, 388, 678–682. [Google Scholar] [CrossRef]
- Egan, K.M.; Lawson, J.A.; Fries, S.; Koller, B.; Rader, D.J.; Smyth, E.M.; Fitzgerald, G.A. COX-2-derived prostacyclin confers atheroprotection on female mice. Science 2004, 306, 1954–1957. [Google Scholar] [CrossRef]
- Dorris, S.L.; Peebles, R.S., Jr. PGI2 as a regulator of inflammatory diseases. Mediators Inflamm. 2012, 2012, 926968. [Google Scholar] [CrossRef]
- Warner, T.D.; Nylander, S.; Whatling, C. Anti-platelet therapy: Cyclo-oxygenase inhibition and the use of aspirin with particular regard to dual anti-platelet therapy. Br. J. Clin. Pharmacol. 2011, 72, 619–633. [Google Scholar] [CrossRef]
- Koh, K.K.; Park, S.M.; Quon, M.J. Leptin and cardiovascular disease: Response to therapeutic interventions. Circulation 2008, 117, 3238–3249. [Google Scholar] [CrossRef]
- Menzaghi, C.; Trischitta, V. The Adiponectin Paradox for All-Cause and Cardiovascular Mortality. Diabetes 2018, 67, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, Q.; Rodriguez, S.; Tan, S.Y.; Seldin, M.M.; McLenithan, J.C.; Jia, W.; Wong, G.W. Thromboxane synthase deficiency improves insulin action and attenuates adipose tissue fibrosis. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E792–E804. [Google Scholar] [CrossRef]
- Wang, W.; Zhong, X.; Guo, J. Role of 2 series prostaglandins in the pathogenesis of type 2 diabetes mellitus and nonalcoholic fatty liver disease (Review). Int. J. Mol. Med. 2021, 47, 1–15. [Google Scholar] [CrossRef]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef]
- Burleigh, M.E.; Babaev, V.R.; Oates, J.A.; Harris, R.C.; Gautam, S.; Riendeau, D.; Marnett, L.J.; Morrow, J.D.; Fazio, S.; Linton, M.F. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in LDL receptor-deficient mice. Circulation 2002, 105, 1816–1823. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tahara, Y.; Matsumoto, M.; Iguchi, M.; Sano, H.; Murayama, T.; Arai, H.; Oida, H.; Yurugi-Kobayashi, T.; Yamashita, J.K.; et al. Roles of thromboxane A2 and prostacyclin in the development of atherosclerosis in apoE-deficient mice. J. Clin. Investig. 2004, 114, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.; Glatt, M.; Kalin, H.; Peskar, B.A. Pharmacological control of prostaglandin and thromboxane release from macrophages. Nature 1978, 274, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Babaev, V.R.; Chew, J.D.; Ding, L.; Davis, S.; Breyer, M.D.; Breyer, R.M.; Oates, J.A.; Fazio, S.; Linton, M.F. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab. 2008, 8, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Richelsen, B. Factors regulating the production of prostaglandin E2 and prostacyclin (prostaglandin I2) in rat and human adipocytes. Biochem. J. 1987, 247, 389–394. [Google Scholar] [CrossRef]
- Ceddia, R.P.; Lee, D.; Maulis, M.F.; Carboneau, B.A.; Threadgill, D.W.; Poffenberger, G.; Milne, G.; Boyd, K.L.; Powers, A.C.; McGuinness, O.P.; et al. The PGE2 EP3 Receptor Regulates Diet-Induced Adiposity in Male Mice. Endocrinology 2016, 157, 220–232. [Google Scholar] [CrossRef]
- Tang, E.H.; Cai, Y.; Wong, C.K.; Rocha, V.Z.; Sukhova, G.K.; Shimizu, K.; Xuan, G.; Vanhoutte, P.M.; Libby, P.; Xu, A. Activation of prostaglandin E2-EP4 signaling reduces chemokine production in adipose tissue. J. Lipid Res. 2015, 56, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Van Rodijnen, W.F.; Korstjens, I.J.; Legerstee, N.; Ter Wee, P.M.; Tangelder, G.J. Direct vasoconstrictor effect of prostaglandin E2 on renal interlobular arteries: Role of the EP3 receptor. Am. J. Physiol. Renal Physiol. 2007, 292, F1094–F1101. [Google Scholar] [CrossRef]
- Gomez, I.; Foudi, N.; Longrois, D.; Norel, X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot. Essent Fat. Acids 2013, 89, 55–63. [Google Scholar] [CrossRef]
- Chan, P.C.; Hsiao, F.C.; Chang, H.M.; Wabitsch, M.; Hsieh, P.S. Importance of adipocyte cyclooxygenase-2 and prostaglandin E2-prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 2016, 30, 2282–2297. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alavez, M.; Klein, I.; Brownell, S.E.; Tabarean, I.V.; Davis, C.N.; Conti, B.; Bartfai, T. Night eating and obesity in the EP3R-deficient mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 3009–3014. [Google Scholar] [CrossRef]
- Jaworski, K.; Ahmadian, M.; Duncan, R.E.; Sarkadi-Nagy, E.; Varady, K.A.; Hellerstein, M.K.; Lee, H.Y.; Samuel, V.T.; Shulman, G.I.; Kim, K.H.; et al. AdPLA ablation increases lipolysis and prevents obesity induced by high-fat feeding or leptin deficiency. Nat. Med. 2009, 15, 159–168. [Google Scholar] [CrossRef]
- Sato, H.; Taketomi, Y.; Ushida, A.; Isogai, Y.; Kojima, T.; Hirabayashi, T.; Miki, Y.; Yamamoto, K.; Nishito, Y.; Kobayashi, T.; et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab. 2014, 20, 119–132. [Google Scholar] [CrossRef]
- Xu, H.; Fu, J.-F.; Miao, Y.-F.; Wang, C.-J.; Han, Q.-F.; Li, S.; Huang, S.-Z.; Du, S.-N.; Qiu, Y.-X.; Yang, J.-C.; et al. Prostaglandin E2 receptor EP3 regulates both adipogenesis and lipolysis in mouse white adipose tissue. J. Mol. Cell Biol. 2016, 8, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Inazumi, T.; Shirata, N.; Morimoto, K.; Takano, H.; Segi-Nishida, E.; Sugimoto, Y. Prostaglandin E(2)-EP4 signaling suppresses adipocyte differentiation in mouse embryonic fibroblasts via an autocrine mechanism. J. Lipid Res. 2011, 52, 1500–1508. [Google Scholar] [CrossRef]
- Yasui, M.; Tamura, Y.; Minami, M.; Higuchi, S.; Fujikawa, R.; Ikedo, T.; Nagata, M.; Arai, H.; Murayama, T.; Yokode, M. The Prostaglandin E2 Receptor EP4 Regulates Obesity-Related Inflammation and Insulin Sensitivity. PLoS ONE 2015, 10, e0136304. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a0006320. [Google Scholar] [CrossRef] [PubMed]
- Inazumi, T.; Yamada, K.; Shirata, N.; Sato, H.; Taketomi, Y.; Morita, K.; Hohjoh, H.; Tsuchiya, S.; Oniki, K.; Watanabe, T.; et al. Prostaglandin E2-EP4 Axis Promotes Lipolysis and Fibrosis in Adipose Tissue Leading to Ectopic Fat Deposition and Insulin Resistance. Cell Rep. 2020, 33, 108265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Cheng, C.K.; Zhang, C.L.; Huang, Y. Interplay Between Oxidative Stress, Cyclooxygenases, and Prostanoids in Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 784–799. [Google Scholar] [CrossRef]
- Cheng, C.-W.; Liu, M.-H.; Tang, H.-Y.; Cheng, M.-L.; Wang, C.-H. Factors associated with elevated plasma phenylalanine in patients with heart failure. Amino Acids 2021, 53, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Demers, L.M.; Harrison, T.S.; Halbert, D.R.; Santen, R.J. Effect of prolonged exercise on plasma prostaglandin levels. Prostaglandins Med. 1981, 6, 413–418. [Google Scholar] [CrossRef]
- Nowak, J.; Wennmalm, A. Effect of exercise on human arterial and regional venous plasma concentrations of prostaglandin E. Prostaglandins Med. 1978, 1, 489–497. [Google Scholar] [CrossRef]
- Wilson, J.R.; Kapoor, S.C. Contribution of prostaglandins to exercise-induced vasodilation in humans. Am. J. Physiol. 1993, 265, H171–H175. [Google Scholar] [CrossRef]
- Boushel, R.; Langberg, H.; Gemmer, C.; Olesen, J.; Crameri, R.; Scheede, C.; Sander, M.; Kjaer, M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J. Physiol. 2002, 543, 691–698. [Google Scholar] [CrossRef]
- Yang, T.; Du, Y. Distinct roles of central and peripheral prostaglandin E2 and EP subtypes in blood pressure regulation. Am. J. Hypertens. 2012, 25, 1042–1049. [Google Scholar] [CrossRef]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Huang, Q.; Fei, X.; Li, S.; Xu, C.; Tu, C.; Jiang, L.; Wo, M. Predicting significance of COX-2 expression of peripheral blood monocyte in patients with coronary artery disease. Ann. Transl. Med. 2019, 7, 483. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Hernandez, A.; Martin-Ventura, J.L.; Sanchez-Galan, E.; Vidal, C.; Ortego, M.; Blanco-Colio, L.M.; Ortega, L.; Tunon, J.; Egido, J. Overexpression of COX-2, Prostaglandin E synthase-1 and prostaglandin E receptors in blood mononuclear cells and plaque of patients with carotid atherosclerosis: Regulation by nuclear factor-kappaB. Atherosclerosis 2006, 187, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Garcia-Cardena, G.; Sukhova, G.K.; Comander, J.; Gimbrone, M.A., Jr.; Libby, P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 2002, 277, 44147–44154. [Google Scholar] [CrossRef]
- Schmid, T.; Brune, B. Prostanoids and Resolution of Inflammation-Beyond the Lipid-Mediator Class Switch. Front. Immunol. 2021, 12, 714042. [Google Scholar] [CrossRef]
- Watanabe, K. Recent reports about enzymes related to the synthesis of prostaglandin (PG) F(2) (PGF(2alpha) and 9alpha, 11beta-PGF(2)). J. Biochem. 2011, 150, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Volat, F.E.; Pointud, J.C.; Pastel, E.; Morio, B.; Sion, B.; Hamard, G.; Guichardant, M.; Colas, R.; Lefrancois-Martinez, A.M.; Martinez, A. Depressed levels of prostaglandin F2alpha in mice lacking Akr1b7 increase basal adiposity and predispose to diet-induced obesity. Diabetes 2012, 61, 2796–2806. [Google Scholar] [CrossRef]
- Milne, G.L.; Musiek, E.S.; Morrow, J.D. F2-isoprostanes as markers of oxidative stress in vivo: An overview. Biomarkers 2005, 10 (Suppl. 1), S10–S23. [Google Scholar] [CrossRef]
- Fujimori, K.; Ueno, T.; Nagata, N.; Kashiwagi, K.; Aritake, K.; Amano, F.; Urade, Y. Suppression of adipocyte differentiation by aldo-keto reductase 1B3 acting as prostaglandin F2alpha synthase. J. Biol. Chem. 2010, 285, 8880–8886. [Google Scholar] [CrossRef]
- Yu, Y.; Lucitt, M.B.; Stubbe, J.; Cheng, Y.; Friis, U.G.; Hansen, P.B.; Jensen, B.L.; Smyth, E.M.; FitzGerald, G.A. Prostaglandin F2alpha elevates blood pressure and promotes atherosclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7985–7990. [Google Scholar] [CrossRef]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Schulze, P.C.; Lee, R.T. Oxidative stress and atherosclerosis. Curr. Atheroscler. Rep. 2005, 7, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Dogne, J.M.; Hanson, J.; de Leval, X.; Pratico, D.; Pace-Asciak, C.R.; Drion, P.; Pirotte, B.; Ruan, K.H. From the design to the clinical application of thromboxane modulators. Curr. Pharm. Des. 2006, 12, 903–923. [Google Scholar] [CrossRef] [PubMed]
- Pratico, D.; Dogne, J.M. Vascular biology of eicosanoids and atherogenesis. Expert Rev. Cardiovasc. Ther. 2009, 7, 1079–1089. [Google Scholar] [CrossRef]
- Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 2015, 157, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Machida, K.; Inoue, H. A turn on and a turn off: BLT1 and BLT2 mechanisms in the lung. Expert Rev. Respir. Med. 2014, 8, 381–383. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mothe-Satney, I.; Filloux, C.; Amghar, H.; Pons, C.; Bourlier, V.; Galitzky, J.; Grimaldi, P.A.; Feral, C.C.; Bouloumie, A.; Van Obberghen, E.; et al. Adipocytes secrete leukotrienes: Contribution to obesity-associated inflammation and insulin resistance in mice. Diabetes 2012, 61, 2311–2319. [Google Scholar] [CrossRef]
- Li, P.; Oh, D.Y.; Bandyopadhyay, G.; Lagakos, W.S.; Talukdar, S.; Osborn, O.; Johnson, A.; Chung, H.; Maris, M.; Ofrecio, J.M.; et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat. Med. 2015, 21, 239–247. [Google Scholar] [CrossRef]
- Chakrabarti, S.K.; Wen, Y.; Dobrian, A.D.; Cole, B.K.; Ma, Q.; Pei, H.; Williams, M.D.; Bevard, M.H.; Vandenhoff, G.E.; Keller, S.R.; et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E175–E187. [Google Scholar] [CrossRef]
- Elias, I.; Ferre, T.; Vila, L.; Munoz, S.; Casellas, A.; Garcia, M.; Molas, M.; Agudo, J.; Roca, C.; Ruberte, J.; et al. ALOX5AP Overexpression in Adipose Tissue Leads to LXA4 Production and Protection Against Diet-Induced Obesity and Insulin Resistance. Diabetes 2016, 65, 2139–2150. [Google Scholar] [CrossRef]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef]
- Filgueiras, L.R.; Serezani, C.H.; Jancar, S. Leukotriene B4 as a Potential Therapeutic Target for the Treatment of Metabolic Disorders. Front. Immunol. 2015, 6, 515. [Google Scholar] [CrossRef] [PubMed]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nagai, Y.; Honda, H.; Okamoto, N.; Yanagibashi, T.; Ogasawara, M.; Yamamoto, S.; Imamura, R.; Takasaki, I.; Hara, H.; et al. Bidirectional crosstalk between neutrophils and adipocytes promotes adipose tissue inflammation. FASEB J. 2019, 33, 11821–11835. [Google Scholar] [CrossRef]
- Sasaki, F.; Yokomizo, T. The leukotriene receptors as therapeutic targets of inflammatory diseases. Int. Immunol. 2019, 31, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Barquissau, V.; Ghandour, R.A.; Ailhaud, G.; Klingenspor, M.; Langin, D.; Amri, E.Z.; Pisani, D.F. Control of adipogenesis by oxylipins, GPCRs and PPARs. Biochimie 2017, 136, 3–11. [Google Scholar] [CrossRef]
- Poeckel, D.; Funk, C.D. The 5-lipoxygenase/leukotriene pathway in preclinical models of cardiovascular disease. Cardiovasc Res. 2010, 86, 243–253. [Google Scholar] [CrossRef]
- Shoieb, S.M.; El-Sherbeni, A.A.; El-Kadi, A.O.S. Subterminal hydroxyeicosatetraenoic acids: Crucial lipid mediators in normal physiology and disease states. Chem. Biol. Interact. 2019, 299, 140–150. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef]
- Muller, D.N.; Schmidt, C.; Barbosa-Sicard, E.; Wellner, M.; Gross, V.; Hercule, H.; Markovic, M.; Honeck, H.; Luft, F.C.; Schunck, W.H. Mouse Cyp4a isoforms: Enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem. J. 2007, 403, 109–118. [Google Scholar] [CrossRef]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals Through G-Protein-Coupled Receptor GPR75 (Gq) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776–1788. [Google Scholar] [CrossRef]
- Gilani, A.; Pandey, V.; Garcia, V.; Agostinucci, K.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Falck, J.R.; Paudyal, M.P.; Capdevila, J.H.; et al. High-fat diet-induced obesity and insulin resistance in CYP4a14(−/−) mice is mediated by 20-HETE. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R934–R944. [Google Scholar] [CrossRef]
- Garbacz, W.G.; Lu, P.; Miller, T.M.; Poloyac, S.M.; Eyre, N.S.; Mayrhofer, G.; Xu, M.; Ren, S.; Xie, W. Hepatic Overexpression of CD36 Improves Glycogen Homeostasis and Attenuates High-Fat Diet-Induced Hepatic Steatosis and Insulin Resistance. Mol. Cell Biol. 2016, 36, 2715–2727. [Google Scholar] [CrossRef]
- Tsai, I.J.; Croft, K.D.; Mori, T.A.; Falck, J.R.; Beilin, L.J.; Puddey, I.B.; Barden, A.E. 20-HETE and F2-isoprostanes in the metabolic syndrome: The effect of weight reduction. Free Radic. Biol. Med. 2009, 46, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Yusuf, R.; Kook, S.; Attar, E.; Lee, D.; Park, B.; Cheng, T.; Scadden, D.T.; Lee, B.C. Purinergic P2Y(1)(4) receptor modulates stress-induced hematopoietic stem/progenitor cell senescence. J. Clin. Investig. 2014, 124, 3159–3171. [Google Scholar] [CrossRef] [PubMed]
- Elshenawy, O.H.; Shoieb, S.M.; Mohamed, A.; El-Kadi, A.O. Clinical Implications of 20-Hydroxyeicosatetraenoic Acid in the Kidney, Liver, Lung and Brain: An Emerging Therapeutic Target. Pharmaceutics 2017, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Gilani, A.; Agostinucci, K.; Hossain, S.; Pascale, J.V.; Garcia, V.; Adebesin, A.M.; Falck, J.R.; Schwartzman, M.L. 20-HETE interferes with insulin signaling and contributes to obesity-driven insulin resistance. Prostaglandins Other Lipid Mediat. 2021, 152, 106485. [Google Scholar] [CrossRef]
- Rocic, P.; Schwartzman, M.L. 20-HETE in the regulation of vascular and cardiac function. Pharmacol. Ther. 2018, 192, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Roman, R.J. GPR75 Identified as the First 20-HETE Receptor: A Chemokine Receptor Adopted by a New Family. Circ Res. 2017, 120, 1696–1698. [Google Scholar] [CrossRef]
- Roman, R.J.; Fan, F. 20-HETE: Hypertension and Beyond. Hypertension 2018, 72, 12–18. [Google Scholar] [CrossRef]
- Shekhar, S.; Varghese, K.; Li, M.; Fan, L.; Booz, G.W.; Roman, R.J.; Fan, F. Conflicting Roles of 20-HETE in Hypertension and Stroke. Int. J. Mol. Sci. 2019, 20, 4520. [Google Scholar] [CrossRef]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, M.; Kurosawa, A.; Tani, Y.; Tsuji, T.; Takeda, S. GPR31 and GPR151 are activated under acidic conditions. J. Biochem. 2019, 166, 317–320. [Google Scholar] [CrossRef]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef]
- Anderson, R.M.; Hernandez-Perez, M.; Tersey, S.; Samala, N.; Maier, B.; Mirmira, R.G. 12-HETE Signaling Through the G Protein-Coupled Receptor 31 (GPR31) Regulates Pancreas Development and Disease. FASEB J. 2017, 31, lb503. [Google Scholar]
- Hernandez-Perez, M.; Kulkarni, A.; Samala, N.; Sorrell, C.; El, K.; Haider, I.; Mukhtar Aleem, A.; Holman, T.R.; Rai, G.; Tersey, S.A.; et al. A 12-lipoxygenase-Gpr31 signaling axis is required for pancreatic organogenesis in the zebrafish. FASEB J. 2020, 34, 14850–14862. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.K.; Morris, M.A.; Grzesik, W.J.; Leone, K.A.; Nadler, J.L. Adipose tissue-specific deletion of 12/15-lipoxygenase protects mice from the consequences of a high-fat diet. Mediat. Inflamm. 2012, 2012, 851798. [Google Scholar] [CrossRef] [PubMed]
- Sears, D.D.; Miles, P.D.; Chapman, J.; Ofrecio, J.M.; Almazan, F.; Thapar, D.; Miller, Y.I. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS ONE 2009, 4, e7250. [Google Scholar] [CrossRef]
- Tersey, S.A.; Maier, B.; Nishiki, Y.; Maganti, A.V.; Nadler, J.L.; Mirmira, R.G. 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol. Cell Biol. 2014, 34, 3735–3745. [Google Scholar] [CrossRef]
- Pell, V.R.; Chouchani, E.T.; Frezza, C.; Murphy, M.P.; Krieg, T. Succinate metabolism: A new therapeutic target for myocardial reperfusion injury. Cardiovasc. Res. 2016, 111, 134–141. [Google Scholar] [CrossRef]
- Feinmark, S.J.; Nardi, M.A.; Harleton, E.; Hu, L.; Pan, R.; Karpatkin, S. The Orphan Receptor GPR31 Is the Platelet and HUVEC Receptor for 12(S)-HETE. Blood 2008, 112, 413. [Google Scholar] [CrossRef]
- Van Doren, L.; Nguyen, N.; Garzia, C.; Fletcher, E.K.; Stevenson, R.; Jaramillo, D.; Kuliopulos, A.; Covic, L. Lipid Receptor GPR31 (G-Protein-Coupled Receptor 31) Regulates Platelet Reactivity and Thrombosis Without Affecting Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e33–e45. [Google Scholar] [CrossRef]
- Calderon-Santiago, M.; Priego-Capote, F.; Galache-Osuna, J.G.; Luque de Castro, M.D. Method based on GC-MS to study the influence of tricarboxylic acid cycle metabolites on cardiovascular risk factors. J. Pharm. Biomed. Anal. 2013, 74, 178–185. [Google Scholar] [CrossRef]
- Atherton, H.J.; Bailey, N.J.; Zhang, W.; Taylor, J.; Major, H.; Shockcor, J.; Clarke, K.; Griffin, J.L. A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR-alpha null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiol. Genom. 2006, 27, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Harada, S.; Kurihara, A.; Fukai, K.; Kuwabara, K.; Sugiyama, D.; Takeuchi, A.; Okamura, T.; Akiyama, M.; Nishiwaki, Y.; et al. Profiling of plasma metabolites in postmenopausal women with metabolic syndrome. Menopause 2016, 23, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Varma, V.R.; Huang, C.W.; An, Y.; Oommen, A.; Tanaka, T.; Ferrucci, L.; Elango, P.; Takebayashi, T.; Harada, S.; et al. Blood Metabolite Signature of Metabolic Syndrome Implicates Alterations in Amino Acid Metabolism: Findings from the Baltimore Longitudinal Study of Aging (BLSA) and the Tsuruoka Metabolomics Cohort Study (TMCS). Int. J. Mol. Sci. 2020, 21, 1249. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Koo, A.N.M.; Prag, H.A.; Krieg, T. Mitochondrial redox and TCA cycle metabolite signaling in the heart. Free Radic. Biol. Med. 2021, 166, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Maurer, J.; Hoene, M.; Weigert, C. Signals from the Circle: Tricarboxylic Acid Cycle Intermediates as Myometabokines. Metabolites 2021, 11, 474. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef]
- De Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal. 2016, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Dressendorfer, R.H. Succinate accumulation in man during exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1976, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [PubMed]
- Aguiar, C.J.; Rocha-Franco, J.A.; Sousa, P.A.; Santos, A.K.; Ladeira, M.; Rocha-Resende, C.; Ladeira, L.O.; Resende, R.R.; Botoni, F.A.; Barrouin Melo, M.; et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun. Signal. 2014, 12, 78. [Google Scholar] [CrossRef]
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef]
- Krzak, G.; Willis, C.M.; Smith, J.A.; Pluchino, S.; Peruzzotti-Jametti, L. Succinate Receptor 1: An Emerging Regulator of Myeloid Cell Function in Inflammation. Trends Immunol. 2021, 42, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef]
- Winther, S.; Trauelsen, M.; Schwartz, T.W. Protective succinate-SUCNR1 metabolic stress signaling gone bad. Cell Metab. 2021, 33, 1276–1278. [Google Scholar] [CrossRef]
- McCreath, K.J.; Espada, S.; Galvez, B.G.; Benito, M.; de Molina, A.; Sepulveda, P.; Cervera, A.M. Targeted disruption of the SUCNR1 metabolic receptor leads to dichotomous effects on obesity. Diabetes 2015, 64, 1154–1167. [Google Scholar] [CrossRef]
- Keiran, N.; Ceperuelo-Mallafre, V.; Calvo, E.; Hernandez-Alvarez, M.I.; Ejarque, M.; Nunez-Roa, C.; Horrillo, D.; Maymo-Masip, E.; Rodriguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef]
- Zhen, Y.; Shu, W.; Hou, X.; Wang, Y. Innate Immune System Orchestrates Metabolic Homeostasis and Dysfunction in Visceral Adipose Tissue During Obesity. Front. Immunol. 2021, 12, 702835. [Google Scholar] [CrossRef]
- Prag, H.A.; Gruszczyk, A.V.; Huang, M.M.; Beach, T.E.; Young, T.; Tronci, L.; Nikitopoulou, E.; Mulvey, J.F.; Ascione, R.; Hadjihambi, A.; et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc. Res. 2021, 117, 1188–1201. [Google Scholar] [CrossRef]
- Osuna-Prieto, F.J.; Martinez-Tellez, B.; Ortiz-Alvarez, L.; Di, X.; Jurado-Fasoli, L.; Xu, H.; Ceperuelo-Mallafre, V.; Nunez-Roa, C.; Kohler, I.; Segura-Carretero, A.; et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc. Diabetol. 2021, 20, 151. [Google Scholar] [CrossRef]
- Yang, L.; Yu, D.; Mo, R.; Zhang, J.; Hua, H.; Hu, L.; Feng, Y.; Wang, S.; Zhang, W.Y.; Yin, N.; et al. The Succinate Receptor GPR91 Is Involved in Pressure Overload-Induced Ventricular Hypertrophy. PLoS ONE 2016, 11, e0147597. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; O’Neill, L.A. Succinate: A metabolic signal in inflammation. Trends Cell Biol. 2014, 24, 313–320. [Google Scholar] [CrossRef]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic Acid (TCA) Cycle Intermediates: Regulators of Immune Responses. Life 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. 2012, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Veledo, S.; Ceperuelo-Mallafre, V.; Vendrell, J. Rethinking succinate: An unexpected hormone-like metabolite in energy homeostasis. Trends Endocrinol. Metab. 2021, 32, 680–692. [Google Scholar] [CrossRef]
- Saeki, K.; Yokomizo, T. Identification, signaling, and functions of LTB4 receptors. Semin. Immunol. 2017, 33, 30–36. [Google Scholar] [CrossRef]
- Li, X.; Xie, L.; Qu, X.; Zhao, B.; Fu, W.; Wu, B.; Wu, J. GPR91, a critical signaling mechanism in modulating pathophysiologic processes in chronic illnesses. FASEB J. 2020, 34, 13091–13105. [Google Scholar] [CrossRef]
- Diehl, J.; Gries, B.; Pfeil, U.; Goldenberg, A.; Mermer, P.; Kummer, W.; Paddenberg, R. Expression and localization of GPR91 and GPR99 in murine organs. Cell Tissue Res. 2016, 364, 245–262. [Google Scholar] [CrossRef]
- Zdzisińska, B.; Żurek, A.; Kandefer-Szerszeń, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Et Ther. Exp. 2017, 65, 21–36. [Google Scholar] [CrossRef]
- Nagaoka, K.; Mulla, J.; Cao, K.; Cheng, Z.; Liu, D.; William, M.; Bay, A.; Hildebrand, G.; Lu, S.; Huang, C.-K. The metabolite, alpha-ketoglutarate inhibits non-alcoholic fatty liver disease progression by targeting lipid metabolism. Liver Res. 2020, 4, 94–100. [Google Scholar] [CrossRef]
- Yuan, Y.; Xu, P.; Jiang, Q.; Cai, X.; Wang, T.; Peng, W.; Sun, J.; Zhu, C.; Zhang, C.; Yue, D.; et al. Exercise-induced alpha-ketoglutaric acid stimulates muscle hypertrophy and fat loss through OXGR1-dependent adrenal activation. EMBO J. 2020, 39, e103304. [Google Scholar] [CrossRef]
- Sousa Fialho, M.D.L.; Abd Jamil, A.H.; Stannard, G.A.; Heather, L.C. Hypoxia-inducible factor 1 signalling, metabolism and its therapeutic potential in cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Visniauskas, B.; Cardenas, P.; Figueroa, S.M.; Vivanco, J.; Salinas-Parra, N.; Araos, P.; Nguyen, Q.M.; Kassan, M.; Amador, C.A.; et al. Alpha-Ketoglutarate Upregulates Collecting Duct (Pro)renin Receptor Expression, Tubular Angiotensin II Formation, and Na(+) Reabsorption During High Glucose Conditions. Front. Cardiovasc. Med. 2021, 8, 644797. [Google Scholar] [CrossRef] [PubMed]
- Omede, A.; Zi, M.; Prehar, S.; Maqsood, A.; Stafford, N.; Mamas, M.; Cartwright, E.; Oceandy, D. The oxoglutarate receptor 1 (OXGR1) modulates pressure overload-induced cardiac hypertrophy in mice. Biochem. Biophys. Res. Commun. 2016, 479, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Omede, A. Role of Alpha-Ketoglutarate Receptor G-Protein Coupled Receptor 99 (GPR99) in Cardiac Hypertrophy. 2015:171.
- An, D.; Zeng, Q.; Zhang, P.; Ma, Z.; Zhang, H.; Liu, Z.; Li, J.; Ren, H.; Xu, D. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox. Biol. 2021, 46, 102088. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, J.; Wu, Z. The Regulatory Role of α-Ketoglutarate Metabolism in Macrophages. Mediat. Inflamm. 2021, 2021, 5577577. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Maekawa, A.; Austen, K.F. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J. Biol. Chem. 2013, 288, 10967–10972. [Google Scholar] [CrossRef]
- Rose, A.J. Amino Acid Nutrition and Metabolism in Health and Disease. Nutrients 2019, 11, 2623. [Google Scholar] [CrossRef]
- Zakaria, N.F.; Hamid, M.; Khayat, M.E. Amino Acid-Induced Impairment of Insulin Signaling and Involvement of G-Protein Coupling Receptor. Nutrients 2021, 13, 2229. [Google Scholar] [CrossRef]
- Zaric, B.L.; Radovanovic, J.N.; Gluvic, Z.; Stewart, A.J.; Essack, M.; Motwalli, O.; Gojobori, T.; Isenovic, E.R. Atherosclerosis Linked to Aberrant Amino Acid Metabolism and Immunosuppressive Amino Acid Catabolizing Enzymes. Front. Immunol. 2020, 11, 551758. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, O.; Shang, J.; Munk, A.; Ekberg, J.P.; Petersen, N.; Engelstoft, M.S.; Egerod, K.L.; Hjorth, S.A.; Wu, M.; Feng, Y.; et al. The aromatic amino acid sensor GPR142 controls metabolism through balanced regulation of pancreatic and gut hormones. Mol. Metab. 2019, 19, 49–64. [Google Scholar] [CrossRef]
- Conigrave, A.D.; Ward, D.T. Calcium-sensing receptor (CaSR): Pharmacological properties and signaling pathways. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 315–331. [Google Scholar] [CrossRef]
- Ueda, Y.; Iwakura, H.; Bando, M.; Doi, A.; Ariyasu, H.; Inaba, H.; Morita, S.; Akamizu, T. Differential role of GPR142 in tryptophan-mediated enhancement of insulin secretion in obese and lean mice. PLoS ONE 2018, 13, e0198762. [Google Scholar] [CrossRef] [PubMed]
- Hajishafiee, M.; Elovaris, R.A.; Jones, K.L.; Heilbronn, L.K.; Horowitz, M.; Poppitt, S.D.; Feinle-Bisset, C. Effects of intragastric administration of L-tryptophan on the glycaemic response to a nutrient drink in men with type 2 diabetes-impacts on gastric emptying, glucoregulatory hormones and glucose absorption. Nutr. Diabetes 2021, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Inubushi, T.; Kamemura, N.; Oda, M.; Sakurai, J.; Nakaya, Y.; Harada, N.; Suenaga, M.; Matsunaga, Y.; Ishidoh, K.; Katunuma, N. L-tryptophan suppresses rise in blood glucose and preserves insulin secretion in type-2 diabetes mellitus rats. J. Nutr. Sci. Vitaminol. 2012, 58, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Dolivo, D.M.; Larson, S.A.; Dominko, T. Tryptophan metabolites kynurenine and serotonin regulate fibroblast activation and fibrosis. Cell Mol. Life Sci. 2018, 75, 3663–3681. [Google Scholar] [CrossRef]
- Liu, L.Z.; Ma, T.; Zhou, J.; Long Hu, Z.; Jun Zhang, X.; Zhen Zhang, H.; Zeng, M.; Liu, J.; Li, L.; Jiang, Y.; et al. Discovery of LY3325656: A GPR142 agonist suitable for clinical testing in human. Bioorg. Med. Chem. Lett. 2020, 30, 126857. [Google Scholar] [CrossRef]
- Laplante, M.A.; Monassier, L.; Freund, M.; Bousquet, P.; Gachet, C. The purinergic P2Y1 receptor supports leptin secretion in adipose tissue. Endocrinology 2010, 151, 2060–2070. [Google Scholar] [CrossRef]
- Cosi, C.; Mannaioni, G.; Cozzi, A.; Carla, V.; Sili, M.; Cavone, L.; Maratea, D.; Moroni, F. G-protein coupled receptor 35 (GPR35) activation and inflammatory pain: Studies on the antinociceptive effects of kynurenic acid and zaprinast. Neuropharmacology 2011, 60, 1227–1231. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Yang, S.; Lu, Z.; Li, G.; Wu, S.; Wu, D.R.; Liu, J.; Zhou, B.; Wang, H.D.; et al. The Beneficial Effects of Edible Kynurenic Acid from Marine Horseshoe Crab (Tachypleus tridentatus) on Obesity, Hyperlipidemia, and Gut Microbiota in High-Fat Diet-Fed Mice. Oxid. Med. Cell. Longev. 2021, 2021, 8874503. [Google Scholar] [CrossRef]
- Quon, T.; Lin, L.C.; Ganguly, A.; Tobin, A.B.; Milligan, G. Therapeutic Opportunities and Challenges in Targeting the Orphan G Protein-Coupled Receptor GPR35. ACS Pharmacol. Transl. Sci. 2020, 3, 801–812. [Google Scholar] [CrossRef]
- Divorty, N.; Mackenzie, A.E.; Nicklin, S.A.; Milligan, G. G protein-coupled receptor 35: An emerging target in inflammatory and cardiovascular disease. Front. Pharmacol. 2015, 6, 41. [Google Scholar] [CrossRef]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed]
- Vander Molen, J.; Frisse, L.M.; Fullerton, S.M.; Qian, Y.; Del Bosque-Plata, L.; Hudson, R.R.; Di Rienzo, A. Population genetics of CAPN10 and GPR35: Implications for the evolution of type 2 diabetes variants. Am. J. Hum. Genet. 2005, 76, 548–560. [Google Scholar] [CrossRef]
- Horikawa, Y.; Oda, N.; Cox, N.J.; Li, X.; Orho-Melander, M.; Hara, M.; Hinokio, Y.; Lindner, T.H.; Mashima, H.; Schwarz, P.E.; et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat. Genet. 2000, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, A.E.; Lappin, J.E.; Taylor, D.L.; Nicklin, S.A.; Milligan, G. GPR35 as a Novel Therapeutic Target. Front. Endocrinol. 2011, 2, 68. [Google Scholar] [CrossRef]
- Agudelo, L.Z.; Ferreira, D.M.S.; Cervenka, I.; Bryzgalova, G.; Dadvar, S.; Jannig, P.R.; Pettersson-Klein, A.T.; Lakshmikanth, T.; Sustarsic, E.G.; Porsmyr-Palmertz, M.; et al. Kynurenic Acid and Gpr35 Regulate Adipose Tissue Energy Homeostasis and Inflammation. Cell Metab. 2018, 27, 378–392.e5. [Google Scholar] [CrossRef] [PubMed]
- McQueen, A.E.; Koliwad, S.K.; Wang, J.C. Fighting obesity by targeting factors regulating beige adipocytes. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Shore, D.M.; Reggio, P.H. The therapeutic potential of orphan GPCRs, GPR35 and GPR55. Front. Pharmacol. 2015, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Nguyen, H.; Meda Venkata, S.P.; Koh, J.Y.; Kowluru, A.; Li, L.; Rossi, N.F.; Chen, W.; Wang, J.M. Novel Role of GPR35 (G-Protein-Coupled Receptor 35) in the Regulation of Endothelial Cell Function and Blood Pressure. Hypertension 2021, 78, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Divorty, N.; Milligan, G.; Graham, D.; Nicklin, S.A. The Orphan Receptor GPR35 Contributes to Angiotensin II-Induced Hypertension and Cardiac Dysfunction in Mice. Am. J. Hypertens. 2018, 31, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; He, L.; Li, Y.; Li, X.; Qiu, C.; Pei, H.; Yang, D. Inhibition of GPR35 Preserves Mitochondrial Function After Myocardial Infarction by Targeting Calpain 1/2. J. Cardiovasc. Pharmacol. 2020, 75, 556–563. [Google Scholar] [CrossRef]
- Ronkainen, V.P.; Tuomainen, T.; Huusko, J.; Laidinen, S.; Malinen, M.; Palvimo, J.J.; Yla-Herttuala, S.; Vuolteenaho, O.; Tavi, P. Hypoxia-inducible factor 1-induced G protein-coupled receptor 35 expression is an early marker of progressive cardiac remodelling. Cardiovasc Res. 2014, 101, 69–77. [Google Scholar] [CrossRef]
- Min, K.D.; Asakura, M.; Liao, Y.; Nakamaru, K.; Okazaki, H.; Takahashi, T.; Fujimoto, K.; Ito, S.; Takahashi, A.; Asanuma, H.; et al. Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem. Biophys. Res. Commun. 2010, 393, 55–60. [Google Scholar] [CrossRef]
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Wirthgen, E.; Hoeflich, A.; Rebl, A.; Gunther, J. Kynurenic Acid: The Janus-Faced Role of an Immunomodulatory Tryptophan Metabolite and Its Link to Pathological Conditions. Front. Immunol. 2017, 8, 1957. [Google Scholar] [CrossRef]
- Hannan, F.M.; Kallay, E.; Chang, W.; Brandi, M.L.; Thakker, R.V. The calcium-sensing receptor in physiology and in calcitropic and noncalcitropic diseases. Nat. Rev. Endocrinol. 2018, 15, 33–51. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; van der Vorst, E.P.C. Calcium-Sensing Receptor (CaSR), Its Impact on Inflammation and the Consequences on Cardiovascular Health. Int. J. Mol. Sci. 2021, 22, 2478. [Google Scholar] [CrossRef] [PubMed]
- Alamshah, A.; Spreckley, E.; Norton, M.; Kinsey-Jones, J.S.; Amin, A.; Ramgulam, A.; Cao, Y.; Johnson, R.; Saleh, K.; Akalestou, E.; et al. l-phenylalanine modulates gut hormone release and glucose tolerance, and suppresses food intake through the calcium-sensing receptor in rodents. Int. J. Obes. 2017, 41, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Tonack, S.; Tang, C.; Offermanns, S. Endogenous metabolites as ligands for G protein-coupled receptors modulating risk factors for metabolic and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H501–H513. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Swierczynski, J.; Sledzinski, T.; Slominska, E.; Smolenski, R.; Sledzinski, Z. Serum phenylalanine concentration as a marker of liver function in obese patients before and after bariatric surgery. Obes. Surg. 2009, 19, 883–889. [Google Scholar] [CrossRef]
- Burrage, L.C.; McConnell, J.; Haesler, R.; O’Riordan, M.A.; Sutton, V.R.; Kerr, D.S.; McCandless, S.E. High prevalence of overweight and obesity in females with phenylketonuria. Mol. Genet. Metab. 2012, 107, 43–48. [Google Scholar] [CrossRef]
- Wang, C.H.; Cheng, M.L.; Liu, M.H. Simplified plasma essential amino acid-based profiling provides metabolic information and prognostic value additive to traditional risk factors in heart failure. Amino Acids 2018, 50, 1739–1748. [Google Scholar] [CrossRef]
- Liou, A.P.; Sei, Y.; Zhao, X.; Feng, J.; Lu, X.; Thomas, C.; Pechhold, S.; Raybould, H.E.; Wank, S.A. The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to L-phenylalanine in acutely isolated intestinal I cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G538–G546. [Google Scholar] [CrossRef]
- Marz, W.; Seelhorst, U.; Wellnitz, B.; Tiran, B.; Obermayer-Pietsch, B.; Renner, W.; Boehm, B.O.; Ritz, E.; Hoffmann, M.M. Alanine to serine polymorphism at position 986 of the calcium-sensing receptor associated with coronary heart disease, myocardial infarction, all-cause, and cardiovascular mortality. J. Clin. Endocrinol. Metab. 2007, 92, 2363–2369. [Google Scholar] [CrossRef]
- Chen, W.S.; Wang, C.H.; Cheng, C.W.; Liu, M.H.; Chu, C.M.; Wu, H.P.; Huang, P.C.; Lin, Y.T.; Ko, T.; Chen, W.H.; et al. Elevated plasma phenylalanine predicts mortality in critical patients with heart failure. ESC Heart Fail. 2020, 7, 2884–2893. [Google Scholar] [CrossRef]
- Hiraiwa, H.; Okumura, T.; Kondo, T.; Kato, T.; Kazama, S.; Kimura, Y.; Ishihara, T.; Iwata, E.; Shimojo, M.; Kondo, S.; et al. Prognostic value of leucine/phenylalanine ratio as an amino acid profile of heart failure. Heart Vessels 2021, 36, 965–977. [Google Scholar] [CrossRef]
- Tan, R.; Li, J.; Liu, F.; Liao, P.; Ruiz, M.; Dupuis, J.; Zhu, L.; Hu, Q. Phenylalanine induces pulmonary hypertension through calcium-sensing receptor activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L1010–L1020. [Google Scholar] [CrossRef]
- Yamamura, A.; Guo, Q.; Yamamura, H.; Zimnicka, A.M.; Pohl, N.M.; Smith, K.A.; Fernandez, R.A.; Zeifman, A.; Makino, A.; Dong, H.; et al. Enhanced Ca(2+)-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ. Res. 2012, 111, 469–481. [Google Scholar] [CrossRef]
- Li, T.; Sun, M.; Yin, X.; Wu, C.; Wu, Q.; Feng, S.; Li, H.; Luan, Y.; Wen, J.; Yan, L.; et al. Expression of the calcium sensing receptor in human peripheral blood T lymphocyte and its contribution to cytokine secretion through MAPKs or NF-kappaB pathways. Mol. Immunol. 2013, 53, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schoneberg, T.; Schaefer, M.; Krugel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef]
- Lopez-Fernandez, I.; Schepelmann, M.; Brennan, S.C.; Yarova, P.L.; Riccardi, D. The calcium-sensing receptor: One of a kind. Exp. Physiol. 2015, 100, 1392–1399. [Google Scholar] [CrossRef] [PubMed]
- Vedel, L.; Nohr, A.C.; Gloriam, D.E.; Brauner-Osborne, H. Pharmacology and function of the orphan GPR139 G protein-coupled receptor. Basic Clin. Pharmacol. Toxicol. 2020, 126 (Suppl. 6), 35–46. [Google Scholar] [CrossRef] [PubMed]
- Nohr, A.C.; Jespers, W.; Shehata, M.A.; Floryan, L.; Isberg, V.; Andersen, K.B.; Aqvist, J.; Gutierrez-de-Teran, H.; Brauner-Osborne, H.; Gloriam, D.E. The GPR139 reference agonists 1a and 7c, and tryptophan and phenylalanine share a common binding site. Sci. Rep. 2017, 7, 1128. [Google Scholar] [CrossRef]
- Austin, J.; Marks, D. Hormonal regulators of appetite. Int. J. Pediatr. Endocrinol. 2009, 2009, 141753. [Google Scholar] [CrossRef] [PubMed]
- Nohr, A.C.; Shehata, M.A.; Hauser, A.S.; Isberg, V.; Mokrosinski, J.; Andersen, K.B.; Farooqi, I.S.; Pedersen, D.S.; Gloriam, D.E.; Brauner-Osborne, H. The orphan G protein-coupled receptor GPR139 is activated by the peptides: Adrenocorticotropic hormone (ACTH), alpha-, and beta-melanocyte stimulating hormone (alpha-MSH, and beta-MSH), and the conserved core motif HFRW. Neurochem. Int. 2017, 102, 105–113. [Google Scholar] [CrossRef]
- Nepomuceno, D.; Kuei, C.; Dvorak, C.; Lovenberg, T.; Liu, C.; Bonaventure, P. Re-evaluation of Adrenocorticotropic Hormone and Melanocyte Stimulating Hormone Activation of GPR139 in Vitro. Front. Pharmacol. 2018, 9, 157. [Google Scholar] [CrossRef]
- Panas, M.W.; Xie, Z.; Panas, H.N.; Hoener, M.C.; Vallender, E.J.; Miller, G.M. Trace amine associated receptor 1 signaling in activated lymphocytes. J. Neuroimmune Pharmacol. 2012, 7, 866–876. [Google Scholar] [CrossRef]
- Eyun, S.I.; Moriyama, H.; Hoffmann, F.G.; Moriyama, E.N. Molecular Evolution and Functional Divergence of Trace Amine-Associated Receptors. PLoS ONE 2016, 11, e0151023. [Google Scholar] [CrossRef]
- Pei, Y.; Asif-Malik, A.; Canales, J.J. Trace Amines and the Trace Amine-Associated Receptor 1: Pharmacology, Neurochemistry, and Clinical Implications. Front. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef]
- Gammone, M.A.; Vicentini, A.; Riccioni, G.; De Girolamo, M.; D’Aulerio, A.; D’Orazio, N. Food-Related Atrial Fibrillation? The Potential Role of Biogenic Amines in “Nutri-Arrhythmias” Genesis. Reports 2019, 2, 1. [Google Scholar] [CrossRef]
- Berry, M.D.; Gainetdinov, R.R.; Hoener, M.C.; Shahid, M. Pharmacology of human trace amine-associated receptors: Therapeutic opportunities and challenges. Pharmacol. Ther. 2017, 180, 161–180. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Kunnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 2016, 5, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Morin, N.; Visentin, V.; Calise, D.; Marti, L.; Zorzano, A.; Testar, X.; Valet, P.; Fischer, Y.; Carpene, C. Tyramine stimulates glucose uptake in insulin-sensitive tissues in vitro and in vivo via its oxidation by amine oxidases. J. Pharmacol. Exp. Ther. 2002, 303, 1238–1247. [Google Scholar] [CrossRef]
- Moore, C.F.; Sabino, V.; Cottone, P. Trace Amine Associated Receptor 1 (TAAR1) Modulation of Food Reward. Front. Pharmacol. 2018, 9, 129. [Google Scholar] [CrossRef]
- Carpene, C.; Abello, V.; Iffiu-Soltesz, Z.; Mercier, N.; Feve, B.; Valet, P. Limitation of adipose tissue enlargement in rats chronically treated with semicarbazide-sensitive amine oxidase and monoamine oxidase inhibitors. Pharmacol. Res. 2008, 57, 426–434. [Google Scholar] [CrossRef]
- Patel, A.; Thompson, A.; Abdelmalek, L.; Adams-Huet, B.; Jialal, I. The relationship between tyramine levels and inflammation in metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 2019, 40. [Google Scholar] [CrossRef] [PubMed]
- Rutigliano, G.; Bandini, L.; Sestito, S.; Chiellini, G. 3-Iodothyronamine and Derivatives: New Allies Against Metabolic Syndrome? Int. J. Mol. Sci. 2020, 21, 2005. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Ford, W.R.; Broadley, K.J.; Herbert, A.A. Vasoconstrictor and vasodilator responses to tryptamine of rat-isolated perfused mesentery: Comparison with tyramine and beta-phenylethylamine. Br. J. Pharmacol. 2012, 165, 2191–2202. [Google Scholar] [CrossRef]
- Colombo, F.; Porro, T.; del Rosso, G.; Bertalero, P.; Orlandi, L.; Libretti, A. Cardiovascular responses to physical exercise and tyramine infusion in hypertensive and normotensive subjects. J. Hum. Hypertens. 1989, 3, 245–249. [Google Scholar] [PubMed]
- Fehler, M. Investigation of Trace Amine Receptors in the Cardiovascular Systems; Cardiff University: Cardiff, UK, 2008. [Google Scholar]
- Nelson, D.A.; Tolbert, M.D.; Singh, S.J.; Bost, K.L. Expression of neuronal trace amine-associated receptor (Taar) mRNAs in leukocytes. J. Neuroimmunol. 2007, 192, 21–30. [Google Scholar] [CrossRef]
- Wasik, A.M.; Millan, M.J.; Scanlan, T.; Barnes, N.M.; Gordon, J. Evidence for functional trace amine associated receptor-1 in normal and malignant B cells. Leuk. Res. 2012, 36, 245–249. [Google Scholar] [CrossRef]
- Babusyte, A.; Kotthoff, M.; Fiedler, J.; Krautwurst, D. Biogenic amines activate blood leukocytes via trace amine-associated receptors TAAR1 and TAAR2. J. Leukoc. Biol. 2013, 93, 387–394. [Google Scholar] [CrossRef]
- Bugda Gwilt, K.; Olliffe, N.; Hoffing, R.A.; Miller, G.M. Trace amine associated receptor 1 (TAAR1) expression and modulation of inflammatory cytokine production in mouse bone marrow-derived macrophages: A novel mechanism for inflammation in ulcerative colitis. Immunopharmacol. Immunotoxicol. 2019, 41, 577–585. [Google Scholar] [CrossRef]
- Christian, S.L.; Berry, M.D. Trace Amine-Associated Receptors as Novel Therapeutic Targets for Immunomodulatory Disorders. Front. Pharmacol. 2018, 9, 680. [Google Scholar] [CrossRef]
- Andersen, G.; Krautwurst, D. Chapter 7—Trace Amine-Associated Receptors in the Cellular Immune System. In Trace Amines and Neurological Disorders; Farooqui, T., Farooqui, A.A., Eds.; Academic Press: San Diego, CA, USA, 2016; pp. 97–105. [Google Scholar]
- Ferragud, A.; Howell, A.D.; Moore, C.F.; Ta, T.L.; Hoener, M.C.; Sabino, V.; Cottone, P. The Trace Amine-Associated Receptor 1 Agonist RO5256390 Blocks Compulsive, Binge-like Eating in Rats. Neuropsychopharmacology 2017, 42, 1458–1470. [Google Scholar] [CrossRef]
- Fitz, J.G. Regulation of cellular ATP release. Trans. Am. Clin. Climatol. Assoc. 2007, 118, 199–208. [Google Scholar] [PubMed]
- Vultaggio-Poma, V.; Sarti, A.C.; Di Virgilio, F. Extracellular ATP: A Feasible Target for Cancer Therapy. Cells 2020, 9, 2496. [Google Scholar] [CrossRef]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef]
- Woodward, H.N.; Anwar, A.; Riddle, S.; Taraseviciene-Stewart, L.; Fragoso, M.; Stenmark, K.R.; Gerasimovskaya, E.V. PI3K, Rho, and ROCK play a key role in hypoxia-induced ATP release and ATP-stimulated angiogenic responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L954–L964. [Google Scholar] [CrossRef]
- Mikolajewicz, N.; Mohammed, A.; Morris, M.; Komarova, S.V. Mechanically stimulated ATP release from mammalian cells: Systematic review and meta-analysis. J. Cell Sci. 2018, 131, jcs223354. [Google Scholar] [CrossRef] [PubMed]
- Losenkova, K.; Zuccarini, M.; Helenius, M.; Jacquemet, G.; Gerasimovskaya, E.; Tallgren, C.; Jalkanen, S.; Yegutkin, G.G. Endothelial cells cope with hypoxia-induced depletion of ATP via activation of cellular purine turnover and phosphotransfer networks. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Reichert, K.P.; Castro, M.F.V.; Assmann, C.E.; Bottari, N.B.; Miron, V.V.; Cardoso, A.; Stefanello, N.; Morsch, V.M.M.; Schetinger, M.R.C. Diabetes and hypertension: Pivotal involvement of purinergic signaling. Biomed. Pharmacother. 2021, 137, 111273. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 2398212818817494. [Google Scholar] [CrossRef]
- Buj, R.; Aird, K.M. Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Front. Endocrinol. 2018, 9, 177. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Tabrizchi, R.; Bedi, S. Pharmacology of adenosine receptors in the vasculature. Pharmacol. Ther. 2001, 91, 133–147. [Google Scholar] [CrossRef]
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular adenosine receptors: Expression, actions and interactions. Pharmacol. Ther. 2013, 140, 92–111. [Google Scholar] [CrossRef]
- Tozzi, M.; Novak, I. Purinergic Receptors in Adipose Tissue As Potential Targets in Metabolic Disorders. Front. Pharmacol. 2017, 8, 878. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, B.; Abraham, A.A.; Ham, J.; Evans, B.A. Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int. J. Obes. 2012, 36, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Swenson, S.; Johnson, J.L.; Partin, M. Characterization of human adipocyte adenosine receptors. Biochem. Biophys. Res. Commun. 1989, 163, 137–142. [Google Scholar] [CrossRef]
- Gnad, T.; Scheibler, S.; von Kugelgen, I.; Scheele, C.; Kilic, A.; Glode, A.; Hoffmann, L.S.; Reverte-Salisa, L.; Horn, P.; Mutlu, S.; et al. Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature 2014, 516, 395–399. [Google Scholar] [CrossRef]
- Faulhaber-Walter, R.; Jou, W.; Mizel, D.; Li, L.; Zhang, J.; Kim, S.M.; Huang, Y.; Chen, M.; Briggs, J.P.; Gavrilova, O.; et al. Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes 2011, 60, 2578–2587. [Google Scholar] [CrossRef]
- Dong, Q.; Ginsberg, H.N.; Erlanger, B.F. Overexpression of the A1 adenosine receptor in adipose tissue protects mice from obesity-related insulin resistance. Diabetes Obes. Metab. 2001, 3, 360–366. [Google Scholar] [CrossRef]
- Gnad, T.; Navarro, G.; Lahesmaa, M.; Reverte-Salisa, L.; Copperi, F.; Cordomi, A.; Naumann, J.; Hochhauser, A.; Haufs-Brusberg, S.; Wenzel, D.; et al. Adenosine/A2B Receptor Signaling Ameliorates the Effects of Aging and Counteracts Obesity. Cell Metab. 2020, 32, 56–70.e7. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Csóka, B.; Pacher, P.; Haskó, G. Adenosine signalling in diabetes mellitus—pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 2015, 11, 228–241. [Google Scholar] [CrossRef]
- Sacramento, J.F.; Martins, F.O.; Rodrigues, T.; Matafome, P.; Ribeiro, M.J.; Olea, E.; Conde, S.V. A 2 Adenosine Receptors Mediate Whole-Body Insulin Sensitivity in a Prediabetes Animal Model: Primary Effects on Skeletal Muscle. Front. Endocrinol. 2020, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Ravid, K. Adenosine, adenosine receptors and their role in glucose homeostasis and lipid metabolism. J. Cell Physiol. 2013, 228, 1703–1712. [Google Scholar] [CrossRef]
- Andersson, O. Role of adenosine signalling and metabolism in beta-cell regeneration. Exp. Cell Res. 2014, 321, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Figler, R.A.; Wang, G.; Srinivasan, S.; Jung, D.Y.; Zhang, Z.; Pankow, J.S.; Ravid, K.; Fredholm, B.; Hedrick, C.C.; Rich, S.S.; et al. Links between insulin resistance, adenosine A2B receptors, and inflammatory markers in mice and humans. Diabetes 2011, 60, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Zhu, C.; Bucher, C.; Blazar, B.R.; Zhang, C.; Chen, J.F.; Linden, J.; Wu, C.; Huo, Y. Inactivation of the adenosine A2A receptor protects apolipoprotein E-deficient mice from atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, A.; Patterson, S.; Ravid, K. The Many Faces of the A2b Adenosine Receptor in Cardiovascular and Metabolic Diseases. J. Cell Physiol. 2015, 230, 2891–2897. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, H.; Koupenova, M.; Carroll, S.H.; Eliades, A.; Freedman, J.E.; Toselli, P.; Ravid, K. A new role for the A2b adenosine receptor in regulating platelet function. J. Thromb. Haemost. 2010, 8, 817–827. [Google Scholar] [CrossRef]
- Darlington, D.N.; Wu, X.; Chang, K.L.; Bynum, J.; Cap, A.P. Regulation of Platelet Function by Adenosine Receptors. Blood 2019, 134, 2348. [Google Scholar] [CrossRef]
- Chen, H.; Koupenova, M.; Yang, D.; Sume, S.S.; Trackman, P.C.; Ravid, K. Regulation of MMP-9 expression by the A2b adenosine receptor and its dependency on TNF-alpha signaling. Exp. Hematol. 2011, 39, 525–530. [Google Scholar] [CrossRef]
- Brown, R.D.; Thoren, P.; Steege, A.; Mrowka, R.; Sallstrom, J.; Skott, O.; Fredholm, B.B.; Persson, A.E. Influence of the adenosine A1 receptor on blood pressure regulation and renin release. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1324–R1329. [Google Scholar] [CrossRef]
- Shen, F.M.; Su, D.F. The effect of adenosine on blood pressure variability in sinoaortic denervated rats is mediated by adenosine A2a-Receptor. J. Cardiovasc. Pharmacol. 2000, 36, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.G.; Navar, L.G. Afferent arteriolar vasodilator effect of adenosine predominantly involves adenosine A2B receptor activation. Am. J. Physiol. Renal. Physiol. 2010, 299, F310–F315. [Google Scholar] [CrossRef]
- Xu, Z.; Park, S.S.; Mueller, R.A.; Bagnell, R.C.; Patterson, C.; Boysen, P.G. Adenosine produces nitric oxide and prevents mitochondrial oxidant damage in rat cardiomyocytes. Cardiovasc. Res. 2005, 65, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.N.; Chen, J.F.; Fredholm, B.B. Physiological roles of A1 and A2A adenosine receptors in regulating heart rate, body temperature, and locomotion as revealed using knockout mice and caffeine. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1141–H1149. [Google Scholar] [CrossRef] [PubMed]
- Ledent, C.; Vaugeois, J.M.; Schiffmann, S.N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.J.; Costentin, J.; Heath, J.K.; Vassart, G.; Parmentier, M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature 1997, 388, 674–678. [Google Scholar] [CrossRef]
- Morgan, J.M.; McCormack, D.G.; Griffiths, M.J.; Morgan, C.J.; Barnes, P.J.; Evans, T.W. Adenosine as a vasodilator in primary pulmonary hypertension. Circulation 1991, 84, 1145–1149. [Google Scholar] [CrossRef]
- Alencar, A.K.; Pereira, S.L.; Montagnoli, T.L.; Maia, R.C.; Kummerle, A.E.; Landgraf, S.S.; Caruso-Neves, C.; Ferraz, E.B.; Tesch, R.; Nascimento, J.H.; et al. Beneficial effects of a novel agonist of the adenosine A2A receptor on monocrotaline-induced pulmonary hypertension in rats. Br. J. Pharmacol. 2013, 169, 953–962. [Google Scholar] [CrossRef]
- Power Coombs, M.R.; Belderbos, M.E.; Gallington, L.C.; Bont, L.; Levy, O. Adenosine modulates Toll-like receptor function: Basic mechanisms and translational opportunities. Expert Rev. Anti. Infect. Ther. 2011, 9, 261–269. [Google Scholar] [CrossRef]
- Chhabra, P.; Linden, J.; Lobo, P.; Okusa, M.D.; Brayman, K.L. The immunosuppressive role of adenosine A2A receptors in ischemia reperfusion injury and islet transplantation. Curr. Diabetes Rev. 2012, 8, 419–433. [Google Scholar] [CrossRef]
- Csoka, B.; Selmeczy, Z.; Koscso, B.; Nemeth, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J.; et al. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef]
- Martin, L.; Pingle, S.C.; Hallam, D.M.; Rybak, L.P.; Ramkumar, V. Activation of the adenosine A3 receptor in RAW 264.7 cells inhibits lipopolysaccharide-stimulated tumor necrosis factor-alpha release by reducing calcium-dependent activation of nuclear factor-kappaB and extracellular signal-regulated kinase 1/2. J. Pharmacol. Exp. Ther. 2006, 316, 71–78. [Google Scholar] [CrossRef]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y. Prostaglandin E2 in remote control of myocardial remodeling. Circulation 2012, 125, 2818–2820. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Eltzschig, H.K.; Fredholm, B.B. Adenosine receptors as drug targets--what are the challenges? Nat. Rev. Drug Discov. 2013, 12, 265–286. [Google Scholar] [CrossRef]
- Strassheim, D.; Verin, A.; Batori, R.; Nijmeh, H.; Burns, N.; Kovacs-Kasa, A.; Umapathy, N.S.; Kotamarthi, J.; Gokhale, Y.S.; Karoor, V.; et al. P2Y Purinergic Receptors, Endothelial Dysfunction, and Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 6855. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Xie, N.; Illes, P.; Di Virgilio, F.; Ulrich, H.; Semyanov, A.; Verkhratsky, A.; Sperlagh, B.; Yu, S.G.; Huang, C.; et al. From purines to purinergic signalling: Molecular functions and human diseases. Signal Transduct. Target. Ther. 2021, 6, 162. [Google Scholar] [CrossRef]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol. Lett. 2019, 205, 16–24. [Google Scholar] [CrossRef]
- Zippel, N.; Limbach, C.A.; Ratajski, N.; Urban, C.; Luparello, C.; Pansky, A.; Kassack, M.U.; Tobiasch, E. Purinergic receptors influence the differentiation of human mesenchymal stem cells. Stem Cells Dev. 2012, 21, 884–900. [Google Scholar] [CrossRef]
- Biver, G.; Wang, N.; Gartland, A.; Orriss, I.; Arnett, T.R.; Boeynaems, J.M.; Robaye, B. Role of the P2Y13 receptor in the differentiation of bone marrow stromal cells into osteoblasts and adipocytes. Stem Cells 2013, 31, 2747–2758. [Google Scholar] [CrossRef]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef]
- Lee, S.C.; Pappone, P.A. Effects of P2 purinergic receptor stimulation in brown adipocytes. Am. J. Physiol. 1997, 273, C679–C686. [Google Scholar] [CrossRef] [PubMed]
- Razzoli, M.; Frontini, A.; Gurney, A.; Mondini, E.; Cubuk, C.; Katz, L.S.; Cero, C.; Bolan, P.J.; Dopazo, J.; Vidal-Puig, A.; et al. Stress-induced activation of brown adipose tissue prevents obesity in conditions of low adaptive thermogenesis. Mol. Metab. 2016, 5, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Pydi, S.P.; Toti, K.S.; Robaye, B.; Idzko, M.; Gavrilova, O.; Wess, J.; Jacobson, K.A. Lack of adipocyte purinergic P2Y6 receptor greatly improves whole body glucose homeostasis. Proc. Natl. Acad. Sci. USA 2020, 117, 30763–30774. [Google Scholar] [CrossRef]
- Goddard, K.E. Consequences of an obesogenic diet can be prevented by knockout of P2Y 6 purinergic receptor in mice. Purinergic Signal. 2021, 1–3. [Google Scholar]
- Burnstock, G.; Novak, I. Purinergic signalling and diabetes. Purinergic Signal. 2013, 9, 307–324. [Google Scholar] [CrossRef]
- Nishimura, A.; Sunggip, C.; Oda, S.; Numaga-Tomita, T.; Tsuda, M.; Nishida, M. Purinergic P2Y receptors: Molecular diversity and implications for treatment of cardiovascular diseases. Pharmacol. Ther. 2017, 180, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Vassort, G. Adenosine 5’-triphosphate: A P2-purinergic agonist in the myocardium. Physiol. Rev. 2001, 81, 767–806. [Google Scholar] [CrossRef]
- Cohen, R.; Shainberg, A.; Hochhauser, E.; Cheporko, Y.; Tobar, A.; Birk, E.; Pinhas, L.; Leipziger, J.; Don, J.; Porat, E. UTP reduces infarct size and improves mice heart function after myocardial infarct via P2Y2 receptor. Biochem. Pharmacol. 2011, 82, 1126–1133. [Google Scholar] [CrossRef]
- Hochhauser, E.; Cohen, R.; Waldman, M.; Maksin, A.; Isak, A.; Aravot, D.; Jayasekara, P.S.; Muller, C.E.; Jacobson, K.A.; Shainberg, A. P2Y2 receptor agonist with enhanced stability protects the heart from ischemic damage in vitro and in vivo. Purinergic Signal. 2013, 9, 633–642. [Google Scholar] [CrossRef]
- Nishida, M.; Ogushi, M.; Suda, R.; Toyotaka, M.; Saiki, S.; Kitajima, N.; Nakaya, M.; Kim, K.M.; Ide, T.; Sato, Y.; et al. Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2Y2 receptor stimulation through S-nitrosylation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2011, 108, 6662–6667. [Google Scholar] [CrossRef]
- Nishida, M.; Sato, Y.; Uemura, A.; Narita, Y.; Tozaki-Saitoh, H.; Nakaya, M.; Ide, T.; Suzuki, K.; Inoue, K.; Nagao, T.; et al. P2Y6 receptor-Galpha12/13 signalling in cardiomyocytes triggers pressure overload-induced cardiac fibrosis. EMBO J. 2008, 27, 3104–3115. [Google Scholar] [CrossRef] [PubMed]
- Horckmans, M.; Esfahani, H.; Beauloye, C.; Clouet, S.; di Pietrantonio, L.; Robaye, B.; Balligand, J.L.; Boeynaems, J.M.; Dessy, C.; Communi, D. Loss of mouse P2Y4 nucleotide receptor protects against myocardial infarction through endothelin-1 downregulation. J. Immunol. 2015, 194, 1874–1881. [Google Scholar] [CrossRef]
- De Oliveira Moreira, D.; Santo Neto, H.; Marques, M.J. P2Y2 purinergic receptors are highly expressed in cardiac and diaphragm muscles of mdx mice, and their expression is decreased by suramin. Muscle Nerve 2017, 55, 116–121. [Google Scholar] [CrossRef]
- Hou, M.; Harden, T.K.; Kuhn, C.M.; Baldetorp, B.; Lazarowski, E.; Pendergast, W.; Moller, S.; Edvinsson, L.; Erlinge, D. UDP acts as a growth factor for vascular smooth muscle cells by activation of P2Y(6) receptors. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H784–H792. [Google Scholar] [CrossRef]
- Benoist, L.; Chadet, S.; Genet, T.; Lefort, C.; Heraud, A.; Danila, M.D.; Muntean, D.M.; Baron, C.; Angoulvant, D.; Babuty, D.; et al. Stimulation of P2Y11 receptor protects human cardiomyocytes against Hypoxia/Reoxygenation injury and involves PKCepsilon signaling pathway. Sci. Rep. 2019, 9, 11613. [Google Scholar] [CrossRef]
- Danila, M.D.; Piollet, M.; Aburel, O.M.; Angoulvant, D.; Lefort, C.; Chadet, S.; Roger, S.; Muntean, M.D.; Ivanes, F. Modulation of P2Y11-related purinergic signaling in inflammation and cardio-metabolic diseases. Eur. J. Pharmacol. 2020, 876, 173060. [Google Scholar] [CrossRef]
- Piollet, M.; Sturza, A.; Chadet, S.; Gabillard-Lefort, C.; Benoist, L.; Muntean, D.M.; Aburel, O.M.; Angoulvant, D.; Ivanes, F. P2Y11 Agonism Prevents Hypoxia/Reoxygenation- and Angiotensin II-Induced Vascular Dysfunction and Intimal Hyperplasia Development. Int. J. Mol. Sci. 2021, 22, 855. [Google Scholar] [CrossRef]
- Wihlborg, A.K.; Wang, L.; Braun, O.O.; Eyjolfsson, A.; Gustafsson, R.; Gudbjartsson, T.; Erlinge, D. ADP receptor P2Y12 is expressed in vascular smooth muscle cells and stimulates contraction in human blood vessels. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1810–1815. [Google Scholar] [CrossRef]
- Sunggip, C.; Nishimura, A.; Shimoda, K.; Numaga-Tomita, T.; Tsuda, M.; Nishida, M. Purinergic P2Y6 receptors: A new therapeutic target of age-dependent hypertension. Pharmacol. Res. 2017, 120, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, S.; Xiang, Q.; Chen, J.; Tian, F.; Xu, J.; Li, X.; Tan, Y.; Liu, L. The Role of a Selective P2Y6 Receptor Antagonist, MRS2578, on the Formation of Angiotensin II-Induced Abdominal Aortic Aneurysms. Biomed. Res. Int. 2020, 2020, 1983940. [Google Scholar] [PubMed]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2Y1 and P2Y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol. Cell Physiol. 2011, 300, C266–C275. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Vitiello, L.; Idzko, M.; la Sala, A. Purinergic signaling in atherosclerosis. Trends Mol. Med. 2015, 21, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.H.; Filep, J.G. Purinergic receptors and atherosclerosis: Emerging role for vessel wall P2Y12. Cardiovasc. Res. 2014, 102, 339–341. [Google Scholar] [CrossRef][Green Version]
- Chen, X.; Qian, S.; Hoggatt, A.; Tang, H.; Hacker, T.A.; Obukhov, A.G.; Herring, P.B.; Seye, C.I. Endothelial Cell-Specific Deletion of P2Y2 Receptor Promotes Plaque Stability in Atherosclerosis-Susceptible ApoE-Null Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 75–83. [Google Scholar] [CrossRef]
- Lichtenstein, L.; Serhan, N.; Espinosa-Delgado, S.; Fabre, A.; Annema, W.; Tietge, U.J.; Robaye, B.; Boeynaems, J.M.; Laffargue, M.; Perret, B.; et al. Increased atherosclerosis in P2Y13/apolipoprotein E double-knockout mice: Contribution of P2Y13 to reverse cholesterol transport. Cardiovasc. Res. 2015, 106, 314–323. [Google Scholar] [CrossRef]
- Lovaszi, M.; Branco Haas, C.; Antonioli, L.; Pacher, P.; Hasko, G. The role of P2Y receptors in regulating immunity and metabolism. Biochem. Pharmacol. 2021, 187, 114419. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; la Sala, A.; Milani, D.; Celeghini, C.; Casciano, F. Purinergic Signaling in Controlling Macrophage and T Cell Functions During Atherosclerosis Development. Front. Immunol. 2020, 11, 617804. [Google Scholar] [CrossRef]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Hechler, B.; Freund, M.; Eckly, A.; Vial, C.; Ohlmann, P.; Dierich, A.; LeMeur, M.; Cazenave, J.P.; Gachet, C. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y(1) receptor-null mice. J. Clin. Investig. 1999, 104, 1731–1737. [Google Scholar] [CrossRef]
- Idzko, M.; Dichmann, S.; Ferrari, D.; Di Virgilio, F.; la Sala, A.; Girolomoni, G.; Panther, E.; Norgauer, J. Nucleotides induce chemotaxis and actin polymerization in immature but not mature human dendritic cells via activation of pertussis toxin-sensitive P2y receptors. Blood 2002, 100, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Erlinge, D. P2Y receptors in health and disease. Adv. Pharmacol. 2011, 61, 417–439. [Google Scholar] [PubMed]
- Salmaso, V.; Jacobson, K.A. Purinergic Signaling: Impact of GPCR Structures on Rational Drug Design. ChemMedChem 2020, 15, 1958–1973. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| GPCR | Physiological/Pathological Action | Tissue Expression |
|---|---|---|
| Short Chain Fatty Acid Receptors | ||
| FFAR2/GPR43 | ↓ fat lipolysis, ↓ insulin sensitivity, ↑ anorectic hormones, GPR43−/− are mice obese on a regular diet and protected from weight gain on HFD | brain and lung tissues, with lesser expression in heart, skeletal muscle, intestine, liver–kidney adipocyte artery, leukocytes. |
| FFAR3/GPR41 | GPR41−/− ↑ insulin secretion, ↑ cardiac hypertrophy ↑, blood pressure | brain and lung tissues, with lesser expression in heart, skeletal muscle, intestine, liver–kidney adipocyte artery, leukocytes |
| Olfr78 | olfr78 ↑ blood pressure ↑ inflammation | Vascular cells |
| Medium Chain Fatty Acid Receptors | ||
| GPR84 | Pro-inflammatory, ↑ diabetes, atherosclerosis, heart failure, and ↑ fatty acid metabolism obesity. ↑ Fibrosis in lung | immune cells, lung, lymph nodes, and adipose tissue |
| Long-Chain Fatty Acid Receptors | ||
| FFAR1/GPR40 | ↑ obesity, ↑ Insulin secretion, adipogenesis. Studies with GPR40−/− mice on fat metabolism controversial may depend on fat and glucose levels suggest a homeostatic role | pancreatic β cells, intestinal cells, adipocytes, and liver, immune cells |
| FFAR4/GPR120 | protective in obesity, blood pressure, atherosclerosis and is anti-inflammatory | pancreatic β cells, intestinal cells, adipocytes, and liver, immune cells |
| GPR119 | GPR119 agonists lowered blood glucose, protective in atherosclerosis, anorectic but lowered metabolism in heart and skeletal muscle | GPR119 also expressed in cardiac and skeletal muscle |
| Ketone Body Receptors | ||
| HCA1/GPR81 | ↑ Insulin sensitivity in mouse models of diabetes regulation of renal vascular resistance by modulation of the endothelin. Possible anti-inflammatory | adipocyte; low kidney, skeletal muscle, and liver levels adipocytes and immune cells, heart, vascular |
| HCA2/GPR109A | ↓ fat accumulation, Agonists protective in systemic and pulmonary hypertension ↓ lipolysis and anti-inflammatory | adipocyte; low kidney, skeletal muscle, and liver levels adipocytes and immune cells, heart, vascular |
| HCA3/GPR109B | GPR109B is expressed only in human’s anti-inflammatory and inhibits adiposity | |
| Bile Acid Receptors | ||
| TGR5 | Bile acid homeostasis, energy homeostasis, insulin signaling, and inflammation. Dysfunction causes cholestatic liver diseases, dyslipidemia, fatty liver diseases, cardiovascular diseases, and diabetes | the small intestine, stomach, liver, lung, placenta, and spleen |
| Ceramide | ||
| SIP1R | ↑ obesity, insulin resistance, hyperglycemia, dyslipidemia, and hypertension. Proinflammatory in macrophages | Macrophages endothelial cells |
| S1P2R | Pancreatic beta cells, Skeletal muscle | |
| Prostanoids | ||
| Prostaglandins | ↓ Protective against obesity-induced inflammation substrate analogs improve insulin sensitivity, protective in diabetic nephropathy, −/− mice show increased intimal hyperplasia, atherosclerosis, and hypercoagulability and thrombus formation | Vascular, T cells, platelets macrophages, pneumocytes, smooth muscle cells, and fibroblasts vascular cells, platelets, macrophagesleukocytes, including granulocytes, T Cells, dendritic macrophages, and vascular smooth muscle cells |
| PGI | ||
| TXA2 | ↑ Obesity, ↑platelet aggregation, modified by insulin sensitivity, inflammatory in macrophages, glucose, insulin resistance, and triglycerides. | |
| PGE2 | ↑ Obesity. EP3 receptor inhibitors reversed obesity-induced tissues inflammation. In the kidney vasculature, EP2 and EP4 ↑vasodilation, whereas EP1 and EP3 ↑ vasoconstriction. Cardiomyocyte-specific deletion of the EP4 ↑cardiac dysfunction after myocardial infarction | |
| PGF | PGF2α suppresses an early phase of adipogenesis. FP−/− mice ↓ blood pressure, coincident with a reduction in plasma renin concentration, angiotensin, and aldosterone | |
| Leukotrienes | LTB4 antagonists and BLT-1−/− mice are protected from HFD-induced insulin resistance and decrease macrophages and T cells infiltration in adipose tissue. Inhibition of BLT1 is protective in atherosclerosis. | |
| BLT1 | ||
| BLT2 | ||
| Hydroxy-eicosatetraenoic acids | agonists promote vascular smooth muscle contraction, endothelial dysfunction, inflammation, and cell proliferation. The 20-HETE antagonist attenuated weight gain and prevented hyperglycemia | T cells, platelets macrophages, pneumocytes, smooth muscle cells, and fibroblasts vascular cells, platelets, macrophages |
| 20-HETE/GPR75 | 12-HETE increases oxidative stress and modulates inflammation via interaction with GPR31. GPR31−/− mice protect obese HFD fed mice from glucose intolerance and improve insulin secretion in cytokine-treated islets. | leukocytes, including granulocytes, T Cells, dendritic macrophages, and vascular smooth muscle cells |
| 12-HETE/GPR31 | ||
| TCA Cycle Metabolites | ||
| GPR91/SUCNR1 | Increased BMI, insulin, increase in adipose tissue protects from liver lipo-toxicity. An increase in the liver promotes atherosclerosis, vasorelaxant, increased in Metabolic syndrome, proinflammatory | white adipose tissue, liver, heart intestine, spleen, and immune system cells, including dendritic cells vascular cells |
| GPR99/Oxgr1 | decreased concentrations of αKG in the right and left ventricles of mice exposed to hypoxia promote cardiac hypertrophy | brain, lung, kidney, heart, and skeletal muscle |
| Amino Acid Receptors | ||
| GPR142 | increased during fasting and decreased in DIO improved insulin sensitivity delayed the onset and progression of diabetes, and is anti-inflammatory | pancreas and the immune system pancreatic islets and skeletal muscle, with relatively higher levels in adult lung, small intestine, colon, and stomach. |
| GPR35 | lipid metabolism, thermogenic, and anti-inflammatory gene expression in adipose tissue | kidney and parathyroid glands and to a lesser extent in lungs, skin, intestine, b |
| CasR | CasR vascular tone, metabolic processes in vascular cells, lung and neuronal development, or cardiac function, promote glucose-induced insulin secretion Pro-inflammatory | |
| GPR139 | GPR139−/− mice are lean, target for obesity and T2D | hypothalamus, pituitary, and habenula rain, and vasculature on immune cells |
| TAAR1 | Increases insulin secretion and glucose tolerance decreased food intake and body weight in a diet-in. increase inflammatory cytokine gene expression in non-polarized and LPS-polarized BMDM Mets and inversely correlated with multiple biomarkers of inflammation and cardiometabolic risk factors such as body mass index and blood pressure | TAAR1 neuronal cells and peripheral organs such as the GI tract immune cells |
| Nucleoside Receptors | ||
| P1R | A1 antilipolytic and is implicated in adipogenesis mediate insulin signaling and age-related changes in adipose tissue. Adenosine ↑ vasodilation and is anti-inflammatory | endothelial cells, vascular smooth muscle cells, liver, adipocytes, and different types of leukocytes. |
| A1R | ||
| A2AR | A2A−/−/ApoE−/− shows an anti-atherosclerotic role for A2A. In macrophages, A2A shows a pro atherosclerotic role, is anti-inflammatory in macrophages. | |
| A2BR | A2B−/− mice develop metabolic syndrome and T2D ↑ inflammation of adipose tissue. ↑ browning of fat, ↓ atherosclerosis is anti-inflammatory in endothelial cells ↓ platelet aggregation. Anti-inflammatory in macrophages | |
| P2R | P2 receptors: All P2 receptors are adipogenic. | endothelial cells, vascular smooth muscle cells, liver adipocytes, and different types of leukocytes Heart, platelet, skeletal muscle, neuron, intestine |
| P2Y1 | P2Y1↑ β-cell apoptosis. P2Y1−/− ↑ insulin secretion. ischemic diseases, pressure overload hypertrophy, and ↓myocardial infarction in the heart | |
| P2Y2 | Pro-atherogenic, proinflammatory | |
| P2Y4 | promote thermogenesis in BAT | Heart, lung, skeletal muscle, spleen, kidney lung, vascular smooth muscle, brain, Liver |
| P2Y6 | Insulin resistance, obesity, hypertension, and electrolyte homeostasis. P2Y6−/− in adipose tissue is insulin-sensitizing, whereas skeletal muscle KO is insulin resistant | |
| P2Y11 | P2Y11 is protective in ischemia, promotes hypertension, pro and anti-inflammatory roles reported, ↓ insulin-stimulated leptin in adipocytes and ↑ lipolysis | Spleen, intestine, Immune cells Heart, lung, spleen, intestine, brain |
| P2Y12 | Pro-atherogenic | |
| P2Y13 | P2Y13−/− mice showed improved metabolic parameters | |
| Natural Metabolite-CAS# | Receptor | Trial Name | Drug | Clinical Trials Relevant to This Review |
|---|---|---|---|---|
| 2-Propionate-butyrate (CAS156-54-7) | GPR43/GPR41 (FFA2 and FFA3) | The Effect of PPI Therapy on Weight, Gut Microbiome, and Expression of GPR41 and GPR43 | Diet questionnaire | NCT02457104 |
| GPR43 (IUPHAR (#226)) | Effect of a Synbiotic on the Gut Microbiota and Adiposity-related Markers in Healthy Overweight Subjects | Dietary Supplement Synbiotic | NCT0215182 | |
| GPR41 (IUPHAR # 227) gene#2865) | Effect of fermentable carbohydrates in glucose homeostasis | Dietary supplement Inulin and cellulose | NCT01841073 | |
| 9-a-linoleic acid (CAS#60-33-3) | GPR40 (FFA1) (IUPHAR-# 311) (gene#2864) | Determination of Safety, Tolerability, Pharmacokinetics, Food Effect and pharmacodynamics of Single and Multiple Doses of P11187 | GPR-40-agonist P11187 | NCT01874366 |
| NCT04703361 | ||||
| Effect of Dietary oils as GPCR agonists on Glucose tolerance | Dietary Supplement Pine nut and olive oil | NCT03774095 | ||
| GPR109A (HCA2) (IUPHAR-# 312) (gene #338442) | Short-term Effect of Extended-release Niacin on Endothelial Function. | Niacin Na-OHB | NCT01942291; | |
| 8-palmitate, PA (CAS#2210-62-0) | GPR120 FFA4 (IUPHAR-# 127) (gene#338557) | Expression of G-protein Coupled Receptor 120 (GPR120) Receptor in Adipose Tissue of German Diabetes Center (GDC) Cohort Subjects | Association of R270H mutant with T2D and expression of GPR120 in T2D | NCT03285750 |
| 10-oleoylglycerol (CAS#111-03-5) | GPR119 (IUPHAR-# 126) (gene#139760) | -Oleoyl Glycerol is a GPR119 Agonist and Signals GLP-1 Release in Humans. | Dietary Supplement: GPR 119 agonist, 2-oleyl glycerol Dietary Supplement: Oleic acid | NCT01043445 |
| Diet oil supplementation induced release of GLP-1 | Diet oil supplementation of olive and carrot oil | NCT01453842 | ||
| 11-Lithocholic acid (434-13-9) | GPBAR1 (TGR5) (IUPHAR #37) (gene#151306) | Effect of Bile Acids on the Secretion of Satiation Peptides in Humans | bile acid (CDCA, chenodeoxycholic acid) oleanolic acid Dietary Supplement: oleic acid | NCT01674946 |
| The Impact of Gall Bladder Emptying and Bile Acids on the Human GLP-1-secretion | Acetaminophen Metformin Colesevelam CCK-8 | NCT01656057 | ||
| Importance of Meal Fat Content and Gall Bladder Emptying for Postprandial GLP-1 Secretion in Type 2 Diabetes Patients | GLP-1 secretion via TGR5 | NCT01374594 | ||
| Bile Acid-induced GLP-secretion. A Study in Cholecystectomized Subjects | GLP-1 secretion via TGR5 in healthy and Cholecystectomized Subjects | NCT01251510 | ||
| Effect of Bile Acids on GLP-1 Secretion | Chenodeoxylic acid Colesevelam | NCT01666223 | ||
| 20-HETE | GPR75 (IUPHAR #115) Gene#10936 | Genetic and Dietary Predictors of Anti-platelet Response | ASPIRIN | Not recruiting yet |
| Sphingosine-1-phosphate, (CAS#26993-30-6) | SIPR1(EDG1) (gene#1901) (IUPHAR#275) | Efficacy, Safety and Tolerability of BAF312 Compared to Placebo in Patients With Intracerebral Hemorrhage (ICH). | BAF312 | NCT03338998 |
| LTB4 | BLT1 IUPHAR#271) | Body Weight, Aspirin Dose and Pro-resolving Mediators | ASPIRIN | Not recruiting yet |
| PGI2-prostacyclin, (CAS # 63859-31-4) | IP (IUPHAR#345) (Gene#5739) | Evaluation of a New Thermostable Formulation of FLOLAN in Japanese Subjects | FLOLAN | NCT02705807 |
| Drug Use Investigation for FLOLAN (Epoprostenol) Injection 0.5mg·1.5mg | FLOLAN | NCT01387191 | ||
| Inhaled Nitric Oxide and Inhaled Prostacyclin After Cardiac Surgery for Heart Transplant or LVAD Placement | Prostacyclin FLOLAN | NCT01717209 | ||
| Epoprostenol in Pulmonary Embolism | Epoprostenol | NCT01014156 | ||
| Thromboxane TxA2 | TXA2-R (Gene#6915) (IUPHAR#346) | Thromboxane Receptor Antagonist to Improve Endothelial Cell Function | ifetroban | NCT03962855 |
| Phenylalanine, and other amino acids. | CaSR (IUPHAR-# 54) (gene#846) | Lipid and Glycogen Metabolism in Patients With Impaired Glucose Tolerance and Calcium Sensing Receptor Mutations | Meal Tolerance Test: Hyperglycemic-hyper insulinemic clamp | NCT02023489 |
| ATP | P2Y12 Gai (IUPHAR#328) (gene#64805) | Aspirin Impact on Platelet Reactivity in Acute Coronary Syndrome Patients on Novel P2Y12 Inhibitors Therapy | Aspirin P2Y12 inhibitors | NCT03190005 |
| Comparison Between P2Y12 Antagonist Monotherapy and Dual Antiplatelet Therapy After DES | Aspirin P2Y12 inhibitors combination | NCT02079194 | ||
| Association Between Genetic Variant Scores and P2Y12 Inhibitor Effects | P2Y12 inhibitors | NCT04580602 | ||
| Aspirin Impact on Platelet Reactivity in Acute Coronary Syndrome Patients on Novel P2Y12 Inhibitors Therapy | P2Y12 inhibitors | NCT02049762 | ||
| Tailoring P2Y12 Inhibiting Therapy in Patients Requiring Oral Anticoagulation After PCI | Ticagrelor Clopidogrel | NCT04483583 | ||
| ComparisoN of ticAgrelor vs. Clopidogrel in endoTHeliAl Function of COPD patieNts | Ticagrelor Clopidogrel Aspirin | NCT02519608 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strassheim, D.; Sullivan, T.; Irwin, D.C.; Gerasimovskaya, E.; Lahm, T.; Klemm, D.J.; Dempsey, E.C.; Stenmark, K.R.; Karoor, V. Metabolite G-Protein Coupled Receptors in Cardio-Metabolic Diseases. Cells 2021, 10, 3347. https://doi.org/10.3390/cells10123347
Strassheim D, Sullivan T, Irwin DC, Gerasimovskaya E, Lahm T, Klemm DJ, Dempsey EC, Stenmark KR, Karoor V. Metabolite G-Protein Coupled Receptors in Cardio-Metabolic Diseases. Cells. 2021; 10(12):3347. https://doi.org/10.3390/cells10123347
Chicago/Turabian StyleStrassheim, Derek, Timothy Sullivan, David C. Irwin, Evgenia Gerasimovskaya, Tim Lahm, Dwight J. Klemm, Edward C. Dempsey, Kurt R. Stenmark, and Vijaya Karoor. 2021. "Metabolite G-Protein Coupled Receptors in Cardio-Metabolic Diseases" Cells 10, no. 12: 3347. https://doi.org/10.3390/cells10123347
APA StyleStrassheim, D., Sullivan, T., Irwin, D. C., Gerasimovskaya, E., Lahm, T., Klemm, D. J., Dempsey, E. C., Stenmark, K. R., & Karoor, V. (2021). Metabolite G-Protein Coupled Receptors in Cardio-Metabolic Diseases. Cells, 10(12), 3347. https://doi.org/10.3390/cells10123347