
Repeated Social Defeat Exaggerates Fibrin-Rich Clot Formation by Enhancing Neutrophil Extracellular Trap Formation via Platelet–Neutrophil Interactions

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
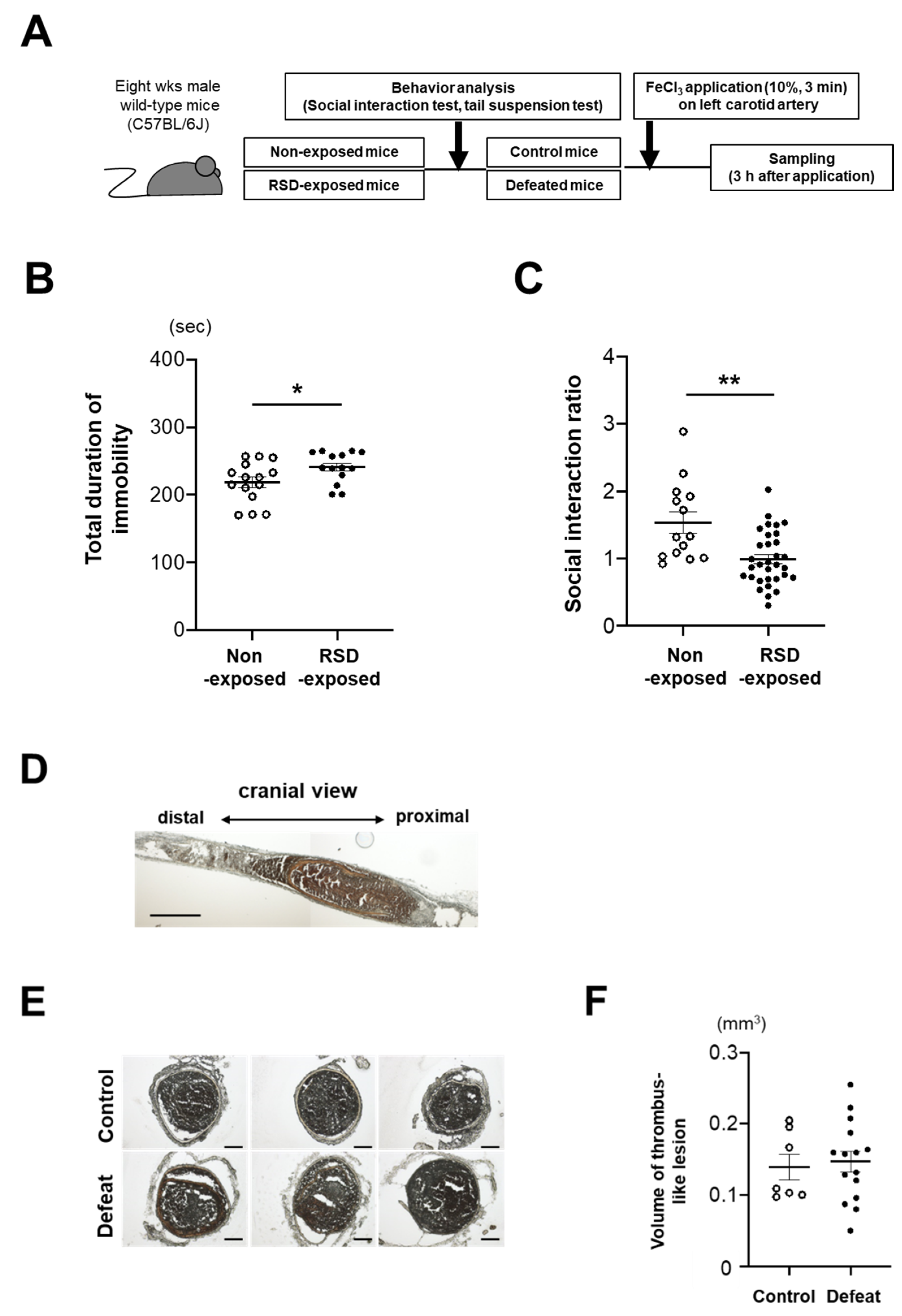
2.1. Repeated Social Defeat Model
2.2. Behavior Analysis
2.3. FeCl3-Induced Clot Formation Model
2.4. Hemodynamic Analysis
2.5. Thrombi Measurement and Histological Analysis
2.6. Immunohistochemical Analysis
2.7. Blood Leukocyte Counts
2.8. Platelets Counts and In Vitro Aggregation
2.9. Preparation of Platelets and P-Selectin Expression Analysis
2.10. Flow Cytometry and Cell Sorting Analysis of Neutrophils
2.11. In Vitro NET Formation Analysis of Mature BM Neutrophils
2.12. Statistical Analysis
3. Results
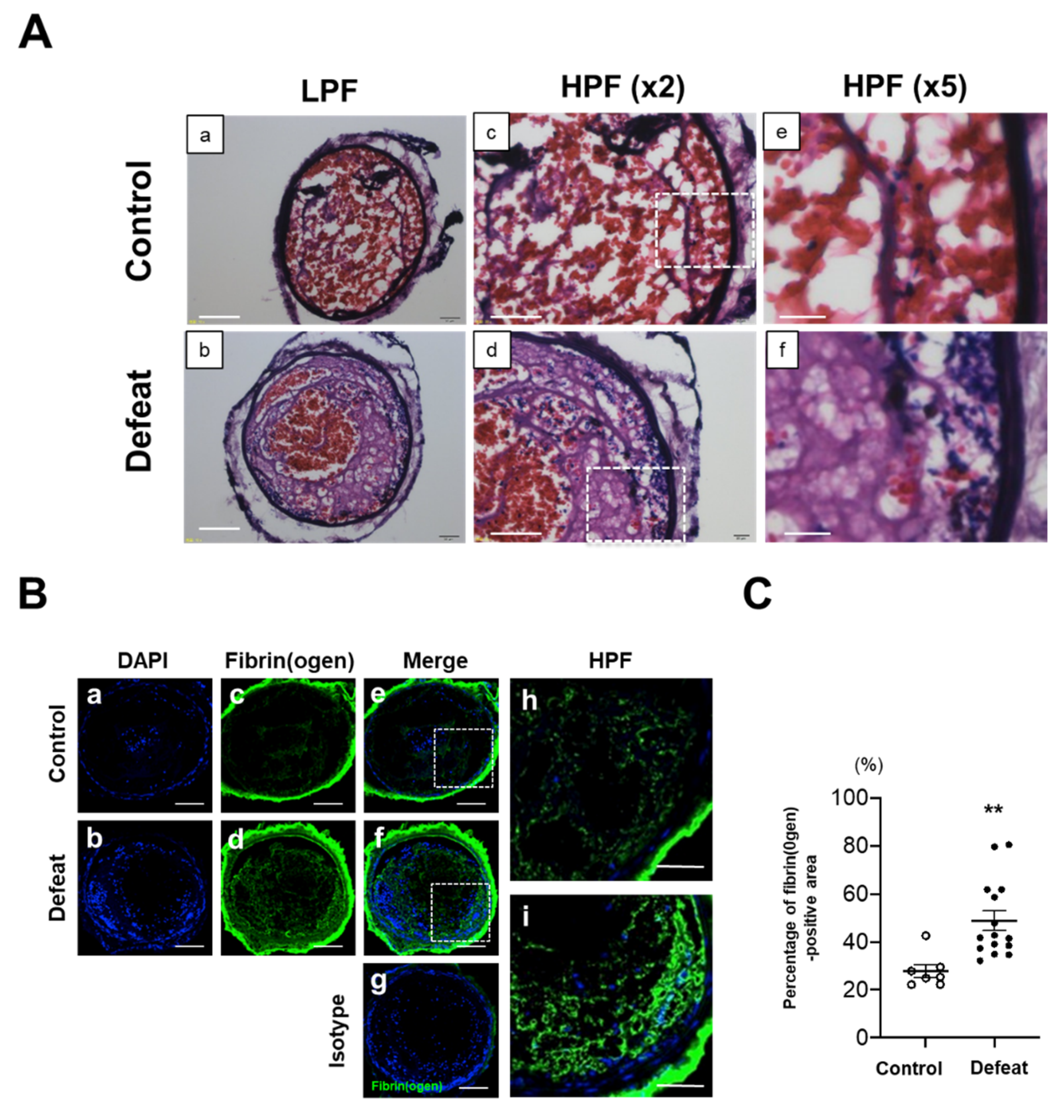
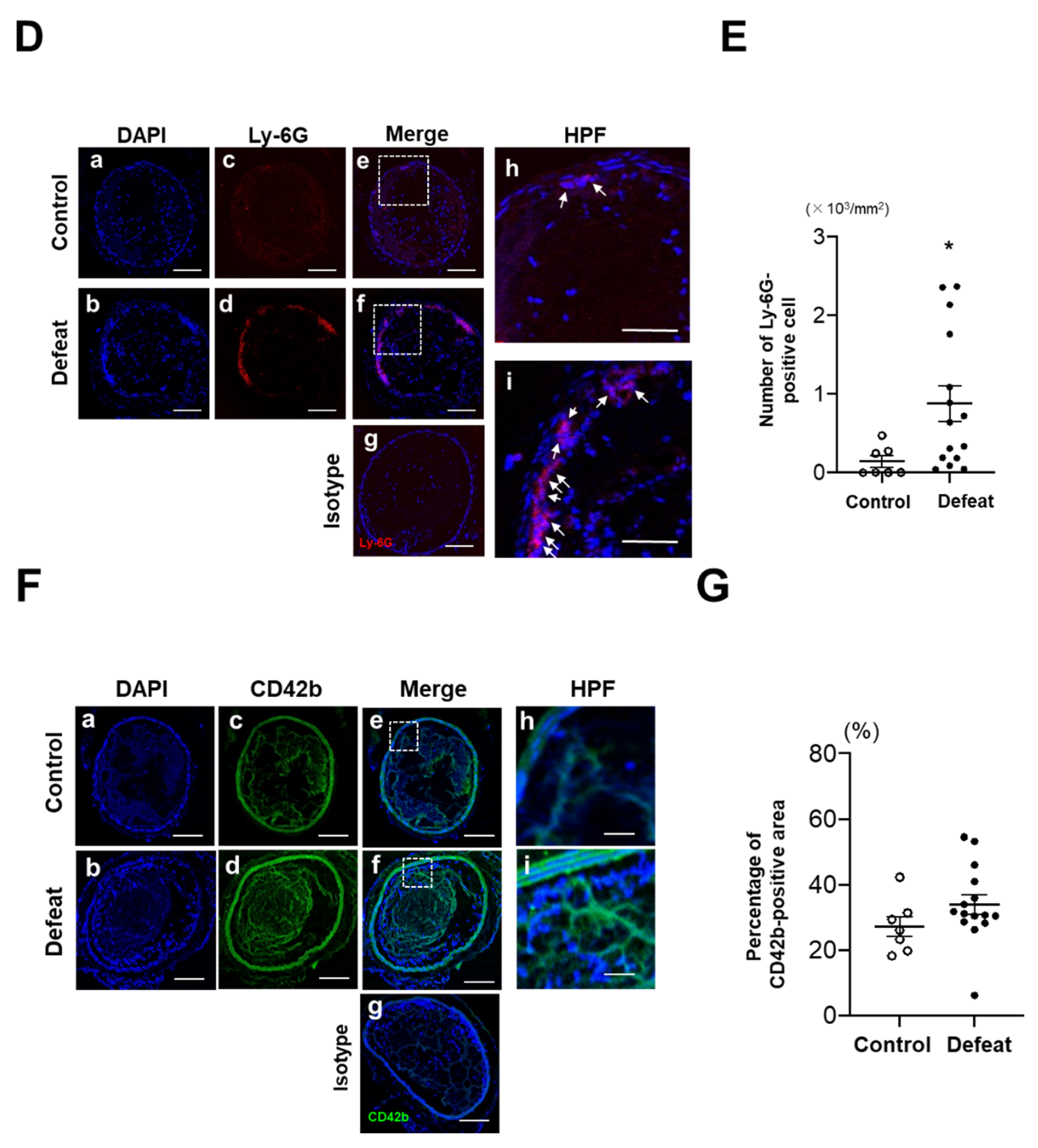
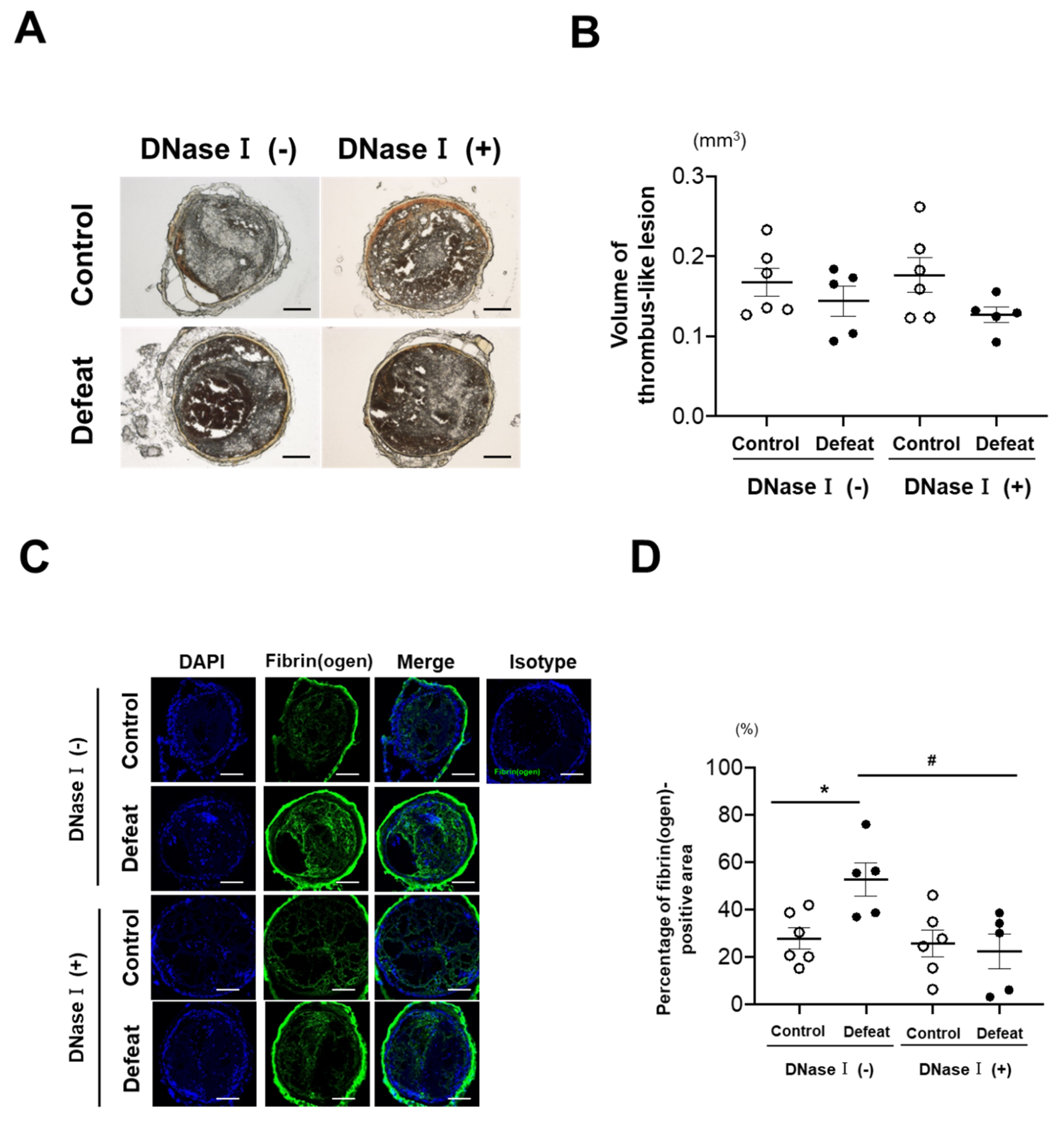
3.1. Fibrin-Rich Clot Formation Was Exaggerated in Defeated Mice
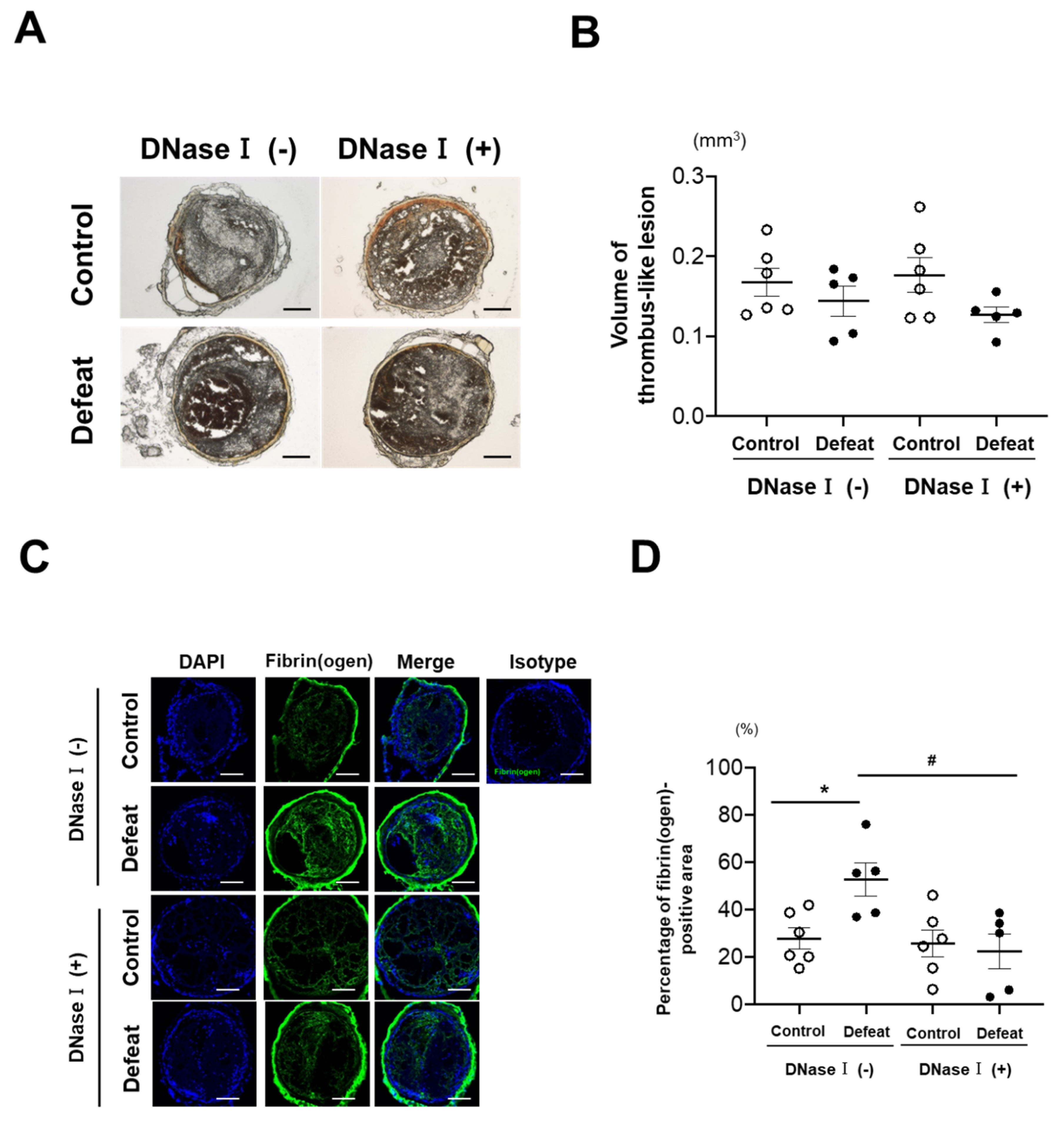
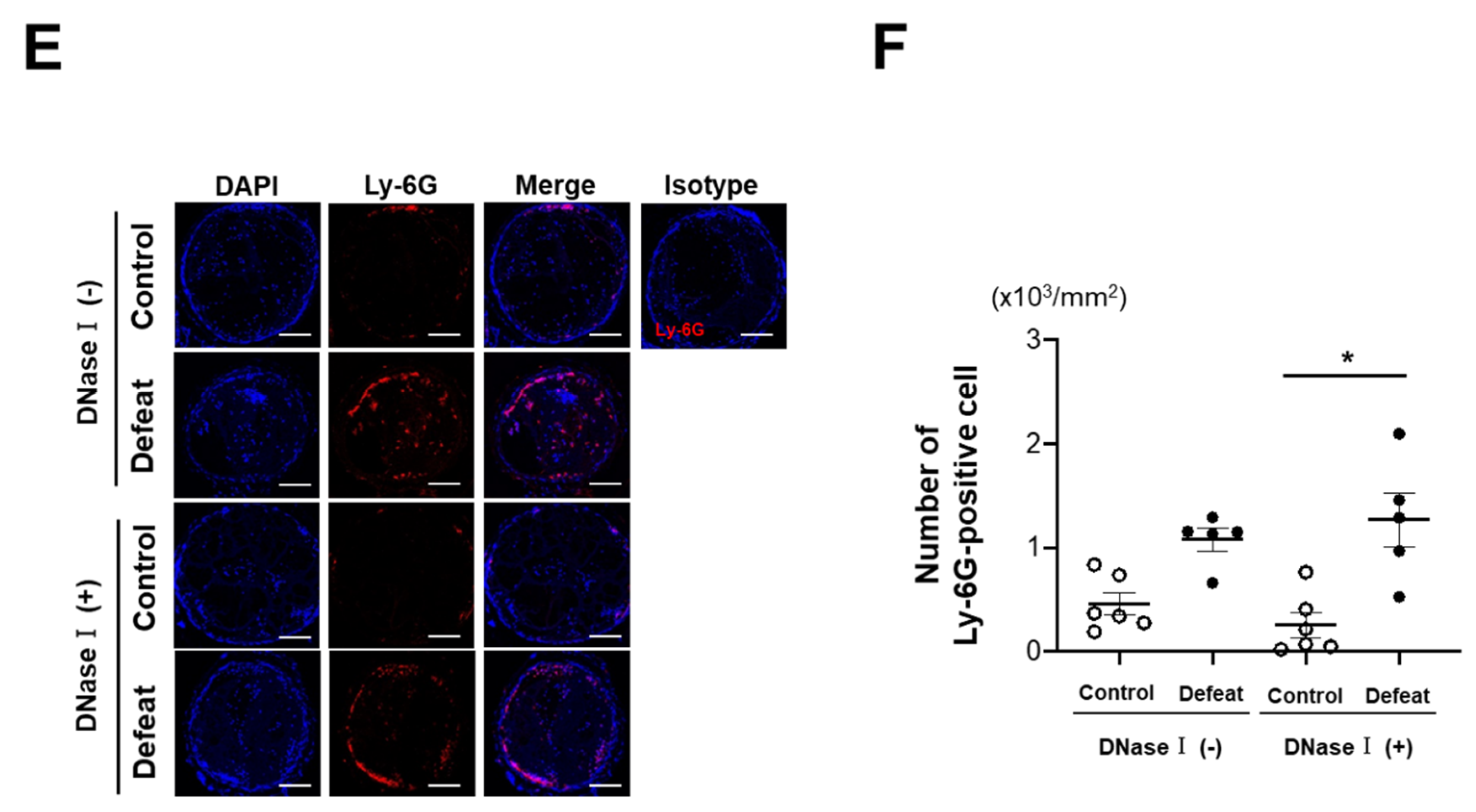
3.2. DNase I Treatment Inhibited Fibrin-Rich Clot Formation in Defeated Mice
3.3. Platelet Aggregation and P-Selectin Expression Were Not Affected by RSD
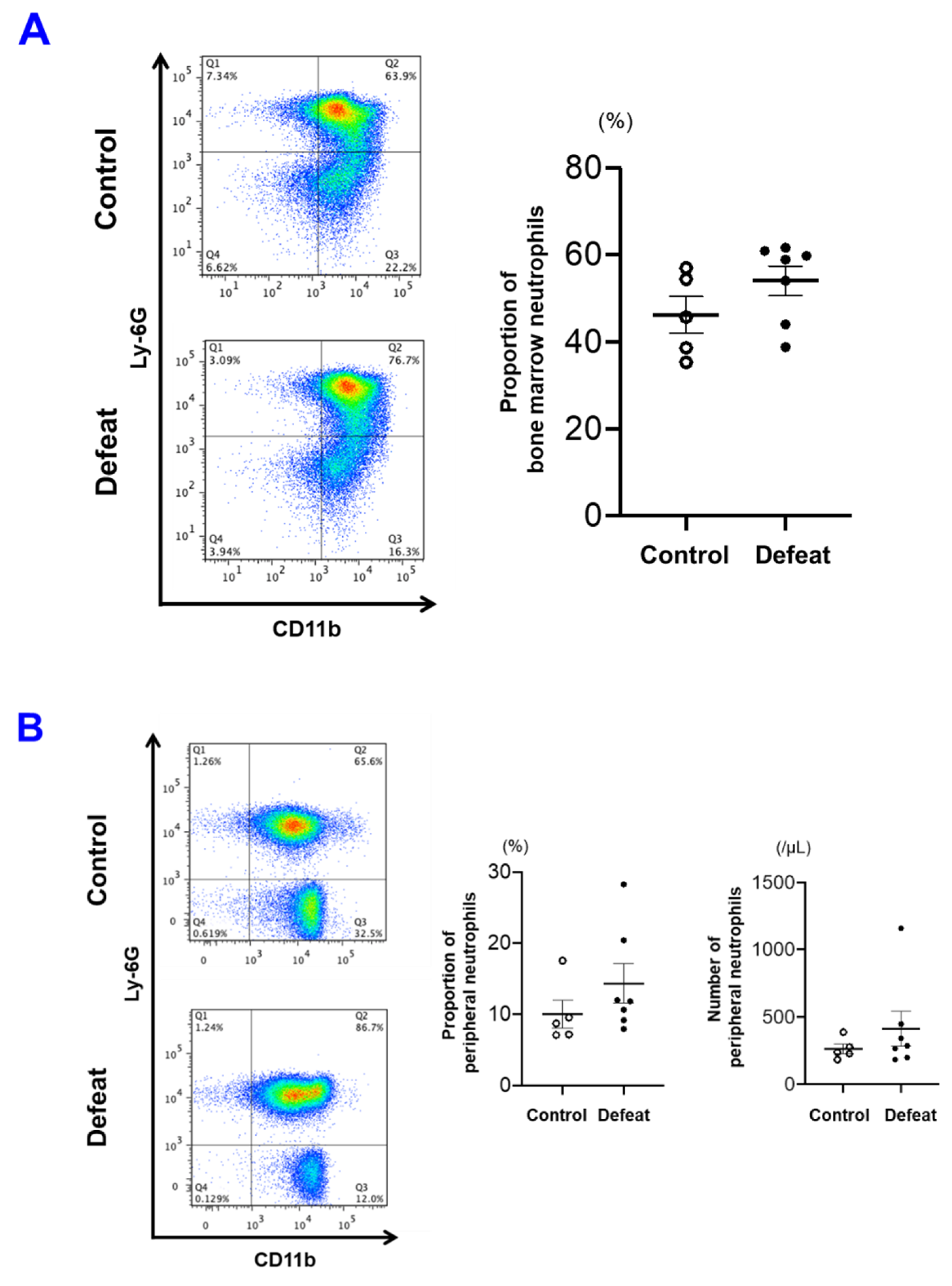
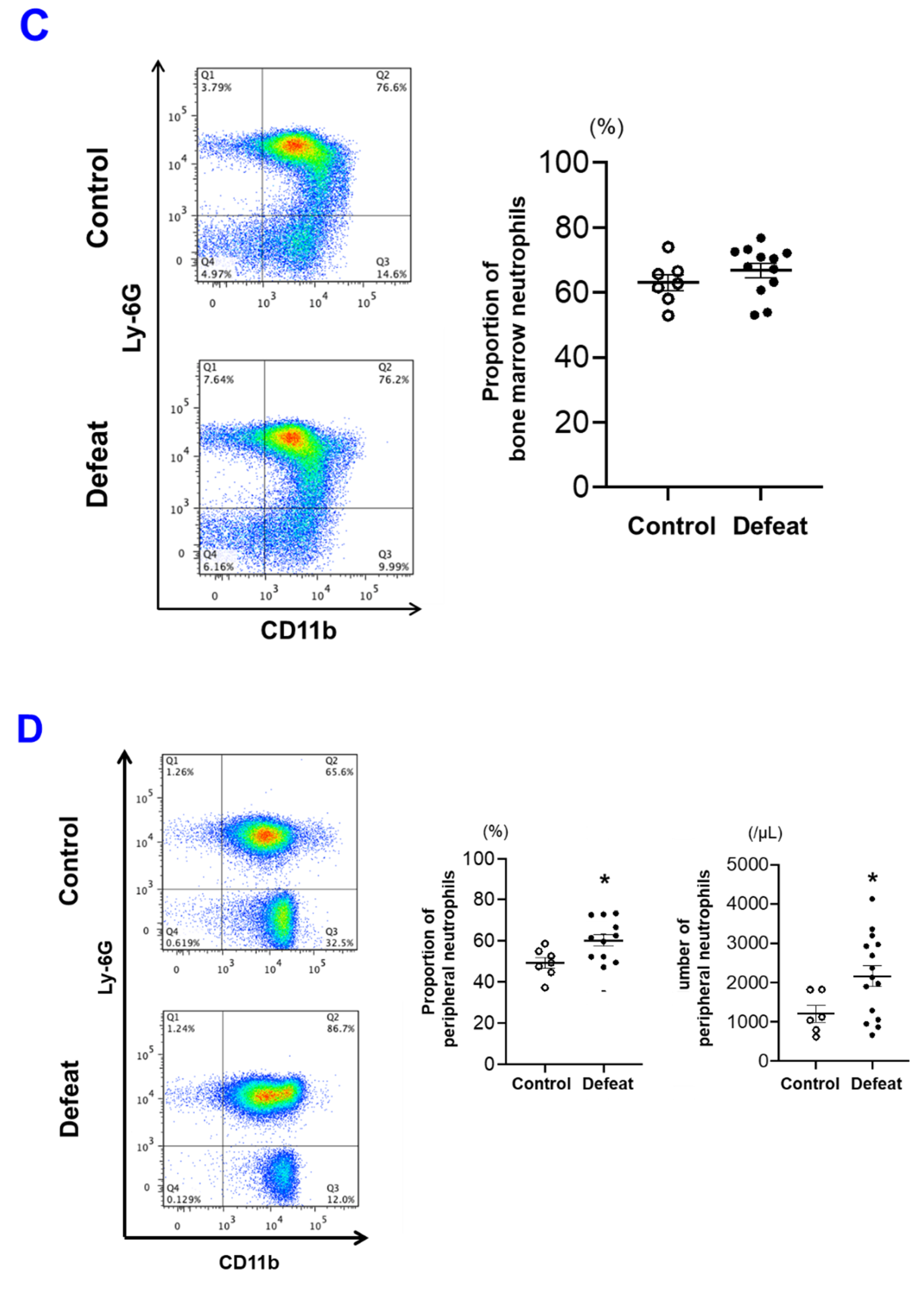
3.4. The Number of PB Neutrophils before FeCl3 Application Was Not Affected by RSD
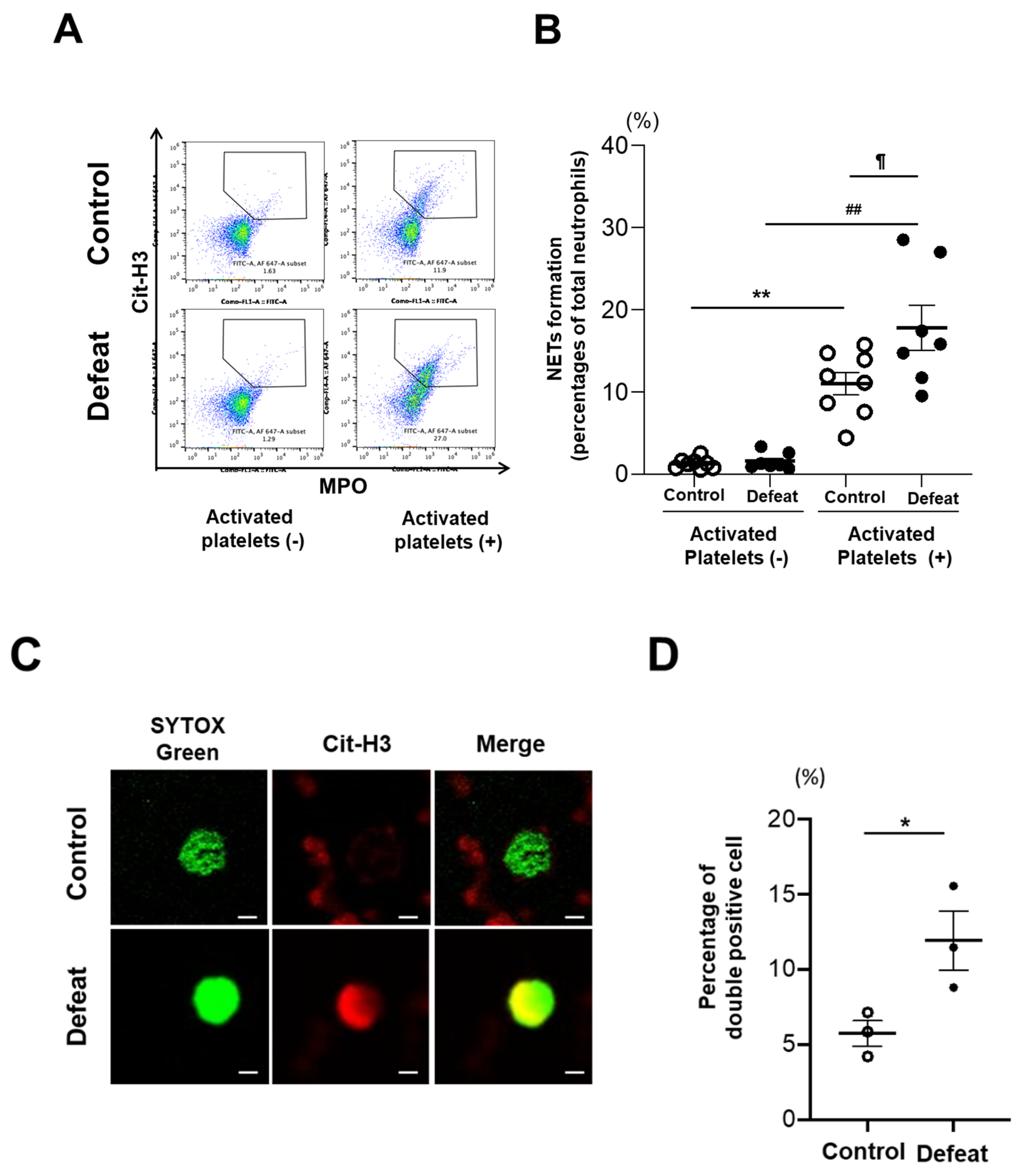
3.5. Activated Platelet-Induced NET Formation Was Augmented in Defeated Mice
3.6. The CD11b/Gpiα Axis Contributed to Augmented NET Formation in Defeated Mice
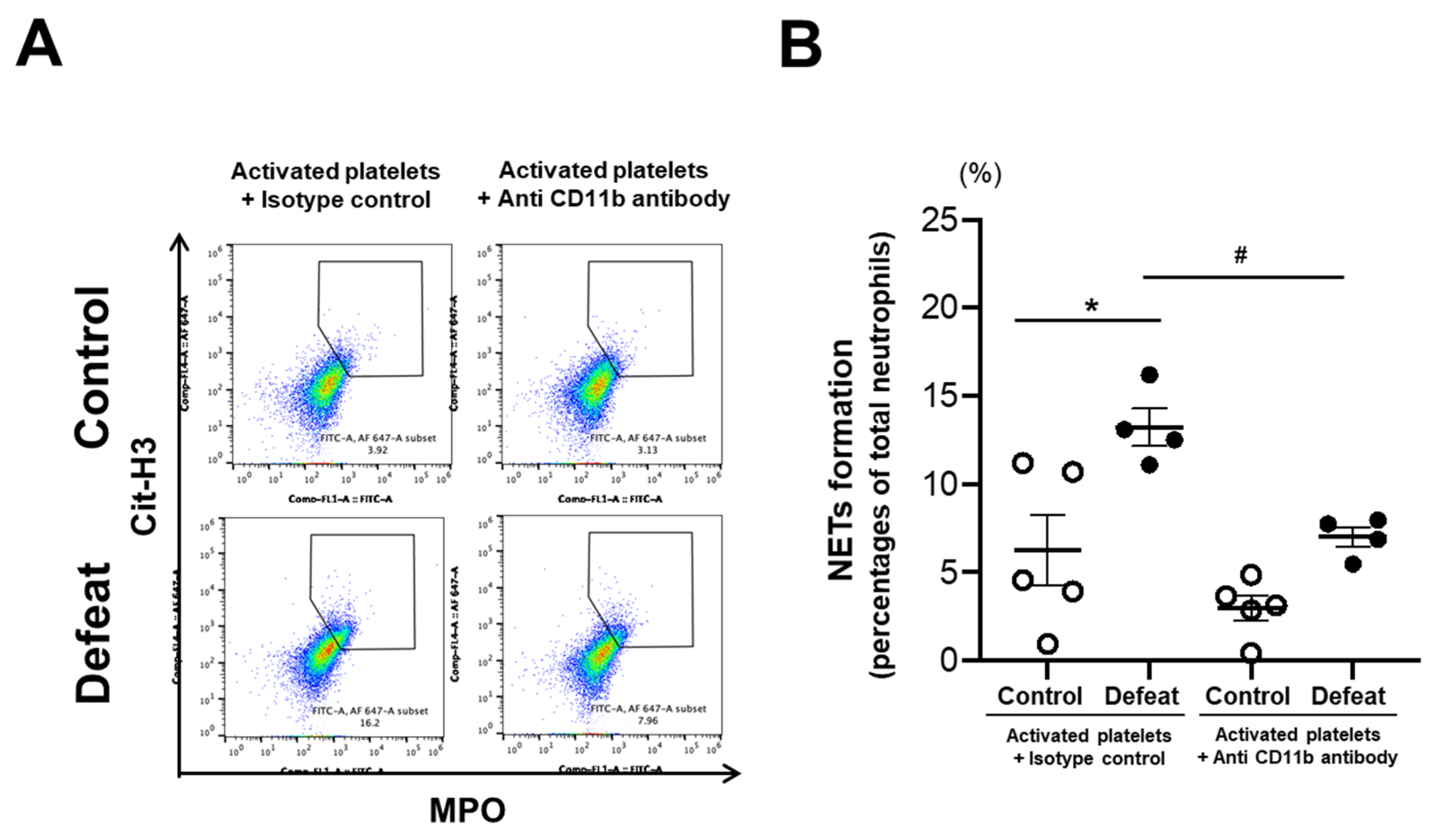
3.7. Augmented NET Formation in Defeated Mice Was Inhibited by Anti-CD11b Antibody Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Walker, E.R.; McGee, R.E.; Druss, B.G. Mortality in mental disorders and global disease burden implications: A systematic review and meta-analysis. JAMA Psychiatry 2015, 72, 334–341. [Google Scholar] [CrossRef]
- Hare, D.L.; Toukhsati, S.R.; Johansson, P.; Jaarsma, T. Depression and cardiovascular disease: A clinical review. Eur. Heart J. 2014, 35, 1365–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtman, J.H.; Froelicher, E.S.; Blumenthal, J.A.; Carney, R.M.; Doering, L.V.; Frasure-Smith, N.; Freedland, K.E.; Jaffe, A.S.; Leifheit-Limson, E.C.; Sheps, D.S.; et al. Depression as a risk factor for poor prognosis among patients with acute coronary syndrome: Systematic review and recommendations: A scientific statement from the American Heart Association. Circulation 2014, 129, 1350–1369. [Google Scholar] [CrossRef] [Green Version]
- Daskalopoulou, M.; George, J.; Walters, K.; Osborn, D.P.; Batty, G.D.; Stogiannis, D.; Rapsomaniki, E.; Pujades-Rodriguez, M.; Denaxas, S.; Udumyan, R.; et al. Depression as a Risk Factor for the Initial Presentation of Twelve Cardiac, Cerebrovascular, and Peripheral Arterial Diseases: Data Linkage Study of 1.9 Million Women and Men. PLoS ONE 2016, 11, e0153838. [Google Scholar] [CrossRef]
- Harshfield, E.L.; Pennells, L.; Schwartz, J.E.; Willeit, P.; Kaptoge, S.; Bell, S.; Shaffer, J.A.; Bolton, T.; Spackman, S.; Wassertheil-Smoller, S.; et al. Emerging Risk Factors Collaboration. Association Between Depressive Symptoms and Incident Cardiovascular Diseases. JAMA 2020, 324, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.I.; Carnethon, M.R.; Matthews, K.A.; McIntyre, R.S.; Miller, G.E.; Raghuveer, G.; Stoney, C.M.; Wasiak, H.; McCrindle, B.W.; American Heart Association Atherosclerosis; et al. Major Depressive Disorder and Bipolar Disorder Predispose Youth to Accelerated Atherosclerosis and Early Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2015, 132, 965–986. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.E.; Banner, J.; Jensen, S.E. Cardiovascular disease in patients with severe mental illness. Nat. Rev. Cardiol. 2021, 18, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; von Zur Muhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Aydin Sunbul, E.; Sunbul, M.; Yanartas, O.; Cengiz, F.; Bozbay, M.; Sari, I.; Gulec, H. Increased Neutrophil/Lymphocyte Ratio in Patients with Depression is Correlated with the Severity of Depression and Cardiovascular Risk Factors. Psychiatry Investig. 2016, 13, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Moschonas, I.C.; Tselepis, A.D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis 2019, 288, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-Specific Deletion of Peptidylarginine Deiminase 4 Mitigates Atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yamada, H.; Wakana, N.; Kikai, M.; Terada, K.; Wada, N.; Motoyama, S.; Saburi, M.; Sugimoto, T.; Kami, D.; et al. Augmented neutrophil extracellular traps formation promotes atherosclerosis development in socially defeated apoE−/− mice. Biochem. Biophys. Res. Commun. 2018, 500, 490–496. [Google Scholar] [CrossRef]
- Kanwar, S.S.; Stone, G.W.; Singh, M.; Virmani, R.; Olin, J.; Akasaka, T.; Narula, J. Acute coronary syndromes without coronary plaque rupture. Nat. Rev. Cardiol. 2016, 13, 257–265. [Google Scholar] [CrossRef]
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160. [Google Scholar] [CrossRef]
- Fahed, A.C.; Jang, I.K. Plaque erosion and acute coronary syndromes: Phenotype, molecular characteristics and future directions. Nat. Rev. Cardiol. 2021, 18, 724–734. [Google Scholar] [CrossRef]
- Golden, S.A.; Covington, H.E., III; Berton, O.; Russo, S.J. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011, 6, 1183–1191. [Google Scholar] [CrossRef]
- Ménard, C.; Hodes, G.E.; Russo, S.J. Pathogenesis of depression: Insights from human and rodent studies. Neuroscience 2016, 321, 138–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berton, O.; McClung, C.A.; Dileone, R.J.; Krishnan, V.; Renthal, W.; Russo, S.J.; Graham, D.; Tsankova, N.M.; Bolanos, C.A.; Rios, M.; et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 2006, 311, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The tail suspension test. J. Vis. Exp. 2012, 59, e3769. [Google Scholar] [CrossRef] [Green Version]
- Novotny, J.; Chandraratne, S.; Weinberger, T.; Philippi, V.; Stark, K.; Ehrlich, A.; Pircher, J.; Konrad, I.; Oberdieck, P.; Titova, A.; et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE 2018, 13, e0190728. [Google Scholar] [CrossRef] [Green Version]
- Séverin, S.; Gratacap, M.P.; Lenain, N.; Alvarez, L.; Hollande, E.; Penninger, J.M.; Gachet, C.; Plantavid, M.; Payrastre, B. Deficiency of Src homology 2 domain-containing inositol 5-phosphatase 1 affects platelet responses and thrombus growth. J. Clin. Investig. 2007, 117, 944–952. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Liu, C.; Victor, A.R.; Cao, D.Y.; Veiras, L.C.; Bernstein, E.A.; Khan, Z.; Giani, J.F.; Cui, X.; Bernstein, K.E.; et al. Tumors exploit CXCR4hiCD62Llo aged neutrophils to facilitate metastatic spread. Oncoimmunology 2021, 10, 1870811. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavillet, M.; Martinod, K.; Renella, R.; Harris, C.; Shapiro, N.I.; Wagner, D.D.; Williams, D.A. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am. J. Hematol. 2015, 90, 1155–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gao, H.; Shi, C.; Erhardt, P.W.; Pavlovsky, A.; Soloviev, D.A.; Bledzka, K.; Ustinov, V.; Zhu, L.; Qin, J.; et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbα. Nat. Commun. 2017, 8, 15559. [Google Scholar] [CrossRef] [Green Version]
- Lisman, T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. 2018, 371, 567–576. [Google Scholar] [CrossRef] [Green Version]
- Longstaff, C.; Varjú, I.; Sótonyi, P.; Szabó, L.; Krumrey, M.; Hoell, A.; Bóta, A.; Varga, Z.; Komorowicz, E.; Kolev, K. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J. Biol. Chem. 2013, 288, 6946–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumaya, W.; Wallentin, L.; James, S.K.; Siegbahn, A.; Gabrysch, K.; Bertilsson, M.; Himmelmann, A.; Ajjan, R.A.; Storey, R.F. Fibrin clot properties independently predict adverse clinical outcome following acute coronary syndrome: A PLATO substudy. Eur. Heart J. 2018, 39, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Alkarithi, G.; Duval, C.; Shi, Y.; Macrae, F.L.; Ariëns, R.A.S. Thrombus Structural Composition in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef]
- Walsh, M.T.; Dinan, T.G.; Condren, R.M.; Ryan, M.; Kenny, D. Depression is associated with an increase in the expression of the platelet adhesion receptor glycoprotein Ib. Life Sci. 2002, 70, 3155–3165. [Google Scholar] [CrossRef]
- Amadio, P.; Zarà, M.; Sandrini, L.; Ieraci, A.; Barbieri, S.S. Depression and Cardiovascular Disease: The Viewpoint of Platelets. Int. J. Mol. Sci. 2020, 21, 7560. [Google Scholar] [CrossRef] [PubMed]
- Flick, M.J.; Du, X.; Witte, D.P.; Jirousková, M.; Soloviev, D.A.; Busuttil, S.J.; Plow, E.F.; Degen, J.L. Leukocyte engagement of fibrin(ogen) via the integrin receptor αMβ2/Mac-1 is critical for host inflammatory response in vivo. J. Clin. Investig. 2004, 113, 1596–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felger, J.C.; Lotrich, F.E. Inflammatory cytokines in depression: Neurobiological mechanisms and therapeutic implications. Neuroscience 2013, 246, 199–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chèvre, R.; A-González, N.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.E.; Scheiermann, C.; et al. Neutrophil ageing is regulated by the microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef]
- Uhl, B.; Vadlau, Y.; Zuchtriegel, G.; Nekolla, K.; Sharaf, K.; Gaertner, F.; Massberg, S.; Krombach, F.; Reichel, C.A. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 2016, 128, 2327–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e10. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, M.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, S.; Opneja, A.; Nayak, L. The role of neutrophils in thrombosis. Thromb. Res. 2018, 170, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Muniz, G.A.; Wood, S.K. Sex Differences in the Inflammatory Consequences of Stress: Implications for Pharmacotherapy. J. Pharmacol. Exp. Ther. 2020, 375, 161–174. [Google Scholar] [CrossRef]
- Smaardijk, V.R.; Lodder, P.; Kop, W.J.; van Gennep, B.; Maas, A.H.E.M.; Mommersteeg, P.M.C. Sex- and Gender-Stratified Risks of Psychological Factors for Incident Ischemic Heart Disease: Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2019, 8, e010859. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugimoto, T.; Yamada, H.; Wada, N.; Motoyama, S.; Saburi, M.; Kubota, H.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T.; et al. Repeated Social Defeat Exaggerates Fibrin-Rich Clot Formation by Enhancing Neutrophil Extracellular Trap Formation via Platelet–Neutrophil Interactions. Cells 2021, 10, 3344. https://doi.org/10.3390/cells10123344
Sugimoto T, Yamada H, Wada N, Motoyama S, Saburi M, Kubota H, Miyawaki D, Wakana N, Kami D, Ogata T, et al. Repeated Social Defeat Exaggerates Fibrin-Rich Clot Formation by Enhancing Neutrophil Extracellular Trap Formation via Platelet–Neutrophil Interactions. Cells. 2021; 10(12):3344. https://doi.org/10.3390/cells10123344
Chicago/Turabian StyleSugimoto, Takeshi, Hiroyuki Yamada, Naotoshi Wada, Shinichiro Motoyama, Makoto Saburi, Hiroshi Kubota, Daisuke Miyawaki, Noriyuki Wakana, Daisuke Kami, Takehiro Ogata, and et al. 2021. "Repeated Social Defeat Exaggerates Fibrin-Rich Clot Formation by Enhancing Neutrophil Extracellular Trap Formation via Platelet–Neutrophil Interactions" Cells 10, no. 12: 3344. https://doi.org/10.3390/cells10123344
APA StyleSugimoto, T., Yamada, H., Wada, N., Motoyama, S., Saburi, M., Kubota, H., Miyawaki, D., Wakana, N., Kami, D., Ogata, T., Ibi, M., & Matoba, S. (2021). Repeated Social Defeat Exaggerates Fibrin-Rich Clot Formation by Enhancing Neutrophil Extracellular Trap Formation via Platelet–Neutrophil Interactions. Cells, 10(12), 3344. https://doi.org/10.3390/cells10123344