
Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Traumatic Brain Injury
2.3. Histology
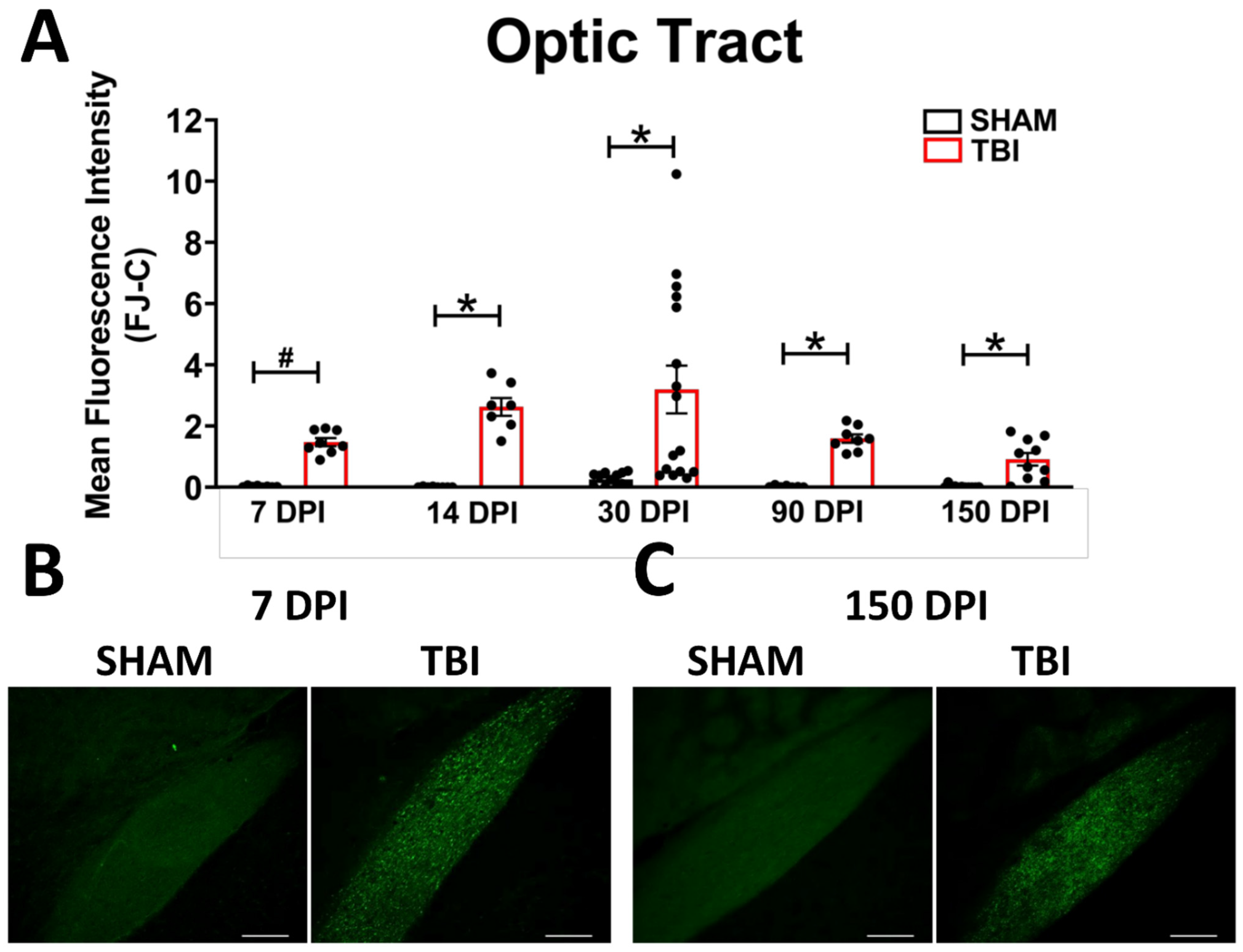
Tissue Collection
2.4. Immunofluorescence
2.5. Image Analysis
2.6. Statistical Analysis
3. Results
3.1. Weight, Righting, and Seizures
3.2. Histology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| TBI | Traumatic Brain Injury | SD | Standard deviation |
| TAI | traumatic axonal injury | OT | Optic Tract |
| iTON | indirect traumatic optic neuropathy | dLGN | dorsal lateral geniculate nucleus |
| WD | Wallerian Degeneration | vLGN | Ventral lateral geniculate nucleus |
| O2 | oxygen | SCN | suprachiasmatic nucleus |
| DPI | Days post-injury | SC | superior colliculi |
| IHC | immunohistochemical | DTN | Dorsal terminal nucleus |
| PBS | phosphate-buffered saline | MTN | medial terminal nucleus |
| FJ-C | Fluoro-Jade C | AON | accessory optomotor nucleus |
| GFAP | glial fibrillary acidic protein | EW | Edinger-Westphal nuclei |
| Iba-1 | ionized calcium-binding adaptor molecule 1 | BoSC | brachium of the superior colliculus |
| ANOVA | Analysis of variance | NOT | nucleus of the optic tract |
| SEM | Standard error of the mean | VC | visual cortex |
| ROI | Region of interest | CNS | Central nervous system |
| M | Mean |
References
- Selassie, A.W.; Zaloshnja, E.; Langlois, J.A.; Miller, T.; Jones, P.; Steiner, C. Incidence of Long-term Disability Following Traumatic Brain Injury Hospitalization, United States, 2003. J. Head Trauma Rehabil. 2008, 23, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Zaloshnja, E.; Miller, T.; Langlois, J.A.; Selassie, A.W. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J. Head Trauma Rehabil 2008, 23, 394–400. [Google Scholar] [CrossRef]
- Finkelstein, E.; Corso, P.S.; Miller, T.R. The Incidence and Economic Burden of Injuries in the United States; Oxford University Press: Oxford, UK; New York, NY, USA, 2006; p. xiii. 187p. [Google Scholar]
- Chen, C.; Peng, J.; Sribnick, E.A.; Zhu, M.; Xiang, H. Trend of Age-Adjusted Rates of Pediatric Traumatic Brain Injury in U.S. Emergency Departments from 2006 to 2013. Int. J. Environ. Res. Public Health 2018, 15, 1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashir, A.; Abebe, Z.A.; Mcinnes, K.A.; Button, E.B.; Tatarnikov, I.; Cheng, W.H.; Haber, M.; Wilkinson, A.; Barron, C.; Diaz-Arrastia, R.; et al. Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Exp. Neurol. 2020, 324, 113116. [Google Scholar] [CrossRef]
- Finnanger, T.G.; Olsen, A.; Skandsen, T.; Lydersen, S.; Vik, A.; Evensen, K.A.I.; Catroppa, C.; Håberg, A.K.; Andersson, S.; Indredavik, M.S. Life after Adolescent and Adult Moderate and Severe Traumatic Brain Injury: Self-Reported Executive, Emotional, and Behavioural Function 2–5 Years after Injury. Behav. Neurol. 2015, 2015, 329241. [Google Scholar] [CrossRef] [Green Version]
- Koliatsos, V.E.; Cernak, I.; Xu, L.; Song, Y.; Savonenko, A.; Crain, B.J.; Eberhart, C.G.; Frangakis, C.E.; Melnikova, T.; Kim, H.; et al. A Mouse Model of Blast Injury to Brain: Initial Pathological, Neuropathological, and Behavioral Characterization. J. Neuropathol. Exp. Neurol. 2011, 70, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, C.E.; Browne, K.D.; Duda, J.E.; Cullen, D.K. Neurons in Subcortical Oculomotor Regions are Vulnerable to Plasma Membrane Damage after Repetitive Diffuse Traumatic Brain Injury in Swine. J. Neurotrauma 2020, 37, 1918–1932. [Google Scholar] [CrossRef] [PubMed]
- Gennarelli, T.A.; Thibault, L.E.; Tipperman, R.; Tomei, G.; Sergot, R.; Brown, M.; Maxwell, W.L.; Graham, D.I.; Adams, J.H.; Irvine, A.; et al. Axonal injury in the optic nerve: A model simulating diffuse axonal injury in the brain. J. Neurosurg. 1989, 71, 244. [Google Scholar] [CrossRef]
- Guley, N.H.; Rogers, J.T.; Del Mar, N.A.; Deng, Y.; Islam, R.M.; D’Surney, L.; Ferrell, J.; Deng, B.; Hines-Beard, J.; Bu, W.; et al. A Novel Closed-Head Model of Mild Traumatic Brain Injury Using Focal Primary Overpressure Blast to the Cranium in Mice. J. Neurotrauma 2016, 33, 403–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu-Wai-Man, P. Traumatic optic neuropathy-Clinical features and management issues. Taiwan J. Ophthalmol. 2015, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Evanson, N.K.; Guilhaume-Correa, F.; Herman, J.P.; Goodman, M.D. Optic tract injury after closed head traumatic brain injury in mice: A model of indirect traumatic optic neuropathy. PLoS ONE 2018, 13, e0197346. [Google Scholar] [CrossRef] [Green Version]
- Guilhaume-Correa, F.; Cansler, S.M.; Shalosky, E.M.; Goodman, M.D.; Evanson, N.K. Greater neurodegeneration and behavioral deficits after single closed head traumatic brain injury in adolescent versus adult male mice. J. Neurosci. Res. 2020, 98, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Hetzer, S.M.; Guilhaume-Correa, F.; Day, D.; Bedolla, A.; Evanson, N.K. Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice. Cells 2021, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.W.; Hills, N.K.; Bakall, B.; Fernandez, B. Indirect Traumatic Optic Neuropathy in Mild Chronic Traumatic Brain Injury. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2005–2011. [Google Scholar] [CrossRef]
- Chan, J.; Tsui, E.; Peh, W.; Fong, D.; Fok, K.; Leung, K.; Yuen, M.; Fung, K. Diffuse axonal injury: Detection of changes in anisotropy of water diffusion by diffusion-weighted imaging. Neuroradiology 2003, 45, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.E.; Barres, B.A. Why Is Wallerian Degeneration in the CNS So Slow? Annu. Rev. Neurosci. 2007, 30, 153–179. [Google Scholar] [CrossRef]
- Marion, C.M.; Radomski, K.L.; Cramer, N.P.; Galdzicki, Z.; Armstrong, R.C. Experimental Traumatic Brain Injury Identifies Distinct Early and Late Phase Axonal Conduction Deficits of White Matter Pathophysiology, and Reveals Intervening Recovery. J. Neurosci 2018, 38, 8723–8736. [Google Scholar] [CrossRef]
- Arfanakis, K.; Haughton, V.M.; Carew, J.D.; Rogers, B.P.; Dempsey, R.J.; Meyerand, M.E. Diffusion Tensor MR Imaging in Diffuse Axonal Injury. Am. J. Neuroradiol. 2002, 23, 794–802. [Google Scholar]
- Wang, J.; Hamm, R.J.; Povlishock, J.T. Traumatic Axonal Injury in the Optic Nerve: Evidence for Axonal Swelling, Disconnection, Dieback, and Reorganization. J. Neurotrauma 2011, 28, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.L.Y.; Povlishock, J.T. The Effect of Traumatic Brain Injury on the Visual System: A Morphologic Characterization of Reactive Axonal Change. J. Neurotrauma 1988, 5, 47–60. [Google Scholar] [CrossRef]
- Turner, J.E.; Glaze, K.A. The early stages of Wallerian degeneration in the severed optic nerve of the newt (Triturus viridescens). Anat. Rec. 1977, 187, 291–310. [Google Scholar] [CrossRef]
- Erb, D.E.; Povlishock, J.T. Neuroplasticity following traumatic brain injury: A study of GABAergic terminal loss and recovery in the cat dorsal lateral vestibular nucleus. Exp. Brain Res. 1991, 83, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Murray, B. Nerve Injury. In Encyclopedia of the Neurological Sciences, 2nd ed.; Aminoff, M.J., Daroff, R.B., Eds.; Academic Press: Oxford, UK, 2014; pp. 333–335. [Google Scholar]
- Chen, Y.-J.; Liang, C.-M.; Tai, M.-C.; Chang, Y.-H.; Lin, T.-Y.; Chung, C.-H.; Lin, F.-H.; Tsao, C.-H.; Chien, W.-C. Longitudinal relationship between traumatic brain injury and the risk of incident optic neuropathy: A 10-year follow-up nationally representative Taiwan survey. Oncotarget 2017, 8, 86924–86933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Giolli, R.A.; Blanks, R.H.; Lui, F. The accessory optic system: Basic organization with an update on connectivity, neurochemistry, and function. Prog. Brain Res. 2006, 151, 407–440. [Google Scholar] [CrossRef] [Green Version]
- Horn, A.K.E.; Adamczyk, C. Reticular Formation: Eye Movements, Gaze and Blinks. In The Human Nervous System, 3rd ed.; Mai, J.K., Paxinos, G., Eds.; Academic Press: San Diego, CA, USA, 2012; Chapter 9; pp. 328–366. [Google Scholar]
- Kozicz, T.; Bittencourt, J.C.; May, P.J.; Reiner, A.; Gamlin, P.D.R.; Palkovits, M.; Horn, A.K.E.; Toledo, C.A.B.; Ryabinin, A.E. The Edinger-Westphal nucleus: A historical, structural, and functional perspective on a dichotomous terminology. J. Comp. Neurol. 2011, 519, 1413–1434. [Google Scholar] [CrossRef] [Green Version]
- Morin, L.P.; Studholme, K.M. Retinofugal projections in the mouse. J. Comp. Neurol 2014, 522, 3733–3753. [Google Scholar] [CrossRef] [Green Version]
- Zubricky, R.D.; Das, J.M. Neuroanatomy, Superior Colliculus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Schröder, H.; Moser, N.; Huggenberger, S. The Mouse Caudate Putamen, Motor System, and Nucleus Accumbens. In Neuroanatomy of the Mouse: An Introduction; Springer International Publishing: Cham, Switzerland, 2020; pp. 305–318. [Google Scholar]
- Seger, C. The visual corticostriatal loop through the tail of the caudate: Circuitry and function. Front. Syst. Neurosci. 2013, 7. [Google Scholar] [CrossRef] [Green Version]
- Yakushin, S.B.; Gizzi, M.; Reisine, H.; Raphan, T.; Büttner-Ennever, J.; Cohen, B. Functions of the nucleus of the optic tract (NOT). II. Control of ocular pursuit. Exp. Brain Res. 2000, 131, 433–447. [Google Scholar] [CrossRef]
- Cansler, S.M.; Guilhaume-Correa, F.; Day, D.; Bedolla, A.; Evanson, N.K. Indirect traumatic optic neuropathy after head trauma in adolescent male mice is associated with behavioral visual impairment, neurodegeneration, and elevated endoplasmic reticulum stress markers at acute and subacute times. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carroll, S.L.; Worley, S.H. Wallerian Degeneration. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Parhamifar, L.; Andersen, H.; Wu, L.; Hall, A.; Hudzech, D.; Moghimi, S.M. Polycation-mediated integrated cell death processes. Adv. Genet. 2014, 88, 353–398. [Google Scholar] [CrossRef]
- Kossmann, T.; Freedman, I.; Morganti-Kossmann, C. SPINAL TRAUMA. In Neurology and Clinical Neuroscience; Schapira, A.H.V., Byrne, E., DiMauro, S., Frackowiak, R.S.J., Johnson, R.T., Mizuno, Y., Samuels, M.A., Silberstein, S.D., Wszolek, Z.K., Eds.; Mosby: Philadelphia, PA, USA, 2007; Chapter 104; pp. 1397–1408. [Google Scholar]
- Maxwell, W.L. Damage to Myelin and Oligodendrocytes: A Role in Chronic Outcomes Following Traumatic Brain Injury? Brain Sci. 2013, 3, 1374–1394. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fox, M.A.; Povlishock, J.T. Diffuse Traumatic Axonal Injury in the Optic Nerve Does Not Elicit Retinal Ganglion Cell Loss. J. Neuropathol. Exp. Neurol. 2013, 72, 768–781. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Dvoriantchikova, G.; Tse, B.C.; Pappas, S.; Chou, T.H.; Tapia, M.; Porciatti, V.; Ivanov, D.; Tse, D.T.; Pelaez, D. A Novel Mouse Model of Traumatic Optic Neuropathy Using External Ultrasound Energy to Achieve Focal, Indirect Optic Nerve Injury. Sci. Rep. 2017, 7, 11779. [Google Scholar] [CrossRef]
- Boehme, N.A.; Hedberg-Buenz, A.; Tatro, N.; Bielecki, M.; Castonguay, W.C.; Scheetz, T.E.; Anderson, M.G.; Dutca, L.M. Axonopathy precedes cell death in ocular damage mediated by blast exposure. Sci. Rep. 2021, 11, 11774. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Colón, A.; Vest, V.; Clark, A.; Cooper, M.L.; Calkins, D.J.; Harrison, F.E.; Rex, T.S. Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, W.L.; Islam, M.N.; Graham, D.I.; Gennarelli, T.A. A qualitative and quantitative analysis of the response of the retinal ganglion cell soma after stretch injury to the adult guinea-pig optic nerve. J. Neurocytol. 1994, 23, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, P.; Zhou, D.; Zhang, J.; Xu, X.; Tang, L. Intravitreal Transplantation of Human Umbilical Cord Blood Stem Cells Protects Rats from Traumatic Optic Neuropathy. PLoS ONE 2013, 8, e69938. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.K.-S.; Weinreb, R.N.; Li, Z.W.; Liu, S.; Lindsey, J.D.; Choi, N.; Liu, L.; Cheung, C.Y.-l.; Ye, C.; Qiu, K.; et al. Long-Term In Vivo Imaging and Measurement of Dendritic Shrinkage of Retinal Ganglion Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1539–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, J.E.; Pease, D.C. Electron microscopic studies of Wallerian degeneration in rat optic nerves. II. Astrocytes, oligodendrocytes and adventitial cells. J. Comp. Neurol. 1970, 140, 207–225. [Google Scholar] [CrossRef]
- Lee, S.Y.; Chung, W.S. The roles of astrocytic phagocytosis in maintaining homeostasis of brains. J. Pharm. Sci 2021, 145, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Oguin, T.H.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzy, S.; Liu, Q.; Fang, Z.; Lule, S.; Wu, L.; Chung, J.Y.; Sarro-Schwartz, A.; Brown-Whalen, A.; Perner, C.; Hickman, S.E.; et al. Time-Dependent Changes in Microglia Transcriptional Networks Following Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13, 307. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.; Mihaila, D.M. Neuroanatomy, Pupillary Light Reflexes and Pathway. Available online: https://www.ncbi.nlm.nih.gov/books/NBK553169/ (accessed on 15 July 2021).
- Baver, S.B.; Pickard, G.E.; Sollars, P.J.; Pickard, G.E. Two types of melanopsin retinal ganglion cell differentially innervate the hypothalamic suprachiasmatic nucleus and the olivary pretectal nucleus. Eur. J. Neurosci. 2008, 27, 1763–1770. [Google Scholar] [CrossRef]
- Hill, C.S.; Coleman, M.P.; Menon, D.K. Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci 2016, 39, 311–324. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhang, J.; Hu, X.; Zhang, L.; Mao, L.; Jiang, X.; Liou, A.K.; Leak, R.K.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J. Cereb. Blood Flow Metab. 2013, 33, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Karve, I.P.; Taylor, J.M.; Crack, P.J. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharm. 2016, 173, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Cekanaviciute, E.; Fathali, N.; Doyle, K.P.; Williams, A.M.; Han, J.; Buckwalter, M.S. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia 2014, 62, 1227–1240. [Google Scholar] [CrossRef] [Green Version]
- Lööv, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 2012, 7, e33090. [Google Scholar] [CrossRef] [Green Version]
- Cekanaviciute, E.; Buckwalter, M.S. Astrocytes: Integrative Regulators of Neuroinflammation in Stroke and Other Neurological Diseases. Neurotherapeutics 2016, 13, 685–701. [Google Scholar] [CrossRef]
- Huang, X.; You, W.; Zhu, Y.; Xu, K.; Yang, X.; Wen, L. Microglia: A Potential Drug Target for Traumatic Axonal Injury. Neural Plast. 2021, 2021, 5554824. [Google Scholar] [CrossRef]
- Wylot, B.; Mieczkowski, J.; Niedziolka, S.; Kaminska, B.; Zawadzka, M. Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination. Cells 2019, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.C.; Jurgens, C.W.D.; Krahe, T.E.; Povlishock, J.T. Adaptive reorganization of retinogeniculate axon terminals in dorsal lateral geniculate nucleus following experimental mild traumatic brain injury. Exp. Neurol. 2017, 289, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimbler, D.E.; Shields, J.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. Activation of P2X7 Promotes Cerebral Edema and Neurological Injury after Traumatic Brain Injury in Mice. PLoS ONE 2012, 7, e41229. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Cui, Y.; Cui, J.Z.; Sun, L.Q.; Cui, C.M.; Zhang, H.A.; Zhu, H.X.; Li, R.; Tian, Y.X.; Gao, J.L. Neuroprotective effects of brilliant blue G on the brain following traumatic brain injury in rats. Mol. Med. Rep. 2015, 12, 2149–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povlishock, J.T.; Kontos, H.A. Continuing Axonal and Vascular Change Following Experimental Brain Trauma. Cent. Nerv. Syst. Trauma 1985, 2, 285–298. [Google Scholar] [CrossRef]
- Wei, E.P.; Dietrich, W.D.; Povlishock, J.T.; Navari, R.M.; Kontos, H.A. Functional, morphological, and metabolic abnormalities of the cerebral microcirculation after concussive brain injury in cats. Circ. Res. 1980, 46, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama Sosa, M.A.; De Gasperi, R.; Janssen, P.L.; Yuk, F.J.; Anazodo, P.C.; Pricop, P.E.; Paulino, A.J.; Wicinski, B.; Shaughness, M.C.; Maudlin-Jeronimo, E.; et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol. Commun. 2014, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Steinman, J.; Cahill, L.S.; Koletar, M.M.; Stefanovic, B.; Sled, J.G. Acute and chronic stage adaptations of vascular architecture and cerebral blood flow in a mouse model of TBI. NeuroImage 2019, 202, 116101. [Google Scholar] [CrossRef] [PubMed]
- Flierl, M.A.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse closed head injury model induced by a weight-drop device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, U.A.; Zeng, Y.; Deyo, D.; Parsley, M.A.; Hawkins, B.E.; Prough, D.S.; DeWitt, D.S. Effects of Mild Blast Traumatic Brain Injury on Cerebral Vascular, Histopathological, and Behavioral Outcomes in Rats. J. Neurotrauma 2017, 35, 375–392. [Google Scholar] [CrossRef] [PubMed]
- McConnell, H.L.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | RRID | Host Species | Immunogen | Manufacturer | Concentration |
|---|---|---|---|---|---|
| GFAP | AB_10013382 | Rabbit | Whole bovine GFAP, isolated from spinal cord. | DAKO (Agilent) CAT: Z033401-2 Lot: 00059585 | 1:2000 |
| Iba-1 | AB_10641962 | Rabbit | Peptide from the c-terminal sequence from rat Iba-1: PTGPPAK KAISELP | Synaptic Systems CAT: 234003 Lot: 234003/9 | 1:2000 |
| Cy3 AffiniPure Donkey anti-Rabbit IgG (H + L) conjugated secondary | AB_2307443 | Donkey | Gamma immunoglobulins, heavy and light chains | Jackson Immuno Research CAT: 711-165-152 Lot: 79424 | 1:500 |
| Time Point | OT | p | dLGN | p | vLGN | p | SC | p |
| 7 DPI | t(14) = −16.94 | p < 0.001 | t(14) = −4.04 | p < 0.001 | t(14) = −5.17 | p < 0.001 | t(11) = −12.46 | p < 0.001 |
| 14 DPI | t(13) = −11.08 | p < 0.001 | U(13) = 0 | p < 0.001 | U(13) = 0 | p < 0.001 | U(13) = 0 | p < 0.001 |
| 30 DPI | U(26) = 18 | p < 0.001 | t(14) =−4.78 | p < 0.001 | U(25) = −39.5 | p = 0.02 | t(26) = −4.12 | p < 0.001 |
| 90 DPI | t(13) = −16.57 | p < 0.001 | t(14) = −0.77 | p = 0.05 | t(13) = −2.47 | p = 0.03 | t(12) = −6.21 | p < 0.001 |
| 150 DPI | t(19) = 6.74 | p < 0.001 | U(15) = 26 | p < 0.001 | t(15) = 0.79 | p = 0.44 | t(15) = −5.60 | p = 0.002 |
| DTN | p | MTN | p | BoSC | p | NOT | p | |
| 7 DPI | U(15) = 0.0 | p = 0.004 | t(14) = −3.44 | p = 0.004 | t(14) = −4.69 | p < 0.001 | U(13) = 22 | p = 0.54 |
| 14 DPI | U(14) = 0 | p < 0.001 | U(12) = 0 | p = 0.001 | t(13) = −4.00 | p = 0.001 | t(13) = −3.27 | p = 0.006 |
| 30 DPI | U(15) = 4 | p = 0.002 | U(15) = 2 | p < 0.001 | U(13) = 14 | p = 0.12 | U(24) = 43 | p = 0.04 |
| 90 DPI | t(13) = −4.95 | p < 0.001 | U(14) = 0 | p < 0.001 | t(12) = −5.09 | p < 0.001 | t(13) = −3.28 | p = 0.006 |
| 150 DPI | t(14) = 0.02 | p = 0.02 | U(17) = 38 | p = 0.89 | t(17) = −5.76 | p < 0.001 | U(19) = 19 | p = 0.02 |
| Time Point | OT | p | dLGN | p | vLGN | p | SCN | p |
| 7 DPI | t(14) = 5.27 | p < 0.001 | t(14) = −1.56 | p = 0.14 | t(14) = 0.12 | p = 0.90 | t(13) = 1.02 | p = 0.33 |
| 14 DPI | t(13) = −2.65 | p = 0.02 | t(13) = 1.13 | p = 0.28 | t(13) = 1.25 | p = 0.23 | t(13) = 1.11 | p = 0.29 |
| 30 DPI | t(14) = 2.07 | p = 0.07 | t(14) = −4.00 | p = 0.001 | t(14) = −3.64 | p = 0.003 | t(13) = −2.28 | p = 0.25 |
| 90 DPI | t(14) = −3.91 | p = 0.002 | t(14) = −1.76 | p = 0.10 | t(14) = −1.09 | p = 0.43 | t(14) = 1.78 | p = 0.10 |
| 150 DPI | t(17) = −4.49 | p < 0.001 | t(18) = 2.51 | p = 0.02 | t(18) = 3.71 | p = 0.002 | t(18) = 2.51 | p = 0.02 |
| SC | p | DTN | p | MTN | p | BoSC | p | |
| 7 DPI | t(13) = −4.12 | p = 0.001 | t(14) = −3.68 | p = 0.002 | t(14) = −3.03 | p = 0.008 | t(14) = −1.43 | p = 0.17 |
| 14 DPI | t(13) = −1.41 | p = 0.18 | t(13) = 0.13 | p = 0.90 | t(13) = 0.95 | p = 0.36 | t(13) = 0.54 | p = 0.02 |
| 30 DPI | t(13) = −3.81 | p = 0.002 | U(14) = 11 | p = 0.03 | U(13) = 21 | p = 0.46 | t(14) = 2.56 | p = 0.59 |
| 90 DPI | t(14) = 2.83 | p = 0.1 | t(14) = −6.63 | p < 0.001 | t(14) = −2.20 | p = 0.04 | t(13) = −2.89 | p = 0.01 |
| 150 DPI | t(18) = −4.51 | p = 0.002 | t(18) = 4.52 | p < 0.001 | t(18) = 5.12 | p < 0.001 | t(18) = 2.40 | p = 0.03 |
| NOT | p | VC | p | SNpr | p | CPu | p | |
| 7 DPI | t(14) = −2.94 | p = 0.01 | t(14) = 0.76 | p = 0.47 | t(14) = −0.69 | p = 0.50 | t(14) = −1.24 | p = 0.23 |
| 14 DPI | t(13) = −0.46 | p = 0.66 | t(13) = −0.74 | p = 0.47 | t(13) = 0.43 | p = 0.67 | t(13) = 0.34 | p = 0.74 |
| 30 DPI | t(14) = 2.56 | p = 0.02 | t(13) = −2.28 | p = 0.04 | t(14) = 1.25 | p = 0.23 | t(14) = 0.98 | p = 0.34 |
| 90 DPI | t(14) = −1.94 | p = 0.07 | t(12) = −0.82 | p = 0.42 | t(13) = −3.12 | p = 0.008 | t(13) = −2.85 | p = 0.01 |
| 150 DPI | t(18) = 1.27 | p = 0.22 | t(18) = 2.32 | p = 0.03 | t(18) = 2.16 | p = 0.04 | t(18) = 2.99 | p = 0.007 |
| Time Point | OT | p | dLGN | p | vLGN | p | SCN | p | SC | p |
| 7 DPI | t(13) = −3.4 | # p = 0.005 | U(13) = 22 | p = 0.33 | t(14) = −0.86 | p = 0.486 | t(14) = −0.16 | p = 0.87 | t(13) = 0.62 | p = 0.55 |
| 14 DPI | t(12) = −2.69 | # p = 0.02 | t(13) = −0.78 | p = 0.45 | t(12) = −0.89 | p = 0.39 | t(13) = 2.89 | # p = 0.01 | t(13) = 0.01 | p = 0.90 |
| 30 DPI | t(11) = −3.04 | # p = 0.01 | t(14) = −0.58 | p = 0.58 | t(14) = −2.42 | # p = 0.03 | t(13) = −0.08 | p = 0.94 | t(13) = −1.29 | p = 0.18 |
| 90 DPI | t(14) = −4.19 | * p < 0.001 | t(14) = −0.40 | p = 0.57 | t(14) = −2.50, | # p = 0.03 | t(13) = 0.78 | p = 0.45 | t(14) = −0.8 | p = 0.44 |
| 150 DPI | t(18) = −3.45 | # p = 0.003 | t(18) = −0.44 | p = 0.70 | t(18) = 1.45 | p = 0.16 | t(18) = −0.33 | p = 0.74 | t(15) = −3.78 | # p = 0.002 |
| DTN | p | MTN | p | EW | p | AON | p | BoSC | p | |
| 7 DPI | t(13) = −3.26 | # p = 0.006 | t(13) = −0.68 | p = 0.51 | U(13) = 19 | p = 0.34 | t(13) = −1.62 | p = 0.13 | t(14) = −1.65 | p = 0.12 |
| 14 DPI | t(13) = −4.99 | * p < 0.001 | t(13) = 1.26 | p = 0.23 | t(13) = 0.03 | p = 0.97 | t(13) = 1.23 | p = 0.24 | t(13) = −2.4 | # p = 0.03 |
| 30 DPI | t(13) = 7.79 | * p < 0.001 | t(11) = −1.85 | p = 0.09 | t(10) = −0.21 | p = 0.84 | T(13) = −2.09 | p = 0.06 | t(22) = 1.80 | p = 0.08 |
| 90 DPI | t(13) = −3.26 | # p = 0.006 | t(13) = −0.68 | p = 0.51 | U(13) = 19 | p = 0.34 | t(13) = −1.62 | p = 0.13 | t(8) = −1.76 | p = 0.06 |
| 150 DPI | U(18) = 16 | # p = 0.01 | U(18) = 37 | p = 0.35 | t(16) = 1.32 | p = 0.20 | t(18) = −0.59 | p = 0.57 | t(17) = 3.1 | # p = 0.006 |
| NOT | p | VC | p | SNpr | p | CPu | p | |||
| 7 DPI | t(14) = −1.33 | p = 0.21 | t(14) = −0.26 | p = 0.80 | U(14) = 22 | p = 0.33 | t(13) = −0.25 | p = 0.8 | ||
| 14 DPI | t(13) = −1.19 | p = 0.47 | t(13) = −0.96 | p = 0.35 | t(13) = −0.32 | p = 0.75 | t(13) = −0.21 | p = 0.83 | ||
| 30 DPI | t(18) = −0.49 | p = 0.63 | U = 42 | p = 0.26 | t(12) = −0.26 | p = 0.79 | t(14) = −1.03 | p = 0.32 | ||
| 90 DPI | t(7) = 0.81 | p = 0.36 | t(14) = −0.48 | p = 0.26 | U(14) = 22 | p = 0.33 | t(13) = −2.44 | # p = 0.03 | ||
| 150 DPI | t(17) = 0.23 | p = 0.82 | t(18) = 0.07 | p = 0.65 | t(18) = −1.38 | p = 0.18 | t(17) = 0.37 | p = 0.72 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hetzer, S.M.; Shalosky, E.M.; Torrens, J.N.; Evanson, N.K. Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses. Cells 2021, 10, 3343. https://doi.org/10.3390/cells10123343
Hetzer SM, Shalosky EM, Torrens JN, Evanson NK. Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses. Cells. 2021; 10(12):3343. https://doi.org/10.3390/cells10123343
Chicago/Turabian StyleHetzer, Shelby M., Emily M. Shalosky, Jordyn N. Torrens, and Nathan K. Evanson. 2021. "Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses" Cells 10, no. 12: 3343. https://doi.org/10.3390/cells10123343
APA StyleHetzer, S. M., Shalosky, E. M., Torrens, J. N., & Evanson, N. K. (2021). Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses. Cells, 10(12), 3343. https://doi.org/10.3390/cells10123343