
Contribution of Human Pluripotent Stem Cell-Based Models to Drug Discovery for Neurological Disorders

{kind=link}
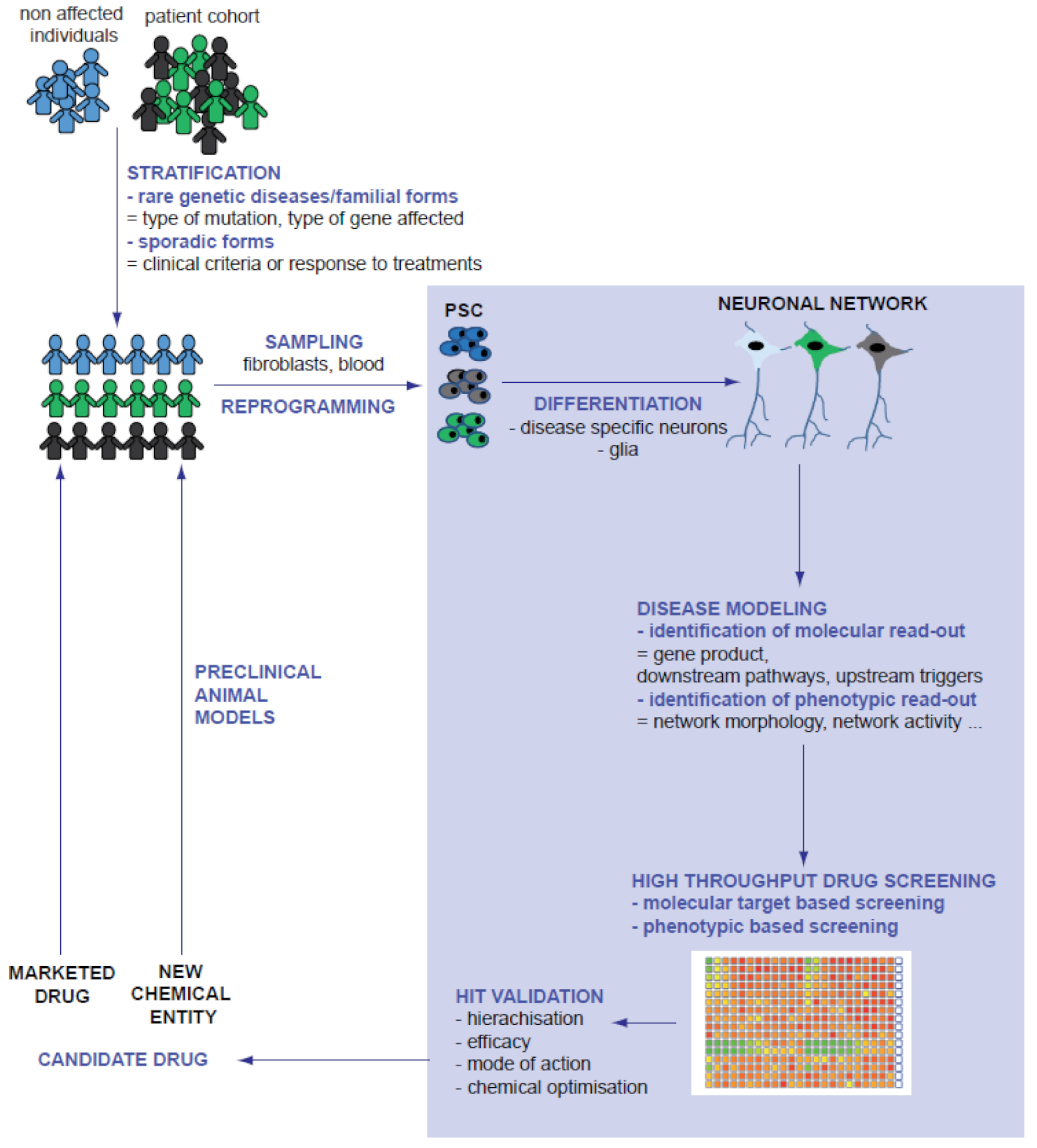{kind=link}
Abstract
1. Introduction
2. Integration of PSC-Derived Models in The Process of Drug Discovery
2.1. Strategies for Drug Discovery
2.2. Pluripotent Stem Cells as Biological Material
3. Paving the Way: Rare Genetic Diseases
3.1. Fragile-X Syndrome (FXS)
3.2. Duplication of a Segment of Chromosome 7 (7Dup)
3.3. Metabolic Disorders
3.4. Cyclin-Dependent Kinase-Like 5 (CDKL5) Deficiency
3.5. Phelan–McDermid Syndrome (PMS)
4. Neurodegenerative Diseases
4.1. Alzheimer’s Disease
4.2. Parkinson’s Disease
4.3. Screenings for Compounds Targeting Several Diseases
5. Psychiatric Disorders
5.1. Strategies Based upon Modulation of Neurogenesis
5.2. Precision Medicine
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef]
- Zecevic, N.; Chen, Y.; Filipovic, R. Contributions of cortical subventricular zone to the development of the human cerebral cortex. J. Comp. Neurol. 2005, 491, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Fietz, S.A.; Kelava, I.; Vogt, J.; Wilsch-Brauninger, M.; Stenzel, D.; Fish, J.L.; Corbeil, D.; Riehn, A.; Distler, W.; Nitsch, R.; et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat. Neurosci. 2010, 13, 690–699. [Google Scholar] [CrossRef]
- Hansen, D.V.; Lui, J.H.; Parker, P.R.; Kriegstein, A.R. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 2010, 464, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Fietz, S.A.; Huttner, W.B. Cortical progenitor expansion, self-renewal and neurogenesis-a polarized perspective. Curr. Opin. Neurobiol. 2011, 21, 23–35. [Google Scholar] [CrossRef]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Cheng, L.; Lei, Q.; Yin, C.; Wang, H.Y.; Jin, K.; Xiang, M. Generation of Urine Cell-Derived Non-integrative Human iPSCs and iNSCs: A Step-by-Step Optimized Protocol. Front. Mol. Neurosci. 2017, 10, 348. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. Induced Pluripotent Stem Cell Generation from Blood Cells Using Sendai Virus and Centrifugation. J. Vis. Exp. JoVE 2016. [Google Scholar] [CrossRef]
- Wang, J.; Gu, Q.; Hao, J.; Bai, D.; Liu, L.; Zhao, X.; Liu, Z.; Wang, L.; Zhou, Q. Generation of induced pluripotent stem cells with high efficiency from human umbilical cord blood mononuclear cells. Genom. Proteom. Bioinform. 2013, 11, 304–311. [Google Scholar] [CrossRef]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Drug discovery: Selecting the optimal approach. Drug Discov. Today 2006, 11, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Bellomo, F.; Medina, D.L.; De Leo, E.; Panarella, A.; Emma, F. High-content drug screening for rare diseases. J. Inherit. Metab. Dis. 2017, 40, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Sapiens, M.A.; Reza-Zaldivar, E.E.; Cevallos, R.R.; Marquez-Aguirre, A.L.; Gazarian, K.; Canales-Aguirre, A.A. A Three-Dimensional Alzheimer’s Disease Cell Culture Model Using iPSC-Derived Neurons Carrying A246E Mutation in PSEN1. Front. Cell. Neurosci. 2020, 14, 151. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Sobue, A.; Kushima, I.; Wang, C.; Arioka, Y.; Kato, H.; Kodama, A.; Kubo, H.; Ito, N.; Sawahata, M.; et al. ARHGAP10, which encodes Rho GTPase-activating protein 10, is a novel gene for schizophrenia risk. Transl. Psychiatry 2020, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Ishikawa, K.I.; Inoshita, T.; Shiba-Fukushima, K.; Saiki, S.; Hatano, T.; Mori, A.; Oji, Y.; Okuzumi, A.; Li, Y.; et al. Identifying Therapeutic Agents for Amelioration of Mitochondrial Clearance Disorder in Neurons of Familial Parkinson Disease. Stem Cell Rep. 2020, 14, 1060–1075. [Google Scholar] [CrossRef]
- Fotis, C.; Antoranz, A.; Hatziavramidis, D.; Sakellaropoulos, T.; Alexopoulos, L.G. Network-based technologies for early drug discovery. Drug Discov. Today 2018, 23, 626–635. [Google Scholar] [CrossRef]
- Caudle, W.M.; Bammler, T.K.; Lin, Y.; Pan, S.; Zhang, J. Using ‘omics’ to define pathogenesis and biomarkers of Parkinson’s disease. Expert Rev. Neurother. 2010, 10, 925–942. [Google Scholar] [CrossRef]
- Lang, C.; Campbell, K.R.; Ryan, B.J.; Carling, P.; Attar, M.; Vowles, J.; Perestenko, O.V.; Bowden, R.; Baig, F.; Kasten, M.; et al. Single-Cell Sequencing of iPSC-Dopamine Neurons Reconstructs Disease Progression and Identifies HDAC4 as a Regulator of Parkinson Cell Phenotypes. Cell Stem Cell 2019, 24, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.J.; Cline, S.G.; Hinds, D.; Jahanbakhsh, S.; Prokop, J.W. Breakdown of multiple sclerosis genetics to identify an integrated disease network and potential variant mechanisms. Physiol. Genom. 2019, 51, 562–577. [Google Scholar] [CrossRef]
- Aldosary, M.; Al-Bakheet, A.; Al-Dhalaan, H.; Almass, R.; Alsagob, M.; Al-Younes, B.; AlQuait, L.; Mustafa, O.M.; Bulbul, M.; Rahbeeni, Z.; et al. Rett Syndrome, a Neurodevelopmental Disorder, Whole-Transcriptome, and Mitochondrial Genome Multiomics Analyses Identify Novel Variations and Disease Pathways. Omics A J. Integr. Biol. 2020, 24, 160–171. [Google Scholar] [CrossRef]
- Neul, J.L.; Skinner, S.A.; Annese, F.; Lane, J.; Heydemann, P.; Jones, M.; Kaufmann, W.E.; Glaze, D.G.; Percy, A.K. Metabolic Signatures Differentiate Rett Syndrome From Unaffected Siblings. Front. Integr. Neurosci. 2020, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Santiago, R.; Carballo-Carbajal, I.; Castellano, G.; Torrent, R.; Richaud, Y.; Sanchez-Danes, A.; Vilarrasa-Blasi, R.; Sanchez-Pla, A.; Mosquera, J.L.; Soriano, J.; et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 2015, 7, 1529–1546. [Google Scholar] [CrossRef]
- Focking, M.; Doyle, B.; Munawar, N.; Dillon, E.T.; Cotter, D.; Cagney, G. Epigenetic Factors in Schizophrenia: Mechanisms and Experimental Approaches. Mol. Neuropsychiatry 2019, 5, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.K.; Saykin, A.J. Pathways to neurodegeneration: Mechanistic insights from GWAS in Alzheimer’s disease, Parkinson’s disease, and related disorders. Am. J. Neurodegener. Dis. 2013, 2, 145–175. [Google Scholar]
- Sadlon, A.; Takousis, P.; Alexopoulos, P.; Evangelou, E.; Prokopenko, I.; Perneczky, R. miRNAs Identify Shared Pathways in Alzheimer’s and Parkinson’s Diseases. Trends Mol. Med. 2019, 25, 662–672. [Google Scholar] [CrossRef]
- Szatmari, B.; Balicza, P.; Nemeth, G.; Molnar, M.J. The Panomics Approach in Neurodegenerative Disorders. Curr. Med. Chem. 2019, 26, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Varma, H.; Lo, D.C.; Stockwell, B.R. High throughput screening for neurodegeneration and complex disease phenotypes. Comb. Chem. High Throughput Screen. 2008, 11, 238–248. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Hwang, E.; Cutler, R.W.; Lee, H.C.; Ko, L.W.; Huang, H.L.; Ho, S.Y. HCS-Neurons: Identifying phenotypic changes in multi-neuron images upon drug treatments of high-content screening. BMC Bioinform. 2013, 14 (Suppl. 16), S12. [Google Scholar] [CrossRef]
- Cooper, D.J.; Zunino, G.; Bixby, J.L.; Lemmon, V.P. Phenotypic screening with primary neurons to identify drug targets for regeneration and degeneration. Mol. Cell. Neurosci. 2017, 80, 161–169. [Google Scholar] [CrossRef][Green Version]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. Selective optimization of side activities: Another way for drug discovery. J. Med. Chem. 2004, 47, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef]
- Schein, C.H. Repurposing approved drugs on the pathway to novel therapies. Med. Res. Rev. 2020, 40, 586–605. [Google Scholar] [CrossRef]
- Roessler, H.I.; Knoers, N.; van Haelst, M.M.; van Haaften, G. Drug Repurposing for Rare Diseases. Trends Pharmacol. Sci. 2021, 42, 255–267. [Google Scholar] [CrossRef]
- Scherman, D.; Fetro, C. Drug repositioning for rare diseases: Knowledge-based success stories. Therapie 2020, 75, 161–167. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef]
- Boissart, C.; Nissan, X.; Giraud-Triboult, K.; Peschanski, M.; Benchoua, A. miR-125 potentiates early neural specification of human embryonic stem cells. Development 2012, 139, 1247–1257. [Google Scholar] [CrossRef]
- Tao, Y.; Zhang, S.C. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell 2016, 19, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Kirkeby, A.; Nelander, J.; Parmar, M. Generating regionalized neuronal cells from pluripotency, a step-by-step protocol. Front. Cell. Neurosci. 2012, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Borghese, L.; Dolezalova, D.; Opitz, T.; Haupt, S.; Leinhaas, A.; Steinfarz, B.; Koch, P.; Edenhofer, F.; Hampl, A.; Brustle, O. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells 2010, 28, 955–964. [Google Scholar] [CrossRef]
- McComish, S.F.; Caldwell, M.A. Generation of defined neural populations from pluripotent stem cells. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2018, 373, 20170214. [Google Scholar] [CrossRef]
- Silva, M.C.; Haggarty, S.J. Human pluripotent stem cell-derived models and drug screening in CNS precision medicine. Ann. New York Acad. Sci. 2020, 1471, 18–56. [Google Scholar] [CrossRef]
- Sarkar, A.; Mei, A.; Paquola, A.C.M.; Stern, S.; Bardy, C.; Klug, J.R.; Kim, S.; Neshat, N.; Kim, H.J.; Ku, M.; et al. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell 2018, 22, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, U.; Gross, A.R.; Hjelm, B.E.; Sequeira, A.; Vawter, M.P.; Tang, J.; Gangalapudi, V.; Wang, Y.; Andres, A.M.; Gottlieb, R.A.; et al. Super-Obese Patient-Derived iPSC Hypothalamic Neurons Exhibit Obesogenic Signatures and Hormone Responses. Cell Stem Cell 2018, 22, 698–712. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Sauvey, C.; Yao, L.; Zarnowska, E.D.; Zhang, S.C. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat. Protoc. 2013, 8, 1670–1679. [Google Scholar] [CrossRef]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013, 12, 559–572. [Google Scholar] [CrossRef]
- Kumar, M.; Kaushalya, S.K.; Gressens, P.; Maiti, S.; Mani, S. Optimized derivation and functional characterization of 5-HT neurons from human embryonic stem cells. Stem Cells Dev. 2009, 18, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhong, X.; Liu, H.; Hao, L.; Huang, C.T.; Sherafat, M.A.; Jones, J.; Ayala, M.; Li, L.; Zhang, S.C. Generation of serotonin neurons from human pluripotent stem cells. Nat. Biotechnol. 2016, 34, 89–94. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Boissart, C.; Poulet, A.; Georges, P.; Darville, H.; Julita, E.; Delorme, R.; Bourgeron, T.; Peschanski, M.; Benchoua, A. Differentiation from human pluripotent stem cells of cortical neurons of the superficial layers amenable to psychiatric disease modeling and high-throughput drug screening. Transl. Psychiatry 2013, 3, e294. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, X.J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef]
- Maury, Y.; Come, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Sances, S.; Bruijn, L.I.; Chandran, S.; Eggan, K.; Ho, R.; Klim, J.R.; Livesey, M.R.; Lowry, E.; Macklis, J.D.; Rushton, D.; et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 2016, 19, 542–553. [Google Scholar] [CrossRef]
- Bradley, R.A.; Shireman, J.; McFalls, C.; Choi, J.; Canfield, S.G.; Dong, Y.; Liu, K.; Lisota, B.; Jones, J.R.; Petersen, A.; et al. Regionally specified human pluripotent stem cell-derived astrocytes exhibit different molecular signatures and functional properties. Development 2019, 146, dev170910. [Google Scholar] [CrossRef] [PubMed]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef]
- Canals, I.; Ginisty, A.; Quist, E.; Timmerman, R.; Fritze, J.; Miskinyte, G.; Monni, E.; Hansen, M.G.; Hidalgo, I.; Bryder, D.; et al. Rapid and efficient induction of functional astrocytes from human pluripotent stem cells. Nat. Methods 2018, 15, 693–696. [Google Scholar] [CrossRef]
- Emdad, L.; D’Souza, S.L.; Kothari, H.P.; Qadeer, Z.A.; Germano, I.M. Efficient differentiation of human embryonic and induced pluripotent stem cells into functional astrocytes. Stem Cells Dev. 2012, 21, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Roybon, L.; Lamas, N.J.; Garcia, A.D.; Yang, E.J.; Sattler, R.; Lewis, V.J.; Kim, Y.A.; Kachel, C.A.; Rothstein, J.D.; Przedborski, S.; et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013, 4, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Gorris, R.; Fischer, J.; Erwes, K.L.; Kesavan, J.; Peterson, D.A.; Alexander, M.; Nothen, M.M.; Peitz, M.; Quandel, T.; Karus, M.; et al. Pluripotent stem cell-derived radial glia-like cells as stable intermediate for efficient generation of human oligodendrocytes. Glia 2015, 63, 2152–2167. [Google Scholar] [CrossRef]
- Wang, S.; Bates, J.; Li, X.; Schanz, S.; Chandler-Militello, D.; Levine, C.; Maherali, N.; Studer, L.; Hochedlinger, K.; Windrem, M.; et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 2013, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Major, T.; Powers, A.; Tabar, V. Derivation of telencephalic oligodendrocyte progenitors from human pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2017, 39, 1H1011–1H1023. [Google Scholar] [CrossRef]
- Espinosa-Jeffrey, A.; Blanchi, B.; Biancotti, J.C.; Kumar, S.; Hirose, M.; Mandefro, B.; Talavera-Adame, D.; Benvenisty, N.; de Vellis, J. Efficient Generation of Viral and Integration-Free Human Induced Pluripotent Stem Cell-Derived Oligodendrocytes. Curr. Protoc. Stem Cell Biol. 2016, 39, 2D1811–2D1828. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293. [Google Scholar] [CrossRef]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef]
- Mancuso, R.; Van Den Daele, J.; Fattorelli, N.; Wolfs, L.; Balusu, S.; Burton, O.; Liston, A.; Sierksma, A.; Fourne, Y.; Poovathingal, S.; et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019, 22, 2111–2116. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Munoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef]
- Russo, F.B.; Freitas, B.C.; Pignatari, G.C.; Fernandes, I.R.; Sebat, J.; Muotri, A.R.; Beltrao-Braga, P.C.B. Modeling the Interplay Between Neurons and Astrocytes in Autism Using Human Induced Pluripotent Stem Cells. Biol. Psychiatry 2018, 83, 569–578. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from alpha-Synuclein Accumulation and Propagation. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 8618–8628. [Google Scholar] [CrossRef]
- Smethurst, P.; Risse, E.; Tyzack, G.E.; Mitchell, J.S.; Taha, D.M.; Chen, Y.R.; Newcombe, J.; Collinge, J.; Sidle, K.; Patani, R. Distinct responses of neurons and astrocytes to TDP-43 proteinopathy in amyotrophic lateral sclerosis. Brain A J. Neurol. 2020, 143, 430–440. [Google Scholar] [CrossRef]
- Ishii, M.N.; Yamamoto, K.; Shoji, M.; Asami, A.; Kawamata, Y. Human induced pluripotent stem cell (hiPSC)-derived neurons respond to convulsant drugs when co-cultured with hiPSC-derived astrocytes. Toxicology 2017, 389, 130–138. [Google Scholar] [CrossRef]
- Gupta, K.; Patani, R.; Baxter, P.; Serio, A.; Story, D.; Tsujita, T.; Hayes, J.D.; Pedersen, R.A.; Hardingham, G.E.; Chandran, S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012, 19, 779–787. [Google Scholar] [CrossRef]
- Zhao, C.; Devlin, A.C.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef]
- Mizuno, G.O.; Wang, Y.; Shi, G.; Wang, Y.; Sun, J.; Papadopoulos, S.; Broussard, G.J.; Unger, E.K.; Deng, W.; Weick, J.; et al. Aberrant Calcium Signaling in Astrocytes Inhibits Neuronal Excitability in a Human Down Syndrome Stem Cell Model. Cell Rep. 2018, 24, 355–365. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, P.; Xue, H.; Peterson, S.E.; Tran, H.T.; McCann, A.E.; Parast, M.M.; Li, S.; Pleasure, D.E.; Laurent, L.C.; et al. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nat. Commun. 2014, 5, 4430. [Google Scholar] [CrossRef]
- Dooves, S.; Nadadhur, A.G.; Gasparotto, L.; Heine, V.M. Co-culture of Human Stem Cell Derived Neurons and Oligodendrocyte Progenitor Cells. Bio-Protoc. 2019, 9, e3350. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Yuan, Q.; Tan, S.; Xiao, Y.; Wang, D.; Khoo, A.T.; Sani, L.; Tran, H.D.; Kim, P.; Chiew, Y.S.; et al. Direct Induction and Functional Maturation of Forebrain GABAergic Neurons from Human Pluripotent Stem Cells. Cell Rep. 2016, 16, 1942–1953. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef]
- Theka, I.; Caiazzo, M.; Dvoretskova, E.; Leo, D.; Ungaro, F.; Curreli, S.; Manago, F.; Dell’Anno, M.T.; Pezzoli, G.; Gainetdinov, R.R.; et al. Rapid generation of functional dopaminergic neurons from human induced pluripotent stem cells through a single-step procedure using cell lineage transcription factors. Stem Cells Transl. Med. 2013, 2, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Hester, M.E.; Murtha, M.J.; Song, S.; Rao, M.; Miranda, C.J.; Meyer, K.; Tian, J.; Boulting, G.; Schaffer, D.V.; Zhu, M.X.; et al. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 1905–1912. [Google Scholar] [CrossRef]
- Efthymiou, A.; Shaltouki, A.; Steiner, J.P.; Jha, B.; Heman-Ackah, S.M.; Swistowski, A.; Zeng, X.; Rao, M.S.; Malik, N. Functional screening assays with neurons generated from pluripotent stem cell-derived neural stem cells. J. Biomol. Screen. 2014, 19, 32–43. [Google Scholar] [CrossRef]
- Pei, Y.; Peng, J.; Behl, M.; Sipes, N.S.; Shockley, K.R.; Rao, M.S.; Tice, R.R.; Zeng, X. Comparative neurotoxicity screening in human iPSC-derived neural stem cells, neurons and astrocytes. Brain Res. 2016, 1638, 57–73. [Google Scholar] [CrossRef]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Aure, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674. [Google Scholar] [CrossRef]
- Odawara, A.; Matsuda, N.; Ishibashi, Y.; Yokoi, R.; Suzuki, I. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Sci. Rep. 2018, 8, 10416. [Google Scholar] [CrossRef]
- Sherman, S.P.; Bang, A.G. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis. Models Mech. 2018, 11, dmm031906. [Google Scholar] [CrossRef]
- Calabrese, B.; Powers, R.M.; Slepian, A.J.; Halpain, S. Post-differentiation Replating of Human Pluripotent Stem Cell-derived Neurons for High-content Screening of Neuritogenesis and Synapse Maturation. J. Vis. Exp. JoVE 2019. [Google Scholar] [CrossRef]
- Little, D.; Luft, C.; Pezzini-Picart, O.; Mosaku, O.; Ketteler, R.; Devine, M.J.; Gissen, P. Seeding Induced Pluripotent Stem Cell-Derived Neurons onto 384-Well Plates. Methods Mol. Biol. 2019, 1994, 159–164. [Google Scholar] [CrossRef]
- Sridharan, B.; Hubbs, C.; Llamosas, N.; Kilinc, M.; Singhera, F.U.; Willems, E.; Piper, D.R.; Scampavia, L.; Rumbaugh, G.; Spicer, T.P. A Simple Procedure for Creating Scalable Phenotypic Screening Assays in Human Neurons. Sci. Rep. 2019, 9, 9000. [Google Scholar] [CrossRef]
- Traub, S.; Heilker, R. hiPS Cell-Derived Neurons for High-Throughput Screening. Methods Mol. Biol. 2019, 1994, 243–263. [Google Scholar] [CrossRef]
- Garcia-Leon, J.A.; Caceres-Palomo, L.; Sanchez-Mejias, E.; Mejias-Ortega, M.; Nunez-Diaz, C.; Fernandez-Valenzuela, J.J.; Sanchez-Varo, R.; Davila, J.C.; Vitorica, J.; Gutierrez, A. Human Pluripotent Stem Cell-Derived Neural Cells as a Relevant Platform for Drug Screening in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6867. [Google Scholar] [CrossRef]
- Boussaad, I.; Cruciani, G.; Bolognin, S.; Antony, P.; Dording, C.M.; Kwon, Y.J.; Heutink, P.; Fava, E.; Schwamborn, J.C.; Kruger, R. Integrated, automated maintenance, expansion and differentiation of 2D and 3D patient-derived cellular models for high throughput drug screening. Sci. Rep. 2021, 11, 1439. [Google Scholar] [CrossRef]
- Pogue, R.E.; Cavalcanti, D.P.; Shanker, S.; Andrade, R.V.; Aguiar, L.R.; de Carvalho, J.L.; Costa, F.F. Rare genetic diseases: Update on diagnosis, treatment and online resources. Drug Discov. Today 2018, 23, 187–195. [Google Scholar] [CrossRef]
- Pasca, S.P.; Portmann, T.; Voineagu, I.; Yazawa, M.; Shcheglovitov, A.; Pasca, A.M.; Cord, B.; Palmer, T.D.; Chikahisa, S.; Nishino, S.; et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011, 17, 1657–1662. [Google Scholar] [CrossRef]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces alpha-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Codazzi, F.; Hu, A.; Rai, M.; Donatello, S.; Salerno Scarzella, F.; Mangiameli, E.; Pelizzoni, I.; Grohovaz, F.; Pandolfo, M. Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet. 2016, 25, 4847–4855. [Google Scholar] [CrossRef]
- Long, Y.; Xu, M.; Li, R.; Dai, S.; Beers, J.; Chen, G.; Soheilian, F.; Baxa, U.; Wang, M.; Marugan, J.J.; et al. Induced Pluripotent Stem Cells for Disease Modeling and Evaluation of Therapeutics for Niemann-Pick Disease Type A. Stem Cells Transl. Med. 2016, 5, 1644–1655. [Google Scholar] [CrossRef]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.T.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells. Stem Cells 2017, 35, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; Troglio, F.; Faga, G.; Fancelli, D.; Shyti, R.; Trattaro, S.; Zanella, M.; D’Agostino, G.; Hughes, J.M.; Cera, M.R.; et al. High-throughput screening identifies histone deacetylase inhibitors that modulate GTF2I expression in 7q11.23 microduplication autism spectrum disorder patient-derived cortical neurons. Mol. Autism 2020, 11, 88. [Google Scholar] [CrossRef]
- Goldson, E.; Hagerman, R.J. The fragile X syndrome. Dev. Med. Child Neurol. 1992, 34, 826–832. [Google Scholar] [CrossRef]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Van der Aa, N.; Rooms, L.; Vandeweyer, G.; van den Ende, J.; Reyniers, E.; Fichera, M.; Romano, C.; Delle Chiaie, B.; Mortier, G.; Menten, B.; et al. Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med Genet. 2009, 52, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I: 10 years later. Gene 2012, 492, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Mervis, C.B.; Dida, J.; Lam, E.; Crawford-Zelli, N.A.; Young, E.J.; Henderson, D.R.; Onay, T.; Morris, C.A.; Woodruff-Borden, J.; Yeomans, J.; et al. Duplication of GTF2I results in separation anxiety in mice and humans. Am. J. Hum. Genet. 2012, 90, 1064–1070. [Google Scholar] [CrossRef]
- Kajihara, R.; Numakawa, T.; Odaka, H.; Yaginuma, Y.; Fusaki, N.; Okumiya, T.; Furuya, H.; Inui, S.; Era, T. Novel Drug Candidates Improve Ganglioside Accumulation and Neural Dysfunction in GM1 Gangliosidosis Models with Autophagy Activation. Stem Cell Rep. 2020, 14, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Ruillier, V.; Tournois, J.; Boissart, C.; Lasbareilles, M.; Mahe, G.; Chatrousse, L.; Cailleret, M.; Peschanski, M.; Benchoua, A. Rescuing compounds for Lesch-Nyhan disease identified using stem cell-based phenotypic screening. JCI Insight 2020, 5, e132094. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef]
- Lesch, M.; Nyhan, W.L. A Familial Disorder of Uric Acid Metabolism and Central Nervous System Function. Am. J. Med. 1964, 36, 561–570. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Visser, J.E.; Harris, J.C.; Verdu, A.; Larovere, L.; Ceballos-Picot, I.; Gonzalez-Alegre, P.; Neychev, V.; Torres, R.J.; Dulac, O.; et al. Delineation of the motor disorder of Lesch-Nyhan disease. Brain A J. Neurol. 2006, 129, 1201–1217. [Google Scholar] [CrossRef]
- Jinnah, H.A.; De Gregorio, L.; Harris, J.C.; Nyhan, W.L.; O’Neill, J.P. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat. Res. 2000, 463, 309–326. [Google Scholar] [CrossRef]
- Allsop, J.; Watts, R.W. Activities of amidophosphoribosyltransferase and purine phosphoribosyltransferases in developing rat brain. Adv. Exp. Med. Biol. 1980, 122A, 361–366. [Google Scholar] [CrossRef]
- Glick, N. Dramatic reduction in self-injury in Lesch-Nyhan disease following S-adenosylmethionine administration. J. Inherit. Metab. Dis. 2006, 29, 687. [Google Scholar] [CrossRef]
- Dolcetta, D.; Parmigiani, P.; Salmaso, L.; Bernardelle, R.; Cesari, U.; Andrighetto, G.; Baschirotto, G.; Nyhan, W.L.; Hladnik, U. Quantitative evaluation of the clinical effects of S-adenosylmethionine on mood and behavior in Lesch-Nyhan patients. NucleosidesNucleotides Nucleic Acids 2013, 32, 174–188. [Google Scholar] [CrossRef]
- Chen, B.C.; Balasubramaniam, S.; McGown, I.N.; O’Neill, J.P.; Chng, G.S.; Keng, W.T.; Ngu, L.H.; Duley, J.A. Treatment of Lesch-Nyhan disease with S-adenosylmethionine: Experience with five young Malaysians, including a girl. Brain Dev. 2014, 36, 593–600. [Google Scholar] [CrossRef]
- Lauber, M.; Plecko, B.; Pfiffner, M.; Nuoffer, J.M.; Haberle, J. The Effect of S-Adenosylmethionine on Self-Mutilation in a Patient with Lesch-Nyhan Disease. JIMD Rep. 2017, 32, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Momosaki, K.; Kido, J.; Matsumoto, S.; Taniguchi, A.; Akiyama, T.; Sawada, T.; Ozasa, S.; Nakamura, K. The Effect of S-Adenosylmethionine Treatment on Neurobehavioral Phenotypes in Lesch-Nyhan Disease: A Case Report. Case Rep. Neurol. 2019, 11, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Negraes, P.D.; Trujillo, C.A.; Yu, N.K.; Wu, W.; Yao, H.; Liang, N.; Lautz, J.D.; Kwok, E.; McClatchy, D.; Diedrich, J.; et al. Altered network and rescue of human neurons derived from individuals with early-onset genetic epilepsy. Mol. Psychiatry 2021. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatric Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Darville, H.; Poulet, A.; Rodet-Amsellem, F.; Chatrousse, L.; Pernelle, J.; Boissart, C.; Heron, D.; Nava, C.; Perrier, A.; Jarrige, M.; et al. Human Pluripotent Stem Cell-derived Cortical Neurons for High Throughput Medication Screening in Autism: A Proof of Concept Study in SHANK3 Haploinsufficiency Syndrome. EBioMedicine 2016, 9, 293–305. [Google Scholar] [CrossRef]
- Costales, J.L.; Kolevzon, A. Phelan-McDermid Syndrome and SHANK3: Implications for Treatment. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2015, 12, 620–630. [Google Scholar] [CrossRef]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsater, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A gradient of severity in cognitive impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef]
- Boeckers, T.M.; Bockmann, J.; Kreutz, M.R.; Gundelfinger, E.D. ProSAP/Shank proteins—a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J. Neurochem. 2002, 81, 903–910. [Google Scholar] [CrossRef]
- Shcheglovitov, A.; Shcheglovitova, O.; Yazawa, M.; Portmann, T.; Shu, R.; Sebastiano, V.; Krawisz, A.; Froehlich, W.; Bernstein, J.A.; Hallmayer, J.F.; et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013, 503, 267–271. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar] [CrossRef]
- Borenstein, A.R.; Copenhaver, C.I.; Mortimer, J.A. Early-life risk factors for Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2006, 20, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; American College of Physicians; American Physiological Society. Alzheimer disease: Mechanistic understanding predicts novel therapies. Ann. Intern. Med. 2004, 140, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity—towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Kimura, J.; Shimizu, K.; Kajima, K.; Yokosuka, A.; Mimaki, Y.; Oku, N.; Ohizumi, Y. Nobiletin Reduces Intracellular and Extracellular beta-Amyloid in iPS Cell-Derived Alzheimer’s Disease Model Neurons. Biol. Pharm. Bull. 2018, 41, 451–457. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 24, 647–657. [Google Scholar] [CrossRef]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S.B. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer’s Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, M.; Mihalik, B.; Turi, Z.; Dinnyes, A.; Kobolak, J. Calcilytic NPS 2143 Reduces Amyloid Secretion and Increases sAbetaPPalpha Release from PSEN1 Mutant iPSC-Derived Neurons. J. Alzheimers Dis. JAD 2019, 72, 885–899. [Google Scholar] [CrossRef] [PubMed]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Teglasi, A.; Bock, I.; Giudice, M.L.; Tancos, Z.; Molnar, K.; Laszlo, L.; et al. Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef]
- Xu, X.; Lei, Y.; Luo, J.; Wang, J.; Zhang, S.; Yang, X.J.; Sun, M.; Nuwaysir, E.; Fan, G.; Zhao, J.; et al. Prevention of beta-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res. 2013, 10, 213–227. [Google Scholar] [CrossRef]
- McLaren, D.; Gorba, T.; Marguerie de Rotrou, A.; Pillai, G.; Chappell, C.; Stacey, A.; Lingard, S.; Falk, A.; Smith, A.; Koch, P.; et al. Automated large-scale culture and medium-throughput chemical screen for modulators of proliferation and viability of human induced pluripotent stem cell-derived neuroepithelial-like stem cells. J. Biomol. Screen. 2013, 18, 258–268. [Google Scholar] [CrossRef]
- Wang, C.; Ward, M.E.; Chen, R.; Liu, K.; Tracy, T.E.; Chen, X.; Xie, M.; Sohn, P.D.; Ludwig, C.; Meyer-Franke, A.; et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Rep. 2017, 9, 1221–1233. [Google Scholar] [CrossRef]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B.; et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 2019, 24, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.G.; Graham, L. The journey: Parkinson’s disease. BMJ 2004, 329, 611–614. [Google Scholar] [CrossRef]
- Gonera, E.G.; van’t Hof, M.; Berger, H.J.; van Weel, C.; Horstink, M.W. Symptoms and duration of the prodromal phase in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 1997, 12, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain A J. Neurol. 1991, 114, 2283–2301. [Google Scholar] [CrossRef]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S.; et al. Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef]
- Mittal, S.; Bjornevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef]
- Oh, Y. Patient-specific pluripotent stem cell-based Parkinson’s disease models showing endogenous alpha-synuclein aggregation. BMB Rep. 2019, 52, 349–359. [Google Scholar] [CrossRef]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Little, D.; Luft, C.; Mosaku, O.; Lorvellec, M.; Yao, Z.; Paillusson, S.; Kriston-Vizi, J.; Gandhi, S.; Abramov, A.Y.; Ketteler, R.; et al. A single cell high content assay detects mitochondrial dysfunction in iPSC-derived neurons with mutations in SNCA. Sci. Rep. 2018, 8, 9033. [Google Scholar] [CrossRef]
- Kondo, T.; Imamura, K.; Funayama, M.; Tsukita, K.; Miyake, M.; Ohta, A.; Woltjen, K.; Nakagawa, M.; Asada, T.; Arai, T.; et al. iPSC-Based Compound Screening and In Vitro Trials Identify a Synergistic Anti-amyloid beta Combination for Alzheimer’s Disease. Cell Rep. 2017, 21, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Tanila, H. The role of BDNF in Alzheimer’s disease. Neurobiol. Dis. 2017, 97, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed]
- Charbord, J.; Poydenot, P.; Bonnefond, C.; Feyeux, M.; Casagrande, F.; Brinon, B.; Francelle, L.; Auregan, G.; Guillermier, M.; Cailleret, M.; et al. High throughput screening for inhibitors of REST in neural derivatives of human embryonic stem cells reveals a chemical compound that promotes expression of neuronal genes. Stem Cells 2013, 31, 1816–1828. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.; Malik, N.; Shah, S.; Zhao, J.; Class, B.; Aguisanda, F.; Southall, N.; Xia, M.; McKew, J.C.; Rao, M.; et al. High-Throughput Phenotypic Screening of Human Astrocytes to Identify Compounds That Protect Against Oxidative Stress. Stem Cells Transl. Med. 2016, 5, 613–627. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Hoing, S.; Rudhard, Y.; Reinhardt, P.; Glatza, M.; Stehling, M.; Wu, G.; Peiker, C.; Bocker, A.; Parga, J.A.; Bunk, E.; et al. Discovery of inhibitors of microglial neurotoxicity acting through multiple mechanisms using a stem-cell-based phenotypic assay. Cell Stem Cell 2012, 11, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, L.; Kordes, S.; Reinhardt, P.; Glatza, M.; Baumann, M.; Drexler, H.C.A.; Menninger, S.; Zischinsky, G.; Eickhoff, J.; Frob, C.; et al. Dual Inhibition of GSK3beta and CDK5 Protects the Cytoskeleton of Neurons from Neuroinflammatory-Mediated Degeneration In Vitro and In Vivo. Stem Cell Rep. 2019, 12, 502–517. [Google Scholar] [CrossRef]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef]
- Miller, J.D.; Ganat, Y.M.; Kishinevsky, S.; Bowman, R.L.; Liu, B.; Tu, E.Y.; Mandal, P.K.; Vera, E.; Shim, J.W.; Kriks, S.; et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 2013, 13, 691–705. [Google Scholar] [CrossRef]
- Steel, Z.; Marnane, C.; Iranpour, C.; Chey, T.; Jackson, J.W.; Patel, V.; Silove, D. The global prevalence of common mental disorders: A systematic review and meta-analysis 1980-2013. Int. J. Epidemiol. 2014, 43, 476–493. [Google Scholar] [CrossRef]
- Vigo, D.; Thornicroft, G.; Atun, R. Estimating the true global burden of mental illness. Lancet. Psychiatry 2016, 3, 171–178. [Google Scholar] [CrossRef]
- Villanueva, R. Neurobiology of major depressive disorder. Neural Plast. 2013, 2013, 873278. [Google Scholar] [CrossRef]
- Meyer, J.H.; Ginovart, N.; Boovariwala, A.; Sagrati, S.; Hussey, D.; Garcia, A.; Young, T.; Praschak-Rieder, N.; Wilson, A.A.; Houle, S. Elevated monoamine oxidase a levels in the brain: An explanation for the monoamine imbalance of major depression. Arch. Gen. Psychiatry 2006, 63, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Sahay, A.; Hen, R. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 2007, 10, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Kempermann, G.; Krebs, J.; Fabel, K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr. Opin. Psychiatry 2008, 21, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Lieberman, J.A. Catching up on schizophrenia: Natural history and neurobiology. Neuron 2000, 28, 325–334. [Google Scholar] [CrossRef]
- Kessler, R.C.; Birnbaum, H.; Demler, O.; Falloon, I.R.; Gagnon, E.; Guyer, M.; Howes, M.J.; Kendler, K.S.; Shi, L.; Walters, E.; et al. The prevalence and correlates of nonaffective psychosis in the National Comorbidity Survey Replication (NCS-R). Biol. Psychiatry 2005, 58, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Insel, T.R. Rethinking schizophrenia. Nature 2010, 468, 187–193. [Google Scholar] [CrossRef]
- Schizophrenia Psychiatric Genome-Wide Association Study, C. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef]
- Goodwin, F.K.; Ghaemi, S.N. The course of bipolar disorder and the nature of agitated depression. Am. J. Psychiatry 2003, 160, 2077–2079. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Schneider, M.R.; DelBello, M.P.; McNamara, R.K.; Strakowski, S.M.; Adler, C.M. Neuroprogression in bipolar disorder. Bipolar Disord. 2012, 14, 356–374. [Google Scholar] [CrossRef] [PubMed]
- Watmuff, B.; Liu, B.; Karmacharya, R. Stem cell-derived neurons in the development of targeted treatment for schizophrenia and bipolar disorder. Pharmacogenomics 2017, 18, 471–479. [Google Scholar] [CrossRef]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef]
- Surget, A.; Saxe, M.; Leman, S.; Ibarguen-Vargas, Y.; Chalon, S.; Griebel, G.; Hen, R.; Belzung, C. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol. Psychiatry 2008, 64, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I. Neuroimaging studies of mood disorder effects on the brain. Biol. Psychiatry 2003, 54, 338–352. [Google Scholar] [CrossRef]
- Steen, R.G.; Mull, C.; McClure, R.; Hamer, R.M.; Lieberman, J.A. Brain volume in first-episode schizophrenia: Systematic review and meta-analysis of magnetic resonance imaging studies. Br. J. Psychiatry J. Ment. Sci. 2006, 188, 510–518. [Google Scholar] [CrossRef]
- Bearden, C.E.; Thompson, P.M.; Dalwani, M.; Hayashi, K.M.; Lee, A.D.; Nicoletti, M.; Trakhtenbroit, M.; Glahn, D.C.; Brambilla, P.; Sassi, R.B.; et al. Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol. Psychiatry 2007, 62, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Yucel, K.; Taylor, V.H.; McKinnon, M.C.; Macdonald, K.; Alda, M.; Young, L.T.; MacQueen, G.M. Bilateral hippocampal volume increase in patients with bipolar disorder and short-term lithium treatment. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Giakoumatos, C.I.; Nanda, P.; Mathew, I.T.; Tandon, N.; Shah, J.; Bishop, J.R.; Clementz, B.A.; Pearlson, G.D.; Sweeney, J.A.; Tamminga, C.A.; et al. Effects of lithium on cortical thickness and hippocampal subfield volumes in psychotic bipolar disorder. J. Psychiatr. Res. 2015, 61, 180–187. [Google Scholar] [CrossRef]
- Tamminga, C.A.; Stan, A.D.; Wagner, A.D. The hippocampal formation in schizophrenia. Am. J. Psychiatry 2010, 167, 1178–1193. [Google Scholar] [CrossRef] [PubMed]
- Mathew, I.; Gardin, T.M.; Tandon, N.; Eack, S.; Francis, A.N.; Seidman, L.J.; Clementz, B.; Pearlson, G.D.; Sweeney, J.A.; Tamminga, C.A.; et al. Medial temporal lobe structures and hippocampal subfields in psychotic disorders: Findings from the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) study. JAMA Psychiatry 2014, 71, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Vadodaria, K.C.; Ji, Y.; Skime, M.; Paquola, A.C.; Nelson, T.; Hall-Flavin, D.; Heard, K.J.; Fredlender, C.; Deng, Y.; Elkins, J.; et al. Altered serotonergic circuitry in SSRI-resistant major depressive disorder patient-derived neurons. Mol. Psychiatry 2019, 24, 808–818. [Google Scholar] [CrossRef]
- Vadodaria, K.C.; Ji, Y.; Skime, M.; Paquola, A.; Nelson, T.; Hall-Flavin, D.; Fredlender, C.; Heard, K.J.; Deng, Y.; Le, A.T.; et al. Serotonin-induced hyperactivity in SSRI-resistant major depressive disorder patient-derived neurons. Mol. Psychiatry 2019, 24, 795–807. [Google Scholar] [CrossRef]
- Viguera, A.C.; Tondo, L.; Baldessarini, R.J. Sex differences in response to lithium treatment. Am. J. Psychiatry 2000, 157, 1509–1511. [Google Scholar] [CrossRef]
- Tohen, M.; Greil, W.; Calabrese, J.R.; Sachs, G.S.; Yatham, L.N.; Oerlinghausen, B.M.; Koukopoulos, A.; Cassano, G.B.; Grunze, H.; Licht, R.W.; et al. Olanzapine versus lithium in the maintenance treatment of bipolar disorder: A 12-month, randomized, double-blind, controlled clinical trial. Am. J. Psychiatry 2005, 162, 1281–1290. [Google Scholar] [CrossRef]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef]
- Santos, R.; Linker, S.B.; Stern, S.; Mendes, A.P.D.; Shokhirev, M.N.; Erikson, G.; Randolph-Moore, L.; Racha, V.; Kim, Y.; Kelsoe, J.R.; et al. Deficient LEF1 expression is associated with lithium resistance and hyperexcitability in neurons derived from bipolar disorder patients. Mol. Psychiatry 2021, 26, 2440–2456. [Google Scholar] [CrossRef]
- Zhao, W.N.; Cheng, C.; Theriault, K.M.; Sheridan, S.D.; Tsai, L.H.; Haggarty, S.J. A high-throughput screen for Wnt/beta-catenin signaling pathway modulators in human iPSC-derived neural progenitors. J. Biomol. Screen. 2012, 17, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Durens, M.; Nestor, J.; Williams, M.; Herold, K.; Niescier, R.F.; Lunden, J.W.; Phillips, A.W.; Lin, Y.C.; Dykxhoorn, D.M.; Nestor, M.W. High-throughput screening of human induced pluripotent stem cell-derived brain organoids. J. Neurosci. Methods 2020, 335, 108627. [Google Scholar] [CrossRef]
- Hendriks, D.; Clevers, H.; Artegiani, B. CRISPR-Cas Tools and Their Application in Genetic Engineering of Human Stem Cells and Organoids. Cell Stem Cell 2020, 27, 705–731. [Google Scholar] [CrossRef]
- Nishiga, M.; Qi, L.S.; Wu, J.C. CRISPRi/a Screening with Human iPSCs. Methods Mol. Biol. 2021, 2320, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Malandraki-Miller, S.; Riley, P.R. Use of artificial intelligence to enhance phenotypic drug discovery. Drug Discov. Today 2021, 26, 887–901. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benchoua, A.; Lasbareilles, M.; Tournois, J. Contribution of Human Pluripotent Stem Cell-Based Models to Drug Discovery for Neurological Disorders. Cells 2021, 10, 3290. https://doi.org/10.3390/cells10123290
Benchoua A, Lasbareilles M, Tournois J. Contribution of Human Pluripotent Stem Cell-Based Models to Drug Discovery for Neurological Disorders. Cells. 2021; 10(12):3290. https://doi.org/10.3390/cells10123290
Chicago/Turabian StyleBenchoua, Alexandra, Marie Lasbareilles, and Johana Tournois. 2021. "Contribution of Human Pluripotent Stem Cell-Based Models to Drug Discovery for Neurological Disorders" Cells 10, no. 12: 3290. https://doi.org/10.3390/cells10123290
APA StyleBenchoua, A., Lasbareilles, M., & Tournois, J. (2021). Contribution of Human Pluripotent Stem Cell-Based Models to Drug Discovery for Neurological Disorders. Cells, 10(12), 3290. https://doi.org/10.3390/cells10123290