
An Insight into GPCR and G-Proteins as Cancer Drivers


Abstract
1. Introduction
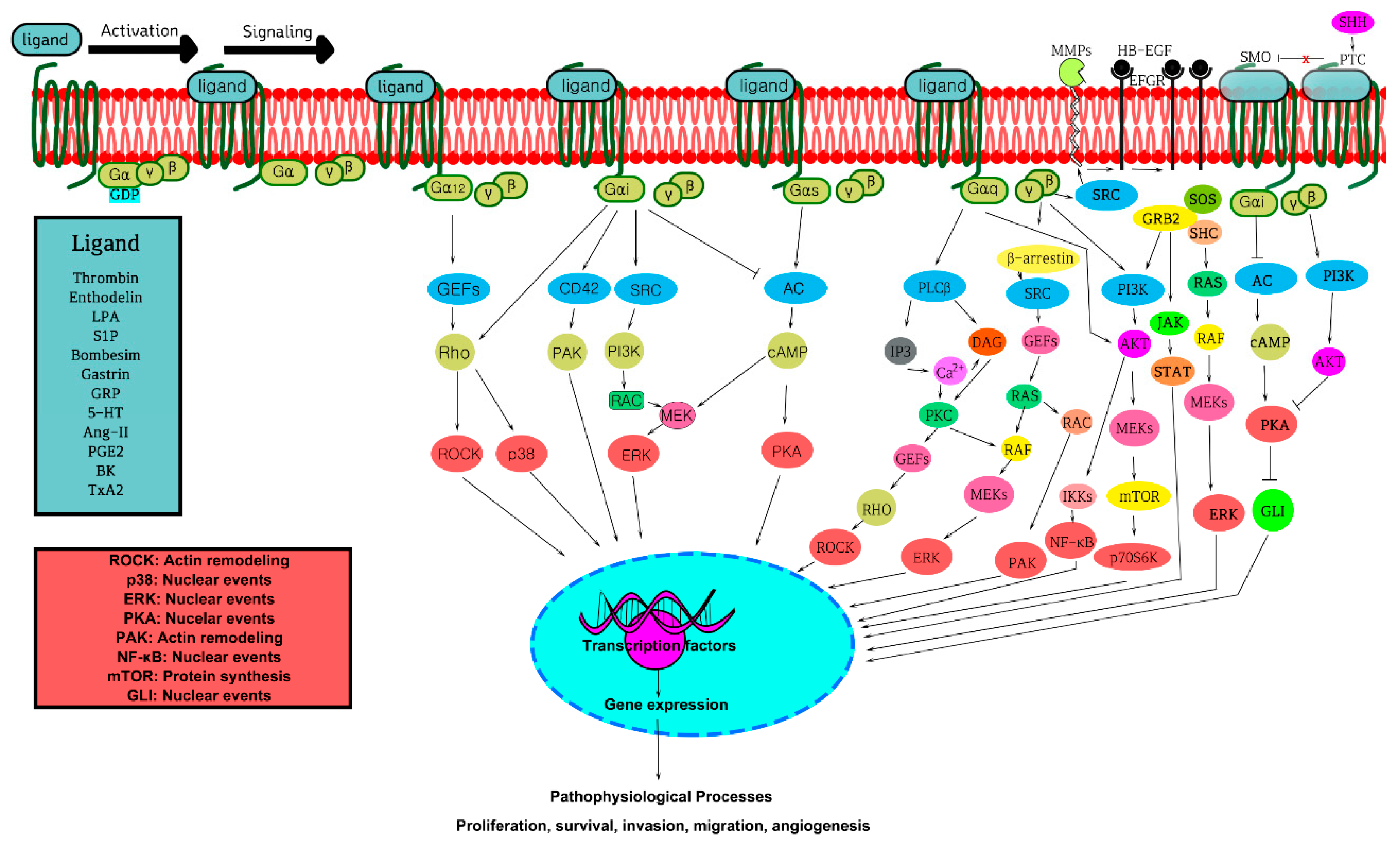
2. GPCRs, GPCR Signaling, and Cross-Talk

3. GPCRs, G-Proteins, and GPCR Signaling Pathways in Oncogenicity
3.1. Aberrant Expression, Mutations, and Activation of GPCRs and G-Proteins in Cancer

{kind=link}
| Receptor (IUPHAR) | Mutations (Amino Acid Changes) | Associations | References |
|---|---|---|---|
| Thyroid-stimulating hormone receptor (TSH receptor) | N-terminal: S281I; ICL3: D619G; A623V; L629F; TM6: F631L; T632I; D633H; ECL2: I568T; ECL3: V656F | a. Activating mutations; b. All mutants activating the cAMP pathway; c. Found in human thyroid carcinoma, breast, lung, and colon cancers | [92,93] |
| Melanocortin 1 receptor (MC1R) | TM2: D84E; TM7: D294H | a. Activating mutations; b. Related with human melanoma and nonmelanoma skin cancers. | [94] |
| ICL2: R151C; R160W | a. Inactivating mutations; b. Changed the relative risk of nomelanoma skin cancer. | [91] | |
| Melanocortin 2 receptor (MC2R) | R137W; S74I; Y254C | a. Activating mutations; b. Involved in adenoma and carcinomas. | [95] |
| Lutropin (LHCG) receptor | TM3: L457R; TM6: D578H; C581R; TM6: A572V; D578Y | a. Activating mutations; b. Found in human Leydig-cell tumor. | [96,97] |
| Smoothened (SMO) receptor | N-terminal: R199W; TM6: D473H; TM7: S533N; W535L; C-terminal: R562Q | a. Activating mutations; b. Found in human sporadic basal cell carcinoma (BCCs), lung and colon, and central nervous system cancers. | [98,99] |
| Follicle-stimulating hormone receptor (FSHR) | ECL2: D576G/N; TM4: D581G/Y; C584R; TM6: H615Y; D619G; A623I/S/V | a. Activating mutations; b. Slightly increasing in basal cAMP production; c. Found in human large intestine cancers, colon. | [100] |
| Brain-specific angiogenesis inhibitors 1–3 (BAI1–BAI3) | BAI1: N-terminal: S927A/D BAI3: GPS domain: G586R; C819Y TSP domain: T420I; A442E; W461L; 7TM domain: A1024P; R1050K; R1124C; C1148F; M1258I; F1378Y; G1404V; N1475T; D1449E; P1510L | a. Activating mutations; b. Found in human squamous lung carcinoma and lung adenocarcinoma. | [101,102] |
| EGF LAG seven-pass (CELSR1–3) | CL1: T838A/P (Gain domain) CL1 and CL3: K561N; D798H; V696L; A760Q; S810L; E811Q (Gain domain) | a. Activating mutations; b. All mutants are especially found in human squamous lung carcinoma and lung adenocarcinoma. | [102,103] |
| Latrophilins (LPHN) | LPHN1: A73D;V696L LPHN2: Q693H LPHN3: H18R; N344I; T442N; K561N; A760G; D798H | a. All mutants were activating mutations; b. All mutants were involved in tumor angiogenesis, invasion, or tumor growth. | [104] |
| Glutamate family of G protein-linked receptors (GRM1–8) | GRM3: N-terminal: G475D; G561E; S610L(ECL1); E767K(ECL2); E870K(C-terminal) GRM8: N-terminal: G49R; L76M; T118I; V150I; W215C; A282D; G523W; S691T; A808M (ECL3) | a. Activating mutations; b. GRM3 is mutated in 7% of human non-small cell lung cancer adenocarcinoma; c. GRM3 mutants are found in human melanoma cancers. a. Activating mutations; b. GRM8 is mutated in 8% of human squamous non-small cell lung cancer and melanoma cancers. | [102,105] |
| Muscarinic receptor | M1: TM2: F77I; TM3: W101A; TM6: E360A; Y381A; ICL3: K362A; N-terminal: I211A; Y212A M3: ECL2: Q207A; C257A; C264A | a. Activating mutations; b. Inactivating mutations; c. M1 mutants are found in human melanoma cancers. a. Inactivating mutations; b. M3 mutants are found in human melanoma cancers. | [106] |
| Lysophosphatidic acid receptor (LPAR) | LPAR1: ICL2: R163W; ICL3: R241Q L LPAR2: ICL2: R146H; ICL3: P230L LPAR3: ICL3: K216A; V219A; TM6: A247V LPAR4: R232H(ICL3) LPAR6: TM4: S154A; TM6: N248Y; TM7: L277P | a. Activating mutations; b. LPAR1 was mutated in human lung, neuroblastoma, and liver cancers. a. Activating mutations; b. LPAR2 was mutated in human colon cancers; a. Activating mutations; b. LPAR3 was mutated in human melanoma cells and osteosarcoma cells. a. Activating mutations; b. LPAR6 was mutated in human melanocarcinoma. | [107,108] |
| Sphingosine 1-phosphate (S1P) receptor | S1PR1: N-terminal: R13G; TM3: R120P; ICL3: T236A; R231K; R233K | a. Inactivating mutations; b. Involved in tumor growth, invasion and metastasis; c. Found in human lung, breast, and prostate cancer. | [109] |
3.2. GPCR-β-Arrestin Signaling in Cancer
3.3. Biased Agonism towards Specific G-Proteins in Cancer
3.4. GPCRs in the Hallmarks of Cancer
3.4.1. GPCRs in Migration, Invasion, and Metastasis
3.4.2. GPCRs in Tumor-Induced Angiogenesis
3.4.3. Inflammation and Immune Cell Evasion in Tumor Microenvironment
3.4.4. Tumor-Suppressor Functions of Some GPCRs
4. Cancer-Associated GPCR-Mediated Signaling Pathways
4.1. Wnt Signaling
4.2. Hippo Signaling Pathway
4.3. PARs and Cancer
5. Key Individual GPCRs and Their Signaling Pathways Involved in Various Cancer
5.1. GPR30
5.2. Lysophosphatidic Acid Receptor (LPAR)
5.3. Angiotensin-II Receptor
5.4. Gastrin Releasing Peptide Receptor (GRPR)
5.5. S1P Receptor
6. GPCRs as Cancer Targets
7. Importance of GPCRomics in Cancer
8. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GPCR | G protein-coupled receptor |
| PAR | Protease-activated receptor |
| LPA | Lysophosphatidic acid |
| WNT | Wingless and Int-1 |
| 7-TM | Seven transmembrane |
| EL | Extracellular loop |
| LGRs | Leucine-rich repeat-containing receptors |
| PLC | PhopholipaseC |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| DAG | Diacylgylcerol |
| IP3 | Inositol 1,4,5-triphosphate |
| PKA | cAMP-dependent kinase |
| PKC | Protein kinase C |
| PKG | cGMP-dependent kinase |
| CAMKs | Calcium-calmodulin regulated kinases |
| GIRK | G-protein-gated inwardly rectifying potassium channel |
| PI3K | Phosphoinositide 3-kinase |
| S1P | Sphingosine-1-phosphate |
| MAPK | Mitogen-activated protein kinase |
| GRKs | G-protein receptor kinases |
| Ang-II | Angiotensin II |
| BK | Bradykinin |
| AT1R | Angiotensin-II type-1 receptor |
| HRH1 | Human histamine receptor H1 |
| GnRH | Gonadotropin-releasing hormone |
| TSHR | Thyroid-stimulating hormone receptor |
| LHCGR | Luteinizing hormone receptor |
| FSHR | Follicle-stimulating hormone receptor |
| SMO | Smoothened |
| PTCH | Patched |
| NSCLC | Squamous non-small cell lung cancer |
| GR | Glutamate receptors |
| AVPR | Arginine vasopressin receptor |
| MC2R | Melanocortin 2 receptor |
| NK | Natural killer |
| SRF | Serum response factor |
| AP-1 | Activating protein 1 |
| PTH | Parathyroid hormone |
| ETAR | Endothelin A receptor |
| MMPs | Matrix metalloproteases |
| ECM | Extracellular matrix |
| VEGF | Vascular endothelial growth factor |
| PGE2 | Prostaglandin E2 |
| COX | Cyclooxygenases |
| NSAIDs | Nonsteroidal anti-inflammatory medications |
| TAMs | Tumor-associated macrophages |
| DLBCL | Diffuse large B cell lymphoma |
| Fz | Frizzled |
| APC | Adenomatis polyposis coli |
| GSK | Glycogen synthase kinase |
| CK1 | Casein kinase1 |
| LRP | Lipoprotein-related protein |
| ET | Endothelin receptor |
| EMT | Extracellular matrix |
| PCP | Planar cell polarity |
| ROCK | Rho-associated kinase |
| YAP | Yes-associated protein |
| TAZ | Transcriptional coactivator with PDZ-binding motif |
| PH | Pleckstrin homology |
| mAChRs | Muscarinic receptors |
| EFGR | Epidermal growth factor receptor |
| PDGF | Platelet-derived growth factor receptor |
| GRP | Gastrin releasing peptide |
| BN | Bombesin |
| HB-EGF | Heparin-bound EGF |
| ER | Estrogen receptor |
| STAT | Signal transducer and activator of transcription |
| ACEis | Angiotensin-converting enzyme inhibitors |
| HIF-2 | Hypoxia-inducible transcription factor 2 |
| OXR | Orexin receptor |
References
- Hua Li, J.; Jain, S.; McMillin, S.M.; Cui, Y.; Gautam, D.; Sakamoto, W.; Lu, H.; Jou, W.; McGuinness, O.P.; Gavrilova, O. A novel experimental strategy to assess the metabolic effects of selective activation of a Gq-coupled receptor in hepatocytes in vivo. Endocrinology 2013, 154, 3539–3551. [Google Scholar] [CrossRef]
- Nickols, H.H.; Conn, P.J. Development of allosteric modulators of GPCRs for treatment of CNS disorders. Neurobiol. Dis. 2014, 61, 55–71. [Google Scholar] [CrossRef]
- Sun, G.C.; Ho, W.Y.; Chen, B.R.; Cheng, P.W.; Cheng, W.H.; Hsu, M.C.; Yeh, T.C.; Hsiao, M.; Lu, P.J.; Tseng, C.J. GPCR dimerization in brainstem nuclei contributes to the development of hypertension. Br. J. Pharmacol. 2015, 172, 2507–2518. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Koch, W.J. Targeting cardiac β-adrenergic signaling via GRK2 inhibition for heart failure therapy. Front. Physiol. 2013, 4, 264. [Google Scholar] [CrossRef]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhuo, X.; Mao, C. G Protein-coupled Receptors in Cancer Stem Cells. Curr. Pharm. Des. 2020, 26, 1952–1963. [Google Scholar] [CrossRef]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta Biomembr. 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef] [PubMed]
- Taussig, R.; Iniguez-Lluhi, J.A.; Gilman, A.G. Inhibition of adenylyl cyclase by Gi alpha. Science 1993, 261, 218–221. [Google Scholar] [CrossRef]
- Rodbell, M.; Birnbaumer, L.; Pohl, S.L.; Krans, H.M.J. The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver: V. An obligatory role of guanyl nucleotides in glucagon action. J. Biol. Chem. 1971, 246, 1877–1882. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Hepler, J.R.; Brown, K.O.; Sternweis, P.C. Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 1991, 251, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Herr, D.; Mutoh, T.; Chun, J. Lysophosphatidic acid (LPA) and its receptors. Curr. Opin. Pharmacol. 2009, 9, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.; Hla, T. Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 2004, 92, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Gutkind, J.S. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J. Biol. Chem. 1998, 273, 1839–1842. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Palmby, T.R.; Simaan, M.; Li, W.; Szabo, R.; Lyons, R.; Martin, D.; Yagi, H.; Fukuhara, S.; Chikumi, H. PDZ-RhoGEF and LARG are essential for embryonic development and provide a link between thrombin and LPA receptors and Rho activation. J. Biol. Chem. 2013, 288, 12232–12243. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-pkb/akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Teoh, C.M.; Tam, J.K.C.; Tran, T. Integrin and GPCR crosstalk in the regulation of ASM contraction signaling in asthma. J. Allergy 2012, 2012, 341282. [Google Scholar] [CrossRef]
- Chaudhary, P.K.; Kim, S. The GRKs Reactome: Role in Cell Biology and Pathology. Int. J. Mol. Sci. 2021, 22, 3375. [Google Scholar] [CrossRef]
- Bagnato, A.; Rosanò, L. New routes in GPCR/β-arrestin-driven signaling in cancer progression and metastasis. Front. Pharmacol. 2019, 10, 114. [Google Scholar] [CrossRef]
- Arakaki, A.K.; Pan, W.-A.; Wedegaertner, H.; Roca-Mercado, I.; Chinn, L.; Gujral, T.S.; Trejo, J. α-Arrestin ARRDC3 tumor suppressor function is linked to GPCR-induced TAZ activation and breast cancer metastasis. J. Cell Sci. 2021, 134, 8. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Waitches, G.; Birchmeier, C.; Fasano, O.; Wigler, M. Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 1986, 45, 711–719. [Google Scholar] [CrossRef]
- Feigin, M.E. Harnessing the genome for characterization of G-protein coupled receptors in cancer pathogenesis. FEBS J. 2013, 280, 4729–4738. [Google Scholar] [CrossRef]
- Li, S.; Huang, S.; Peng, S.-B. Overexpression of G protein-coupled receptors in cancer cells: Involvement in tumor progression. Int. J. Oncol. 2005, 27, 1329–1338. [Google Scholar] [CrossRef]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- Kawanabe, Y.; Okamoto, Y.; Nozaki, K.; Hashimoto, N.; Miwa, S.; Masaki, T. Molecular Mechanism for Endothelin-1–Induced Stress-Fiber Formation: Analysis of G Proteins Using a Mutant EndothelinA Receptor. Mol. Pharmacol. 2002, 61, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.J.; Park, S.; Cho, K.; Sohn, J.; Lee, J.; Kim, Y.; Kang, J.; Park, C.; Han, J.; Lee, H. Correction: The Rho/ROCK pathway for lysophosphatidic acid-induced proteolytic enzyme expression and ovarian cancer cell invasion. Oncogene 2019, 38, 5108–5110. [Google Scholar] [CrossRef]
- Contos, J.J.; Ishii, I.; Chun, J. Lysophosphatidic acid receptors. Mol. Pharmacol. 2000, 58, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Gwak, J.; Cho, M.; Ryu, M.-J.; Lee, J.-H.; Kim, S.K.; Kim, Y.H.; Lee, G.W.; Yun, M.-Y.; Cuong, N.M. Murrayafoline A attenuates the Wnt/β-catenin pathway by promoting the degradation of intracellular β-catenin proteins. Biochem. Biophys. Res. Commun. 2010, 391, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Bian, D.; Mahanivong, C.; Yu, J.; Frisch, S.; Pan, Z.; Ye, R.; Huang, S. The G 12/13-RhoA signaling pathway contributes to efficient lysophosphatidic acid-stimulated cell migration. Oncogene 2006, 25, 2234–2244. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, A.; Hart, S.; Fischer, O.M.; Ullrich, A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003, 22, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Kim, N.-H.; Ji, L.; Kim, S.-H.; Lee, J.; Rhee, H.J. Lysophosphatidic acid activates β-catenin/T cell factor signaling, which contributes to the suppression of apoptosis in H19-7 cells. Mol. Med. Rep. 2013, 8, 1729–1733. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic acid initiates epithelial to mesenchymal transition and induces β-catenin-mediated transcription in epithelial ovarian carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Krüppel-like factor 5. J. Biol. Chem. 2007, 282, 15541–15549. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.-X.; Karin, M.; Pan, D. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Cuevas, B.; Russo, A.; Johnson, G.; Trejo, J. Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 2008, 27, 4434–4445. [Google Scholar] [CrossRef]
- Arora, P.; Ricks, T.K.; Trejo, J. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J. Cell Sci. 2007, 120, 921–928. [Google Scholar] [CrossRef]
- Tsopanoglou, N.E.; Maragoudakis, M.E. Role of Thrombin in Angiogenesis and Tumor Progression. Semin. Thromb. Hemost. 2004, 30, 63–69. [Google Scholar]
- Clevers, H. Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef]
- Lai, S.-L.; Chien, A.J.; Moon, R.T. Wnt/Fz signaling and the cytoskeleton: Potential roles in tumorigenesis. Cell Res. 2009, 19, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Gugger, M.; White, R.; Song, S.; Waser, B.; Cescato, R.; Riviere, P.; Reubi, J.C. GPR87 is an overexpressed G-protein coupled receptor in squamous cell carcinoma of the lung. Dis. Markers 2008, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lustig, B.; Behrens, J. The Wnt signaling pathway and its role in tumor development. J. Cancer Res. Clin. Oncol. 2003, 129, 199–221. [Google Scholar] [CrossRef] [PubMed]
- Birch, M.; Carron, J.; Scott, M.; Fraser, W.; Gallagher, J. Parathyroid hormone (PTH)/PTH-related protein (PTHrP) receptor expression and mitogenic responses in human breast cancer cell lines. Br. J. Cancer 1995, 72, 90–95. [Google Scholar] [CrossRef][Green Version]
- Schwartz, G.G. Prostate cancer, serum parathyroid hormone, and the progression of skeletal metastases. Cancer Epidemiol. Prevent. Biomark. 2008, 17, 478–483. [Google Scholar] [CrossRef]
- Cojoc, M.; Peitzsch, C.; Trautmann, F.; Polishchuk, L.; Telegeev, G.D.; Dubrovska, A. Emerging targets in cancer management: Role of the CXCL12/CXCR4 axis. OncoTargets Ther. 2013, 6, 1347. [Google Scholar]
- Growcott, J.W. Preclinical anticancer activity of the specific endothelin A receptor antagonist ZD4054. Anti-Cancer Drugs 2009, 20, 83–88. [Google Scholar] [CrossRef]
- Smollich, M.; Götte, M.; Fischgräbe, J.; Macedo, L.F.; Brodie, A.; Chen, S.; Radke, I.; Kiesel, L.; Wülfing, P. ETAR antagonist ZD4054 exhibits additive effects with aromatase inhibitors and fulvestrant in breast cancer therapy, and improves in vivo efficacy of anastrozole. Breast Cancer Res. Treat. 2010, 123, 345–357. [Google Scholar] [CrossRef]
- Rosanò, L.; Cianfrocca, R.; Tocci, P.; Spinella, F.; Di Castro, V.; Spadaro, F.; Salvati, E.; Biroccio, A.; Natali, P.; Bagnato, A. β-arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced β-catenin signaling. Oncogene 2013, 32, 5066–5077. [Google Scholar] [CrossRef]
- Rosanò, L.; Spinella, F.; Bagnato, A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2013, 13, 637–651. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Subbaramaiah, K. Targeting cyclooxygenase-2 in human neoplasia: Rationale and promise. Cancer Cell 2003, 4, 431–436. [Google Scholar] [CrossRef]
- Hull, M.A.; Ko, S.C.; Hawcroft, G. Prostaglandin EP receptors: Targets for treatment and prevention of colorectal cancer? Mol. Cancer Ther. 2004, 3, 1031–1039. [Google Scholar] [PubMed]
- O’callaghan, G.; Houston, A. Prostaglandin E2 and the EP receptors in malignancy: Possible therapeutic targets? Br. J. Pharmacol. 2015, 172, 5239–5250. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE 2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef]
- Taub, J.S.; Guo, R.; Leeb-Lundberg, L.F.; Madden, J.F.; Daaka, Y. Bradykinin receptor subtype 1 expression and function in prostate cancer. Cancer Res. 2003, 63, 2037–2041. [Google Scholar]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.-X.; Li, W.; Xu, T.-R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Maehama, T.; Mogi, C.; Tomura, H.; Ishiuchi, S.; Yoshimoto, Y.; Kurose, H.; Okajima, F. S1P2 receptors mediate inhibition of glioma cell migration through Rho signaling pathways independent of PTEN. Biochem. Biophys. Res. Commun. 2008, 366, 963–968. [Google Scholar] [CrossRef]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef]
- Malchinkhuu, E.; Sato, K.; Horiuchi, Y.; Mogi, C.; Ohwada, S.; Ishiuchi, S.; Saito, N.; Kurose, H.; Tomura, H.; Okajima, F. Role of p38 mitogen-activated kinase and c-Jun terminal kinase in migration response to lysophosphatidic acid and sphingosine-1-phosphate in glioma cells. Oncogene 2005, 24, 6676–6688. [Google Scholar] [CrossRef]
- Kinoshita, J.; Fushida, S.; Harada, S.; Yagi, Y.; Fujita, H.; Kinami, S.; Ninomiya, I.; Fujimura, T.; Kayahara, M.; Yashiro, M. Local angiotensin II-generation in human gastric cancer: Correlation with tumor progression through the activation of ERK1/2, NF-κB and survivin. Int. J. Oncol. 2009, 34, 1573–1582. [Google Scholar] [CrossRef]
- Uemura, H.; Ishiguro, H.; Nagashima, Y.; Sasaki, T.; Nakaigawa, N.; Hasumi, H.; Kato, S.; Kubota, Y. Antiproliferative activity of angiotensin II receptor blocker through cross-talk between stromal and epithelial prostate cancer cells. Mol. Cancer Ther. 2005, 4, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Chao, C.; Ives, K.; Hellmich, M.R. Regulation of bombesin-stimulated cyclooxygenase-2 expression in prostate cancer cells. BMC Mol. Biol. 2011, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Grabowska, M.M.; Forestier, I.S.; Mirosevich, J.; Case, T.C.; Chung, D.H.; Cates, J.M.; Matusik, R.J.; Manning, H.C.; Jin, R. Activation of GRP/GRP-R signaling contributes to castration-resistant prostate cancer progression. Oncotarget 2016, 7, 61955. [Google Scholar] [CrossRef]
- Demenais, F.; Mohamdi, H.; Chaudru, V.; Goldstein, A.M.; Newton Bishop, J.A.; Bishop, D.; Kanetsky, P.A.; Hayward, N.; Gillanders, E.; Elder, D.E. Association of MC1R variants and host phenotypes with melanoma risk in CDKN2A mutation carriers: A GenoMEL study. J. Natl. Cancer Inst. 2010, 102, 1568–1583. [Google Scholar] [CrossRef]
- Rios, C.; Jordan, B.; Gomes, I.; Devi, L. G-protein-coupled receptor dimerization: Modulation of receptor function. Pharmacol. Ther. 2001, 92, 71–87. [Google Scholar] [CrossRef]
- Hoshino, K.; Ishiguro, H.; Teranishi, J.I.; Yoshida, S.I.; Umemura, S.; Kubota, Y.; Uemura, H. Regulation of androgen receptor expression through angiotensin II type 1 receptor in prostate cancer cells. Prostate 2011, 71, 964–975. [Google Scholar] [CrossRef]
- Guha, S.; Lunn, J.A.; Santiskulvong, C.; Rozengurt, E. Neurotensin stimulates protein kinase C-dependent mitogenic signaling in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2003, 63, 2379–2387. [Google Scholar]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Iguchi, T.; Sakata, K.; Yoshizaki, K.; Tago, K.; Mizuno, N.; Itoh, H. Orphan G protein-coupled receptor GPR56 regulates neural progenitor cell migration via a Gα12/13 and Rho pathway. J. Biol. Chem. 2008, 283, 14469–14478. [Google Scholar] [CrossRef]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Halmos, G.; Arencibia, J.M.; Schally, A.V.; Davis, R.; Bostwick, D.G. High incidence of receptors for luteinizing hormone-releasing hormone (LHRH) and LHRH receptor gene expression in human prostate cancers. J. Urol. 2000, 163, 623–629. [Google Scholar] [CrossRef]
- Pommerville, P.J.; de Boer, J.G. GnRH antagonists in the treatment of advanced prostate cancer. Can. J. Urol. 2010, 17, 5063–5070. [Google Scholar] [PubMed]
- Yu, B.; Ruman, J.; Christman, G. The role of peripheral gonadotropin-releasing hormone receptors in female reproduction. Fertil. Steril. 2011, 95, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; De Lellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a seven membrane–spanning estrogen receptor, in primary breast cancer and its association with clinicopathologic determinants of tumor progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef]
- Smith, H.O.; Arias-Pulido, H.; Kuo, D.Y.; Howard, T.; Qualls, C.R.; Lee, S.-J.; Verschraegen, C.F.; Hathaway, H.J.; Joste, N.E.; Prossnitz, E.R. GPR30 predicts poor survival for ovarian cancer. Gynecol. Oncol. 2009, 114, 465–471. [Google Scholar] [CrossRef]
- Smith, H.O.; Leslie, K.K.; Singh, M.; Qualls, C.R.; Revankar, C.M.; Joste, N.E.; Prossnitz, E.R. GPR30: A novel indicator of poor survival for endometrial carcinoma. Am. J. Obst. Gynecol. 2007, 196, 386.e1–386.e11. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.-M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.-W.; Hynes, M.; Goddard, A. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- O’hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Duzenli, D.; Saglar, E.; Deniz, F.; Azal, O.; Erdem, B.; Mergen, H. Mutations in the AVPR2, AVP-NPII, and AQP2 genes in Turkish patients with diabetes insipidus. Endocrine 2012, 42, 664–669. [Google Scholar] [CrossRef]
- Huang, L.; Li, W.; Tang, W.; Lu, G. A novel AVPR2 missense mutation in a Chinese boy with severe inherited nephrogenic diabetes insipidus. J. Pediatr. Endocrinol. Metab. 2011, 24, 807–809. [Google Scholar] [CrossRef]
- Sasaki, S.; Chiga, M.; Kikuchi, E.; Rai, T.; Uchida, S. Hereditary nephrogenic diabetes insipidus in Japanese patients: Analysis of 78 families and report of 22 new mutations in AVPR2 and AQP2. Clin. Exp. Nephrol. 2013, 17, 338–344. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Akahori, M.; Itabashi, T.; Nishino, J.; Yoshitake, K.; Furuno, M.; Ikeo, K.; Okada, T.; Tsuneoka, H. RHO mutations (p. W126L and p. A346P) in two Japanese families with autosomal dominant retinitis pigmentosa. J. Ophthalmol. 2014, 2012, 210947. [Google Scholar] [CrossRef]
- Rossmiller, B.P.; Ryals, R.C.; Lewin, A.S. Gene therapy to rescue retinal degeneration caused by mutations in rhodopsin. In Rhodopsin; Springer: Berlin/Heidelberg, Germany, 2015; pp. 391–410. [Google Scholar]
- Aza-Carmona, M.; Barreda-Bonis, A.C.; Guerrero-Fernández, J.; González-Casado, I.; Gracia, R.; Heath, K.E. Familial glucocorticoid deficiency due to compound heterozygosity of two novel MC2R mutations. J. Pediatr. Endocrinol. Metab. 2011, 24, 395–397. [Google Scholar] [CrossRef]
- Fridmanis, D.; Petrovska, R.; Pjanova, D.; Schiöth, H.B.; Klovins, J. Replacement of short segments within transmembrane domains of MC2R disrupts retention signal. J. Mol. Endocrinol. 2014, 53, 201–215. [Google Scholar] [CrossRef]
- Switonski, M.; Mankowska, M.; Salamon, S. Family of melanocortin receptor (MCR) genes in mammals—mutations, polymorphisms and phenotypic effects. J. Appl. Genet. 2013, 54, 461–472. [Google Scholar] [CrossRef]
- Turan, S.; Hughes, C.; Atay, Z.; Guran, T.; Haliloglu, B.; Clark, A.J.; Bereket, A.; Metherell, L.A. An atypical case of familial glucocorticoid deficiency without pigmentation caused by coexistent homozygous mutations in MC2R (T152K) and MC1R (R160W). J. Clin. Endocrinol. Metabol. 2012, 97, E771–E774. [Google Scholar] [CrossRef]
- Bonomi, M.; Proverbio, M.C.; Weber, G.; Chiumello, G.; Beck-Peccoz, P.; Persani, L. Hyperplastic Pituitary Gland, High Serum Glycoprotein Hormoneα-Subunit, and Variable Circulating Thyrotropin (TSH) Levels as Hallmark of Central Hypothyroidism due to Mutations of the TSHβ Gene. J. Clin. Endocrinol. Metabol. 2001, 86, 1600–1604. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miyai, K. Congenital Thyrotropin Deficiency—From Discovery to Molecular Biology, Postgenome and Preventive Medicine. Endocr. J. 2007, 54, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.N.; Bishop, D.T. The genetics of susceptibility to cutaneous melanoma. Drugs Today 2005, 41, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Flück, C.E.; Martens, J.W.; Conte, F.A.; Miller, W.L. Clinical, genetic, and functional characterization of adrenocorticotropin receptor mutations using a novel receptor assay. J. Clin. Endocrinol. Metabol. 2002, 87, 4318–4323. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Duranteau, L.; Carel, J.-C.; Monroe, J.; Doyle, D.A.; Shenker, A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N. Engl. J. Med. 1999, 341, 1731–1736. [Google Scholar] [CrossRef]
- Powlson, A.S.; Challis, B.G.; Halsall, D.J.; Schoenmakers, E.; Gurnell, M. Nephrogenic syndrome of inappropriate antidiuresis secondary to an activating mutation in the arginine vasopressin receptor AVPR2. Clin. Endocrinol. 2016, 85, 306–312. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.-P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Tao, Y.-X. Constitutive activation of G protein-coupled receptors and diseases: Insights into mechanisms of activation and therapeutics. Pharmacol. Ther. 2008, 120, 129–148. [Google Scholar] [CrossRef]
- Cork, S.M.; Van Meir, E.G. Emerging roles for the BAI1 protein family in the regulation of phagocytosis, synaptogenesis, neurovasculature, and tumor development. J. Mol. Med. 2011, 89, 743–752. [Google Scholar] [CrossRef]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.; Copp, A.J.; Stanier, P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Meza-Aguilar, D.G.; Boucard, A.A. Latrophilins updated. Biomol. Concepts 2014, 5, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Elia, J.; Glessner, J.T.; Wang, K.; Takahashi, N.; Shtir, C.J.; Hadley, D.; Sleiman, P.M.; Zhang, H.; Kim, C.E.; Robison, R. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat. Genet. 2012, 44, 78–84. [Google Scholar] [CrossRef]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Tashiro, M.; Fukudo, S.; Yanai, K.; Utsumi, A.; Kano, M.; Karahasi, M.; Endo, Y.; Morisita, J.; Sato, Y. Increased brain histamine H1 receptor binding in patients with anorexia nervosa. Biolo. Psychiatry 2009, 65, 329–335. [Google Scholar] [CrossRef]
- Raza, S.I.; Muhammad, D.; Jan, A.; Ali, R.H.; Hassan, M.; Ahmad, W.; Rashid, S. In silico analysis of missense mutations in LPAR6 reveals abnormal phospholipid signaling pathway leading to hypotrichosis. PLoS ONE 2014, 9, e104756. [Google Scholar] [CrossRef]
- Obinata, H.; Gutkind, S.; Stitham, J.; Okuno, T.; Yokomizo, T.; Hwa, J.; Hla, T. Individual variation of human S1P1 coding sequence leads to heterogeneity in receptor function and drug interactions[S]. J. Lipid Res. 2014, 55, 2665–2675. [Google Scholar] [CrossRef]
- Arang, N.; Gutkind, J.S. G Protein-Coupled receptors and heterotrimeric G proteins as cancer drivers. FEBS Lett. 2020, 594, 4201–4232. [Google Scholar] [CrossRef]
- van Biesen, T.; Luttrell, L.M.; Hawes, B.E.; Lefkowitz, R.J. Mitogenic signaling via G protein-coupled receptors. Endocr. Rev. 1996, 17, 698–714. [Google Scholar] [CrossRef]
- Kalinec, G.; Nazarali, A.; Hermouet, S.; Xu, N.; Gutkind, J. Mutated alpha subunit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol. Cell. Biol. 1992, 12, 4687–4693. [Google Scholar] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Ayturk, U.M.; Couto, J.A.; Hann, S.; Mulliken, J.B.; Williams, K.L.; Huang, A.Y.; Fishman, S.J.; Boyd, T.K.; Kozakewich, H.P.; Bischoff, J. Somatic activating mutations in GNAQ and GNA11 are associated with congenital hemangioma. Am. J. Hum. Genet. 2016, 98, 789–795. [Google Scholar] [CrossRef]
- Shirley, M.D.; Tang, H.; Gallione, C.J.; Baugher, J.D.; Frelin, L.P.; Cohen, B.; North, P.E.; Marchuk, D.A.; Comi, A.M.; Pevsner, J. Sturge–Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013, 368, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Küsters-Vandevelde, H.V.; van Grunsven, I.A.; Küsters, B.; van Dijk, M.R.; Groenen, P.J.; Wesseling, P.; Blokx, W.A. Improved discrimination of melanotic schwannoma from melanocytic lesions by combined morphological and GNaQ mutational analysis. Acta Neuropathol. 2010, 120, 755–764. [Google Scholar] [CrossRef]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein–coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the α chain of G s and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune–Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, X.; Xue, W.; Zhang, Y.; Li, C.; Song, Y.; Mei, M.; Lu, L.; Wang, Y.; Zhou, Z. Recurrent GNAQ mutation encoding T96S in natural killer/T cell lymphoma. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Rao, R.; Salloum, R.; Xin, M.; Lu, Q.R. The G protein Gαs acts as a tumor suppressor in sonic hedgehog signaling-driven tumorigenesis. Cell Cycle 2016, 15, 1325–1330. [Google Scholar] [CrossRef]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 1–12. [Google Scholar]
- He, X.; Zhang, L.; Chen, Y.; Remke, M.; Shih, D.; Lu, F.; Wang, H.; Deng, Y.; Yu, Y.; Xia, Y. The G protein α subunit Gα s is a tumor suppressor in Sonic hedgehog−driven medulloblastoma. Nat. Med. 2014, 20, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Marinissen, M.J.; Servitja, J.-M.; Offermanns, S.; Simon, M.I.; Gutkind, J.S. Thrombin protease-activated receptor-1 signals through Gq-and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J. Biol. Chem. 2003, 278, 46814–46825. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, N.; Tsim, S.-T.; Dermott, J.M.; Onesime, D. Regulation of cell proliferation by G proteins. Oncogene 1998, 17, 1383–1394. [Google Scholar] [CrossRef]
- Pace, A.M.; Wong, Y.H.; Bourne, H.R. A mutant alpha subunit of Gi2 induces neoplastic transformation of Rat-1 cells. Proc. Natl. Acad. Sci. USA 1991, 88, 7031–7035. [Google Scholar] [CrossRef]
- Garcia-Marcos, M.; Ghosh, P.; Farquhar, M.G. Molecular basis of a novel oncogenic mutation in GNAO1. Oncogene 2011, 30, 2691–2696. [Google Scholar] [CrossRef]
- Lyons, J.; Landis, C.A.; Harsh, G.; Vallar, L.; Grunewald, K.; Feichtinger, H.; Duh, Q.-Y.; Clark, O.H.; Kawasaki, E.; Bourne, H.R. Two G protein oncogenes in human endocrine tumors. Science 1990, 249, 655–659. [Google Scholar] [CrossRef]
- Chan, A.; Fleming, T.; McGovern, E.; Chedid, M.; Miki, T.; Aaronson, S. Expression cDNA cloning of a transforming gene encoding the wild-type G alpha 12 gene product. Mol. Cell. Biol. 1993, 13, 762–768. [Google Scholar]
- Juneja, J.; Casey, P.J. Role of G12 proteins in oncogenesis and metastasis. Br. J. Pharmacol. 2009, 158, 32–40. [Google Scholar] [CrossRef]
- Fukuhara, S.; Marinissen, M.J.; Chiariello, M.; Gutkind, J.S. Signaling from G Protein-coupled Receptors to ERK5/Big MAPK 1 Involves Gαq and Gα12/13 Families of Heterotrimeric G Proteins: Evidence for the Existence of a Novel Ras and Rho-Independent pathway. J. Biol. Chem. 2000, 275, 21730–21736. [Google Scholar] [CrossRef] [PubMed]
- Meigs, T.E.; Fields, T.A.; McKee, D.D.; Casey, P.J. Interaction of Gα12 and Gα13 with the cytoplasmic domain of cadherin provides a mechanism for β-catenin release. Proc. Natl. Acad. Sci. USA 2001, 98, 519–524. [Google Scholar] [PubMed]
- Radhika, V.; Dhanasekaran, N. Transforming G proteins. Oncogene 2001, 20, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Moeller, B.J.; Juneja, J.; Booden, M.A.; Der, C.J.; Daaka, Y.; Dewhirst, M.W.; Fields, T.A.; Casey, P.J. The G12 family of heterotrimeric G proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8173–8178. [Google Scholar] [CrossRef]
- Kelly, P.; Stemmle, L.N.; Madden, J.F.; Fields, T.A.; Daaka, Y.; Casey, P.J. A role for the G12 family of heterotrimeric G proteins in prostate cancer invasion. J. Biol. Chem. 2006, 281, 26483–26490. [Google Scholar] [CrossRef]
- Daaka, Y. G proteins in cancer: The prostate cancer paradigm. Sci. STKE 2004, 2004, re2. [Google Scholar] [CrossRef]
- O’Hayre, M.; Inoue, A.; Kufareva, I.; Wang, Z.; Mikelis, C.M.; Drummond, R.A.; Avino, S.; Finkel, K.; Kalim, K.; DiPasquale, G. Inactivating mutations in GNA13 and RHOA in Burkitt’s lymphoma and diffuse large B-cell lymphoma: A tumor suppressor function for the Gα 13/RhoA axis in B cells. Oncogene 2016, 35, 3771–3780. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef]
- Morin, R.D.; Mungall, K.; Pleasance, E.; Mungall, A.J.; Goya, R.; Huff, R.D.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Schmitz, R.; Green, J.A.; Xiao, W.; Larsen, A.B.; Braun, S.E.; An, J.; Xu, Y.; Rosenwald, A.; Ott, G. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature 2014, 516, 254–258. [Google Scholar] [CrossRef]
- Liu, S.-C.; Jen, Y.-M.; Jiang, S.S.; Chang, J.-L.; Hsiung, C.A.; Wang, C.-H.; Juang, J.-L. Gα12-mediated pathway promotes invasiveness of nasopharyngeal carcinoma by modulating actin cytoskeleton reorganization. Cancer Res. 2009, 69, 6122–6130. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Lu, E.; Cyster, J.G. The G protein–coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. J. Exp. Med. 2015, 212, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Hong, M.; Choi, M.; Kim, Y.; Chang, W.; Maeng, C.; Park, S.; Lee, S.; Do, I.-G.; Jo, J.-S. The impact of activated p-AKT expression on clinical outcomes in diffuse large B-cell lymphoma: A clinicopathological study of 262 cases. Ann. Oncol. 2014, 25, 182–188. [Google Scholar] [CrossRef]
- Hurst, J.H.; Hooks, S.B. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem. Pharmacol. 2009, 78, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- DiGiacomo, V.; Maziarz, M.; Luebbers, A.; Norris, J.M.; Laksono, P.; Garcia-Marcos, M. Probing the mutational landscape of regulators of G protein signaling proteins in cancer. Sci. Signal. 2020, 13, 617. [Google Scholar] [CrossRef]
- Sethakorn, N.; Dulin, N.O. RGS expression in cancer: Oncomining the cancer microarray data. J. Recept. Signal Transduct. 2013, 33, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Qutob, N.; Masuho, I.; Alon, M.; Emmanuel, R.; Cohen, I.; Di Pizio, A.; Madore, J.; Elkahloun, A.; Ziv, T.; Levy, R. Correction: RGS7 is recurrently mutated in melanoma and promotes migration and invasion of human cancer cells. Sci. Rep. 2019, 9, 4523. [Google Scholar] [CrossRef] [PubMed]
- Maity, B.; Stewart, A.; O’Malley, Y.; Askeland, R.W.; Sugg, S.L.; Fisher, R.A. Regulator of G protein signaling 6 is a novel suppressor of breast tumor initiation and progression. Carcinogenesis 2013, 34, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Platt, L.T.; Maity, B.; Ahlers, K.E.; Luo, Z.; Lin, Z.; Chakravarti, B.; Ibeawuchi, S.-R.; Askeland, R.W.; Bondaruk, J. RGS6 is an essential tumor suppressor that prevents bladder carcinogenesis by promoting p53 activation and DNMT1 downregulation. Oncotarget 2016, 7, 69159. [Google Scholar] [CrossRef]
- Tang, X.; Sun, Z.; Runne, C.; Madsen, J.; Domann, F.; Henry, M.; Lin, F.; Chen, S. A critical role of Gβγ in tumorigenesis and metastasis of breast cancer. J. Biol. Chem. 2011, 286, 13244–13254. [Google Scholar] [CrossRef]
- Vázquez-Prado, J.; Bracho-Valdés, I.; Cervantes-Villagrana, R.D.; Reyes-Cruz, G. Gβγ pathways in cell polarity and migration linked to oncogenic GPCR signaling: Potential relevance in tumor microenvironment. Mol. Pharmacol. 2016, 90, 573–586. [Google Scholar] [CrossRef]
- Song, Q.; Ji, Q.; Li, Q. The role and mechanism of β-arrestins in cancer invasion and metastasis. Int. J. Mol. Med. 2018, 41, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Zoudilova, M.; Kumar, P.; Ge, L.; Wang, P.; Bokoch, G.M.; DeFea, K.A. β-Arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 2007, 282, 20634–20646. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; DeFea, K. β-arrestin-dependent actin reorganization: Bringing the right players together at the leading edge. Mol. Pharmacol. 2011, 80, 760–768. [Google Scholar] [CrossRef]
- Ma, X.; Espana-Serrano, L.; Kim, W.-J.; Purayil, H.T.; Nie, Z.; Daaka, Y. βArrestin1 regulates the guanine nucleotide exchange factor RasGRF2 expression and the small GTPase Rac-mediated formation of membrane protrusion and cell motility. J. Biol. Chem. 2014, 289, 13638–13650. [Google Scholar] [CrossRef]
- Kang, J.; Shi, Y.; Xiang, B.; Qu, B.; Su, W.; Zhu, M.; Zhang, M.; Bao, G.; Wang, F.; Zhang, X. A nuclear function of β-arrestin1 in GPCR signaling: Regulation of histone acetylation and gene transcription. Cell 2005, 123, 833–847. [Google Scholar] [CrossRef]
- Shi, Y.; Feng, Y.; Kang, J.; Liu, C.; Li, Z.; Li, D.; Cao, W.; Qiu, J.; Guo, Z.; Bi, E. Critical regulation of CD4+ T cell survival and autoimmunity by β-arrestin 1. Nat. Immunol. 2007, 8, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Hoeppner, C.Z.; Cheng, N.; Richard, D.Y. Identification of a nuclear localization sequence in β-arrestin-1 and its functional implications. J. Biol. Chem. 2012, 287, 8932–8943. [Google Scholar] [CrossRef]
- Yang, Y.; Guo, Y.; Tan, S.; Ke, B.; Tao, J.; Liu, H.; Jiang, J.; Chen, J.; Chen, G.; Wu, B. β-Arrestin1 enhances hepatocellular carcinogenesis through inflammation-mediated Akt signalling. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Czogalla, B.; Partenheimer, A.; Jeschke, U.; von Schönfeldt, V.; Mayr, D.; Mahner, S.; Burges, A.; Simoni, M.; Melli, B.; Benevelli, R. β-arrestin 2 is a prognostic factor for survival of ovarian cancer patients upregulating cell proliferation. Front. Endocrinol. 2020, 11, 658. [Google Scholar] [CrossRef]
- Whalen, E.J.; Rajagopal, S.; Lefkowitz, R.J. Therapeutic potential of β-arrestin-and G protein-biased agonists. Trends Mol. Med. 2011, 17, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Rosanò, L.; Cianfrocca, R.; Masi, S.; Spinella, F.; Di Castro, V.; Biroccio, A.; Salvati, E.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. β-Arrestin links endothelin A receptor to β-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2806–2811. [Google Scholar] [CrossRef] [PubMed]
- Bostanabad, S.Y.; Noyan, S.; Dedeoglu, B.G.; Gurdal, H. Overexpression of β-Arrestins inhibits proliferation and motility in triple negative breast cancer cells. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-Y.; Hu, S.-S.; Wu, J.-J.; Huang, Q.; Ma, Y.; Wang, Q.-T.; Chen, J.-Y.; Wei, W. Down-regulation of β-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Cong, L.; Qiu, Z.-Y.; Zhao, Y.; Wang, W.-B.; Wang, C.-X.; Shen, H.-C.; Han, J.-Q. Loss of β-arrestin-2 and activation of CXCR2 correlate with lymph node metastasis in non-small cell lung cancer. J. Cancer 2017, 8, 2785. [Google Scholar] [CrossRef][Green Version]
- Duan, X.; Kong, Z.; Liu, Y.; Zeng, Z.; Li, S.; Wu, W.; Ji, W.; Yang, B.; Zhao, Z.; Zeng, G. β-Arrestin2 contributes to cell viability and proliferation via the down-regulation of FOXO1 in castration-resistant prostate cancer. J. Cell. Physiol. 2015, 230, 2371–2381. [Google Scholar] [CrossRef]
- Lakshmikanthan, V.; Zou, L.; Kim, J.I.; Michal, A.; Nie, Z.; Messias, N.C.; Benovic, J.L.; Daaka, Y. Identification of βArrestin2 as a corepressor of androgen receptor signaling in prostate cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 9379–9384. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef]
- Ge, L.; Shenoy, S.K.; Lefkowitz, R.J.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both β-arrestin-1 and-2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef]
- Ray, S.; Maunsell, J.H. Different origins of gamma rhythm and high-gamma activity in macaque visual cortex. PLoS Biol. 2011, 9, e1000610. [Google Scholar] [CrossRef]
- Gol, S.; Pena, R.N.; Rothschild, M.F.; Tor, M.; Estany, J. A polymorphism in the fatty acid desaturase-2 gene is associated with the arachidonic acid metabolism in pigs. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Heitzler, D.; Durand, G.; Gallay, N.; Rizk, A.; Ahn, S.; Kim, J.; Violin, J.D.; Dupuy, L.; Gauthier, C.; Piketty, V. Competing G protein-coupled receptor kinases balance G protein and β-arrestin signaling. Mol. Syst. Biol. 2012, 8, 590. [Google Scholar] [CrossRef] [PubMed]
- Nogues, L.; Palacios-Garcia, J.; Reglero, C.; Rivas, V.; Neves, M.; Ribas, C.; Penela, P.; Mayor, F., Jr. G Protein-Coupled Receptor Kinases (GRKs) in Tumorigenesis and Cancer Progression: GPCR Regulators and Signaling Hubs. Semin. Cancer Biol. 2018, 48, 78–90. [Google Scholar]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 2011, 336, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Bradley, L.; Ambdukar, I.; Gutkind, J.S. A mutant alpha subunit of G12 potentiates the eicosanoid pathway and is highly oncogenic in NIH 3T3 cells. Proc. Natl. Acad. Sci. USA 1993, 90, 6741–6745. [Google Scholar] [CrossRef]
- Jiang, H.; Wu, D.; Simon, M.I. The transforming activity of activated Gα12. FEBS Lett. 1993, 330, 319–322. [Google Scholar] [CrossRef]
- Kelly, P.; Casey, P.J.; Meigs, T.E. Biologic functions of the G12 subfamily of heterotrimeric g proteins: Growth, migration, and metastasis. Biochemistry 2007, 46, 6677–6687. [Google Scholar] [CrossRef]
- Lee, S.; Yang, J.; Cho, I.; Kim, W.; Cho, M.; Lee, C.; Kim, S. The gep oncogenes, Gα 12 and Gα 13, upregulate the transforming growth factor-β1 gene. Oncogene 2009, 28, 1230–1240. [Google Scholar] [CrossRef]
- Seo, H.; Kim, M.; Choi, Y.; Lee, C.-K.; Ka, H. Analysis of lysophosphatidic acid (LPA) receptor and LPA-induced endometrial prostaglandin-endoperoxide synthase 2 expression in the porcine uterus. Endocrinology 2008, 149, 6166–6175. [Google Scholar] [CrossRef]
- Wang, P.; Wu, X.; Chen, W.; Liu, J.; Wang, X. The lysophosphatidic acid (LPA) receptors their expression and significance in epithelial ovarian neoplasms. Gynecol. Oncol. 2007, 104, 714–720. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef]
- Bodor, E.; Offermanns, S. Nicotinic acid: An old drug with a promising future. Br. J. Pharmacol. 2008, 153, S68–S75. [Google Scholar] [CrossRef]
- Soto, A.G.; Smith, T.H.; Chen, B.; Bhattacharya, S.; Cordova, I.C.; Kenakin, T.; Vaidehi, N.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop 2 regulates G-protein signaling bias. Proc. Natl. Acad. Sci. USA 2015, 112, E3600–E3608. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yanagisawa, M.; Takuwat, Y.; Miyazakit, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Robinson, J.E.; Zhu, H.; DiBerto, J.F.; Polepally, P.R.; Zjawiony, J.K.; Nichols, D.E.; Malanga, C.; Roth, B.L. The G protein–biased κ-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. [Google Scholar] [CrossRef]
- Bagnato, A.; Salani, D.; Di Castro, V.; Wu-Wong, J.R.; Tecce, R.; Nicotra, M.R.; Venuti, A.; Natali, P.G. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: Evidence for an autocrine role in tumor growth. Cancer Res. 1999, 59, 720–727. [Google Scholar]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Inhibition of cyclooxygenase-1 and-2 expression by targeting the endothelin a receptor in human ovarian carcinoma cells. Clin. Cancer Res. 2004, 10, 4670–4679. [Google Scholar] [CrossRef] [PubMed]
- Vickers, N.J. Animal communication: When i’m calling you, will you answer too? Curr. Biol. 2017, 27, R713–R715. [Google Scholar] [CrossRef]
- Takahashi, A.; Kato, K.; Kuboyama, A.; Inoue, T.; Tanaka, Y.; Kuhara, A.; Kinoshita, K.; Takeda, S.; Wake, N. Induction of senescence by progesterone receptor-B activation in response to cAMP in ovarian cancer cells. Gynecol. Oncol. 2009, 113, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Follin-Arbelet, V.; Torgersen, M.L.; Naderi, E.H.; Misund, K.; Sundan, A.; Blomhoff, H.K. Death of multiple myeloma cells induced by cAMP-signaling involves downregulation of Mcl-1 via the JAK/STAT pathway. Cancer Lett. 2013, 335, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Guleng, B.; Tateishi, K.; Ohta, M.; Kanai, F.; Jazag, A.; Ijichi, H.; Tanaka, Y.; Washida, M.; Morikane, K.; Fukushima, Y. Blockade of the stromal cell–derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor–independent manner. Cancer Res. 2005, 65, 5864–5871. [Google Scholar] [CrossRef] [PubMed]
- Rich, J.N. Cancer stem cells in radiation resistance. Cancer Res. 2007, 67, 8980–8984. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Yagi, H.; Tan, W.; Dillenburg-Pilla, P.; Armando, S.; Amornphimoltham, P.; Simaan, M.; Weigert, R.; Molinolo, A.A.; Bouvier, M.; Gutkind, J.S. A synthetic biology approach reveals a CXCR4-G13-Rho signaling axis driving transendothelial migration of metastatic breast cancer cells. Sci. Signal. 2011, 4, 200222. [Google Scholar] [CrossRef]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef]
- Tang, X.; Jin, R.; Qu, G.; Wang, X.; Li, Z.; Yuan, Z.; Zhao, C.; Siwko, S.; Shi, T.; Wang, P. GPR116, an adhesion G-protein–coupled receptor, promotes breast cancer metastasis via the Gαq-p63RhoGEF-Rho GTPase pathway. Cancer Res. 2013, 73, 6206–6218. [Google Scholar] [CrossRef]
- Nag, J.K.; Malka, H.; Appasamy, P.; Sedley, S.; Bar-Shavit, R. GPCR Partners as Cancer Driver Genes: Association with PH-Signal Proteins in a Distinctive Signaling Network. Int. J. Mol. Sci. 2021, 22, 8985. [Google Scholar] [CrossRef]
- Moore, B.B.; Keane, M.P.; Addison, C.L.; Arenberg, D.A.; Strieter, R.M. CXC chemokine modulation of angiogenesis: The importance of balance between angiogenic and angiostatic members of the family. J. Investig. Med. 1998, 46, 113–120. [Google Scholar]
- Richard, D.E.; Vouret-Craviari, V.; Pouysségur, J. Angiogenesis and G-protein-coupled receptors: Signals that bridge the gap. Oncogene 2001, 20, 1556–1562. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; DuBois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Iñiguez, M.A.; Rodríguez, A.; Volpert, O.V.; Fresno, M.; Redondo, J.M. Cyclooxygenase-2: A therapeutic target in angiogenesis. Trends Mol. Med. 2003, 9, 73–78. [Google Scholar] [CrossRef]
- Brown, J.R.; DuBois, R.N. COX-2: A molecular target for colorectal cancer prevention. J. Clin. Oncol. 2005, 23, 2840–2855. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; DuBois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef]
- Hull, S.C.; Gooding, H.; Klein, A.P.; Warshauer-Baker, E.; Metosky, S.; Wilfond, B.S. Genetic research involving human biological materials: A need to tailor current consent forms. IRB Ethics Hum. Res. 2004, 26, 3. [Google Scholar] [CrossRef]
- Hansen-Petrik, M.B.; McEntee, M.F.; Jull, B.; Shi, H.; Zemel, M.B.; Whelan, J. Prostaglandin E2 protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in ApcMin/+ mice. Cancer Res. 2002, 62, 403–408. [Google Scholar]
- Sonoshita, M.; Takaku, K.; Sasaki, N.; Sugimoto, Y.; Ushikubi, F.; Narumiya, S.; Oshima, M.; Taketo, M.M. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc Δ716 knockout mice. Nat. Med. 2001, 7, 1048–1051. [Google Scholar] [CrossRef]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-ß-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef]
- Shao, J.; Jung, C.; Liu, C.; Sheng, H. Prostaglandin E2 stimulates the β-catenin/T cell factor-dependent transcription in colon cancer. J. Biol. Chem. 2005, 280, 26565–26572. [Google Scholar] [CrossRef]
- Rollins, B.J. Inflammatory chemokines in cancer growth and progression. Eur. J. Cancer 2006, 42, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.; Gravel, R.; Collu, R. Identification of G protein α subunit mutations in human growth hormone (GH)-and GH/prolactin-secreting pituitary tumors by single-strand conformation polymorphism (SSCP) analysis. Mol. Cell. Endocrinol. 1992, 87, 125–129. [Google Scholar] [CrossRef]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Green, J.A.; Suzuki, K.; Cho, B.; Willison, L.D.; Palmer, D.; Allen, C.D.; Schmidt, T.H.; Xu, Y.; Proia, R.L.; Coughlin, S.R. The sphingosine 1-phosphate receptor S1P 2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 2011, 12, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Miele, M.E.; Hicks, D.J.; Phillips, K.K.; Trent, J.M.; Weissman, B.E.; Welch, D.R. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996, 88, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; Van Wichen, D.; De Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Van Amerongen, R. Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a007914. [Google Scholar] [CrossRef]
- Rasola, A.; Fassetta, M.; De Bacco, F.; D’alessandro, L.; Gramaglia, D.; Di Renzo, M.F.; Comoglio, P. A positive feedback loop between hepatocyte growth factor receptor and β-catenin sustains colorectal cancer cell invasive growth. Oncogene 2007, 26, 1078–1087. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Liu, Z.-R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from β-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef]
- Birdsey, G.M.; Shah, A.V.; Dufton, N.; Reynolds, L.E.; Almagro, L.O.; Yang, Y.; Aspalter, I.M.; Khan, S.T.; Mason, J.C.; Dejana, E. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/β-catenin signaling. Dev. Cell 2015, 32, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Katanaev, V.L.; Ponzielli, R.; Sémériva, M.; Tomlinson, A. Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 2005, 120, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; DeCostanzo, A.J.; Liu, X.; Wang, H.-Y.; Hallagan, S.; Moon, R.T.; Malbon, C.C. G protein signaling from activated rat frizzled-1 to the β-catenin-Lef-Tcf pathway. Science 2001, 292, 1718–1722. [Google Scholar] [CrossRef] [PubMed]
- Slusarski, D.C.; Corces, V.G.; Moon, R.T. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 1997, 390, 410–413. [Google Scholar] [CrossRef]
- Major, M.B.; Roberts, B.S.; Berndt, J.D.; Marine, S.; Anastas, J.; Chung, N.; Ferrer, M.; Yi, X.; Stoick-Cooper, C.L.; Von Haller, P.D. New regulators of Wnt/β-catenin signaling revealed by integrative molecular screening. Sci. Signal. 2008, 1, 2000037. [Google Scholar] [CrossRef]
- Regard, J.B.; Cherman, N.; Palmer, D.; Kuznetsov, S.A.; Celi, F.S.; Guettier, J.-M.; Chen, M.; Bhattacharyya, N.; Wess, J.; Coughlin, S.R. Wnt/β-catenin signaling is differentially regulated by Gα proteins and contributes to fibrous dysplasia. Proc. Natl. Acad. Sci. USA 2011, 108, 20101–20106. [Google Scholar] [CrossRef]
- Miller, E.; Yang, J.; DeRan, M.; Wu, C.; Su, A.I.; Bonamy, G.M.; Liu, J.; Peters, E.C.; Wu, X. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 2012, 19, 955–962. [Google Scholar] [CrossRef]
- Mo, J.-S.; Yu, F.-X.; Gong, R.; Brown, J.H.; Guan, K.-L. Regulation of the Hippo–YAP pathway by protease-activated receptors (PARs). Genes Dev. 2012, 26, 2138–2143. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef]
- Yu, F.-X.; Luo, J.; Mo, J.-S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo–YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Bork, P.; Einbond, A.; Kastury, K.; Druck, T.; Negrini, M.; Huebner, K.; Lehman, D. Characterization of the Mammalian YAP (Yes-associated Protein) Gene and Its Role in Defining a Novel Protein Module, the WW Domain∗. J. Biol. Chem. 1995, 270, 14733–14741. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhang, Y.; Park, H.W.; Jewell, J.L.; Chen, Q.; Deng, Y.; Pan, D.; Taylor, S.S.; Lai, Z.-C.; Guan, K.-L. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013, 27, 1223–1232. [Google Scholar] [CrossRef]
- Kim, M.; Kim, M.; Lee, S.; Kuninaka, S.; Saya, H.; Lee, H.; Lee, S.; Lim, D.S. cAMP/PKA signalling reinforces the LATS–YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 2013, 32, 1543–1555. [Google Scholar] [CrossRef]
- Iglesias-Bartolome, R.; Torres, D.; Marone, R.; Feng, X.; Martin, D.; Simaan, M.; Chen, M.; Weinstein, L.S.; Taylor, S.S.; Molinolo, A.A. Inactivation of a Gα s–PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat. Cell Biol. 2015, 17, 793–803. [Google Scholar] [CrossRef]
- Gong, R.; Hong, A.W.; Plouffe, S.W.; Zhao, B.; Liu, G.; Yu, F.-X.; Xu, Y.; Guan, K.-L. Opposing roles of conventional and novel PKC isoforms in Hippo-YAP pathway regulation. Cell Res. 2015, 25, 985–988. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.-S.; Lu, W.; Lu, S. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M. Alternative Wnt signaling activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Xu, Q.; Zhao, Y.; Stevens, J.V.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin receptor and GPCR crosstalk stimulates YAP via PI3K and PKD in pancreatic cancer cells. Mol. Cancer Res. 2017, 15, 929–941. [Google Scholar] [CrossRef]
- Feng, R.; Gong, J.; Wu, L.; Wang, L.; Zhang, B.; Liang, G.; Zheng, H.; Xiao, H. MAPK and Hippo signaling pathways crosstalk via the RAF-1/MST-2 interaction in malignant melanoma. Oncol. Rep. 2017, 38, 1199–1205. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hansen, L.S.; Lillelund, C.; Andersen, C.; Gehl, J.; Christensen, J.F.; Pedersen, B.K.; Hojman, P. Exercise-induced catecholamines activate the hippo tumor suppressor pathway to reduce risks of breast cancer development. Cancer Res. 2017, 77, 4894–4904. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Nakagawa, K.; Yang, Z.; Ikeda, M.; Withanage, K.; Ishigami-Yuasa, M.; Okuno, Y.; Hata, S.; Nishina, H.; Hata, Y. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J. Biochem. 2011, 150, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Guan, K.-L. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol. Sci. 2013, 34, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.N.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Onken, M.D.; Makepeace, C.M.; Kaltenbronn, K.M.; Kanai, S.M.; Todd, T.D.; Wang, S.; Broekelmann, T.J.; Rao, P.K.; Cooper, J.A.; Blumer, K.J. Targeting nucleotide exchange to inhibit constitutively active G protein α subunits in cancer cells. Sci. Signal. 2018, 11, 546. [Google Scholar] [CrossRef]
- Annala, S.; Feng, X.; Shridhar, N.; Eryilmaz, F.; Patt, J.; Yang, J.; Pfeil, E.M.; Cervantes-Villagrana, R.D.; Inoue, A.; Häberlein, F. Direct targeting of Gαq and Gα11 oncoproteins in cancer cells. Sci. Signal. 2019, 12, 573. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 mediates MAPK pathway activation in GNAQ mutant uveal melanoma. Cancer Cell 2017, 31, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef]
- Borcherding, D.C.; Tong, W.; Hugo, E.; Barnard, D.; Fox, S.; LaSance, K.; Shaughnessy, E.; Ben-Jonathan, N. Expression and therapeutic targeting of dopamine receptor-1 (D1R) in breast cancer. Oncogene 2016, 35, 3103–3113. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Puente, X.S.; Gutiérrez-Fernández, A.; Ordóñez, G.R.; Hillier, L.W.; López-Otín, C. Comparative genomic analysis of human and chimpanzee proteases. Genomics 2005, 86, 638–647. [Google Scholar] [CrossRef]
- Burger, M.M. Proteolytic enzymes initiating cell division and escape from contact inhibition of growth. Nature 1970, 227, 170–171. [Google Scholar] [CrossRef]
- Chen, L.B.; Buchanan, J.M. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc. Natl. Acad. Sci. USA 1975, 72, 131–135. [Google Scholar] [CrossRef]
- Carney, D.H.; Cunningham, D.D. Initiation of chick cell division by trypsin action at the cell surface. Nature 1977, 268, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.H.; Cunningham, D.D. Cell surface action of thrombin is sufficient to initiate division of chick cells. Cell 1978, 14, 811–823. [Google Scholar] [CrossRef]
- Coughlin, S.R. Protease-activated receptors start a family. Proc. Natl. Acad. Sci. USA 1994, 91, 9200. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.-K.H.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506. [Google Scholar] [CrossRef]
- Kahn, M.L.; Zheng, Y.-W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Xu, W.-f.; Andersen, H.; Whitmore, T.E.; Presnell, S.R.; Yee, D.P.; Ching, A.; Gilbert, T.; Davie, E.W.; Foster, D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA 1998, 95, 6642–6646. [Google Scholar] [CrossRef]
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212. [Google Scholar] [CrossRef]
- Coughlin, S.R. How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11023–11027. [Google Scholar] [CrossRef]
- Arakaki, A.K.; Pan, W.-A.; Trejo, J. GPCRs in cancer: Protease-activated receptors, endocytic adaptors and signaling. Int. J. Mol. Sci. 2018, 19, 1886. [Google Scholar] [CrossRef]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-Activated Receptors (PAR1 and PAR2) Contribute to Tumor Cell Motility and Metastasis11NIH grants CA85405 (BM Mueller), HL16411 (W. Ruf), and HL60742 (W. Ruf). Mol. Cancer Res. 2004, 2, 395–402. [Google Scholar]
- Jiang, P.; De Li, S.; Li, Z.G.; Zhu, Y.C.; Yi, X.J.; Li, S.M. The expression of protease-activated receptors in esophageal carcinoma cells: The relationship between changes in gene expression and cell proliferation, apoptosis in vitro and growing ability in vivo. Cancer Cell Int. 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Versteeg, H.H.; Schillert, A.; Yokota, N.; Petersen, L.C.; Mueller, B.M.; Ruf, W. Cooperation of tissue factor cytoplasmic domain and PAR2 signaling in breast cancer development. Blood 2010, 116, 6106–6113. [Google Scholar] [CrossRef]
- Parisis, N.; Metodieva, G.; Metodiev, M.V. Pseudopodial and β-arrestin-interacting proteomes from migrating breast cancer cells upon PAR2 activation. J. Proteom. 2013, 80, 91–106. [Google Scholar] [CrossRef]
- Hu, L.; Xia, L.; Zhou, H.; Wu, B.; Mu, Y.; Wu, Y.; Yan, J. TF/FVIIa/PAR2 promotes cell proliferation and migration via PKCα and ERK-dependent c-Jun/AP-1 pathway in colon cancer cell line SW620. Tumor Biol. 2013, 34, 2573–2581. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Ellies, L.G.; Andrade-Gordon, P.; Mueller, B.M.; Ruf, W. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer Res. 2008, 68, 7219–7227. [Google Scholar] [CrossRef]
- Srinivasan, S.; Ranga, R.S.; Burikhanov, R.; Han, S.-S.; Chendil, D. Par-4-dependent apoptosis by the dietary compound withaferin A in prostate cancer cells. Cancer Res. 2007, 67, 246–253. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, G.; Jiang, P.; Xiang, Y.; Li, W.; Lee, W.; Zhang, Y. Decreased expression of protease-activated receptor 4 in human gastric cancer. Int. J. Biochem. Cell Biol. 2011, 43, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.V.; Pan, T.-C.; Ruth, J.; Feng, Y.; Zhou, A.; Pant, D.; Grimley, J.S.; Wandless, T.J.; DeMichele, A.; Chodosh, L.A. Par-4 downregulation promotes breast cancer recurrence by preventing multinucleation following targeted therapy. Cancer Cell 2013, 24, 30–44. [Google Scholar] [CrossRef]
- Nagai, M.A.; Gerhard, R.; Salaorni, S.; Fregnani, J.H.T.G.; Nonogaki, S.; Netto, M.M.; Soares, F.A. Down-regulation of the candidate tumor suppressor gene PAR-4 is associated with poor prognosis in breast cancer. Int. J. Oncol. 2010, 37, 41–49. [Google Scholar] [CrossRef]
- Gratio, V.; Walker, F.; Lehy, T.; Laburthe, M.; Darmoul, D. Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src and ErbB-2 kinase. Int. J. Cancer 2009, 124, 1517–1525. [Google Scholar] [CrossRef]
- Kancharla, A.; Maoz, M.; Jaber, M.; Agranovich, D.; Peretz, T.; Grisaru-Granovsky, S.; Uziely, B.; Bar-Shavit, R. PH motifs in PAR 1&2 endow breast cancer growth. Nat. Commun. 2015, 6, 1–12. [Google Scholar]
- Gschwind, A.; Prenzel, N.; Ullrich, A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002, 62, 6329–6336. [Google Scholar]
- Kalmes, A.; Vesti, B.R.; Daum, G.N.; Abraham, J.A.; Clowes, A.W. Heparin blockade of thrombin-induced smooth muscle cell migration involves inhibition of epidermal growth factor (EGF) receptor transactivation by heparin-binding EGF-like growth factor. Circ. Res. 2000, 87, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mueller, B.M. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochem. Biophys. Res. Commun. 2006, 344, 1263–1270. [Google Scholar] [CrossRef]
- Su, S.; Li, Y.; Luo, Y.; Sheng, Y.; Su, Y.; Padia, R.; Pan, Z.; Dong, Z.; Huang, S. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene 2009, 28, 3047–3057. [Google Scholar] [CrossRef]
- Lidfeldt, J.; Bendahl, P.-O.; Forsare, C.; Malmström, P.; Fernö, M.; Belting, M. Protease activated receptors 1 and 2 correlate differently with breast cancer aggressiveness depending on tumor ER status. PLoS ONE 2015, 10, e0134932. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Mueller, B.M. Thrombin generation and the pathogenesis of cancer. Semin. Thromb. Hemost. 2006, 32, 61–68. [Google Scholar]
- Grisaru-Granovsky, S.; Kumar Nag, J.; Zakar, L.; Rudina, T.; Lal Gupta, C.; Maoz, M.; Kozlova, D.; Bar-Shavit, R. PAR1&2 driven placenta EVT invasion act via LRP5/6 as coreceptors. FASEB J. 2020, 34, 15701–15717. [Google Scholar] [PubMed]
- Almendro, V.; García-Recio, S.; Gascón, P. Tyrosine kinase receptor transactivation associated to G protein-coupled receptors. Curr. Drug Targets 2010, 11, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.E.; Qian, Y.; Liu, G.; Chen, H.; Chen, X. p53, a target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J. Biol. Chem. 2012, 287, 30117–30127. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef]
- Ge, C.; Yu, M.; Zhang, C. G protein-coupled receptor 30 mediates estrogen-induced proliferation of primordial germ cells via EGFR/Akt/β-catenin signaling pathway. Endocrinology 2012, 153, 3504–3516. [Google Scholar] [CrossRef]
- Luo, L.-J.; Liu, F.; Lin, Z.-K.; Xie, Y.-F.; Xu, J.-L.; Tong, Q.-C.; Shu, R. Genistein regulates the IL-1 beta induced activation of MAPKs in human periodontal ligament cells through G protein-coupled receptor 30. Arch. Biochem. Biophys. 2012, 522, 9–16. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Chen, Z.; Zhang, K.; Yang, X.; Wu, Y.; Chen, X.; Huang, H.; Liu, H.; Cai, S.; Du, J. The activation of G protein-coupled receptor 30 (GPR30) inhibits proliferation of estrogen receptor-negative breast cancer cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1428. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Lappano, R.; Madeo, A.; Chimento, A.; Prossnitz, E.R.; Cappello, A.R.; Dolce, V.; Abonante, S.; Pezzi, V.; Maggiolini, M. G-Protein–Coupled Receptor 30 and Estrogen Receptor-α Are Involved in the Proliferative Effects Induced by Atrazine in Ovarian Cancer Cells. Environ. Health Perspect. 2008, 116, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Albanito, L.; Madeo, A.; Rago, V.; Carpino, A.; Musti, A.M.; Picard, D.; Andò, S.; Maggiolini, M. 17β-Estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein-coupled receptor GPR30. Mol. Pharmacol. 2006, 70, 1414–1423. [Google Scholar] [CrossRef]
- Vivacqua, A.; Bonofiglio, D.; Recchia, A.G.; Musti, A.M.; Picard, D.; Andò, S.; Maggiolini, M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol. 2006, 20, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Abram, P. Fulvestrant, a new treatment option for advanced breast cancer: Tolerability versus existing agents. Ann. Oncol. 2006, 17, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Agha, R.A.; Sevdalis, N. Surveillance and quality improvement in the United Kingdom: Is there a meeting point? Surgeon 2014, 12, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.C.; Sun, H.; Wang, D.; Rusovici, R.; Castleberry, A.; Hall, R.A.; Shim, H. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am. J. Physiol. Cell Physiol. 2005, 289, C2–C11. [Google Scholar] [CrossRef]
- Shida, D.; Kitayama, J.; Yamaguchi, H.; Okaji, Y.; Tsuno, N.H.; Watanabe, T.; Takuwa, Y.; Nagawa, H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003, 63, 1706–1711. [Google Scholar] [PubMed]
- Yang, M.; Zhong, W.W.; Srivastava, N.; Slavin, A.; Yang, J.; Hoey, T.; An, S. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the β-catenin pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 6027–6032. [Google Scholar] [CrossRef] [PubMed]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin–LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Choi, J.W.; Lim, S.; Oh, Y.-S.; Kim, E.-K.; Kim, S.-H.; Kim, Y.-H.; Heo, K.; Kim, J.; Kim, J.K.; Yang, Y.R. Subtype-specific role of phospholipase C-β in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell. Signal. 2010, 22, 1153–1161. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef]
- Komachi, M.; Sato, K.; Tobo, M.; Mogi, C.; Yamada, T.; Ohta, H.; Tomura, H.; Kimura, T.; Im, D.S.; Yanagida, K. Orally active lysophosphatidic acid receptor antagonist attenuates pancreatic cancer invasion and metastasis in vivo. Cancer Sci. 2012, 103, 1099–1104. [Google Scholar] [CrossRef]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef]
- David, M.; Ribeiro, J.; Descotes, F.; Serre, C.-M.; Barbier, M.; Murone, M.; Clézardin, P.; Peyruchaud, O. Targeting lysophosphatidic acid receptor type 1 with Debio 0719 inhibits spontaneous metastasis dissemination of breast cancer cells independently of cell proliferation and angiogenesis. Int. J. Oncol. 2012, 40, 1133–1141. [Google Scholar] [CrossRef]
- Sidduri, A.; Budd, D.C.; Fuentes, M.E.; Lambros, T.; Ren, Y.; Roongta, V.; Schoenfeld, R.C.; Gillespie, P.; Stevenson, C.S.; Truitt, T. Discovery of novel non-carboxylic acid 5-amino-4-cyanopyrazole derivatives as potent and highly selective LPA1R antagonists. Bioorgan. Med. Chem. Lett. 2014, 24, 4450–4454. [Google Scholar] [CrossRef] [PubMed]
- Budd, D.; Qian, Y.; Schoenfeld, R.; Sidduri, A. Preparation of Substituted Cyanopyrazole Compounds as Lysophosphatidic Acid (LPA) Antagonists; F. Hoffmann-La Roche AG: Bassel, Switzerland, 2014. [Google Scholar]
- Erickson, J.; Goddard, J.G.; Kiefer, M. Methods for Detecting Compounds which Modulate the Activity of an LPA Receptor. U.S. Patents 6,485,922, 26 November 2002. [Google Scholar]
- Llona-Minguez, S.; Ghassemian, A.; Helleday, T. Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res. 2015, 58, 51–75. [Google Scholar] [CrossRef]
- Acconcia, F. The network of angiotensin receptors in breast cancer. Cells 2020, 9, 1336. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.; Rodewald, M.; Fraser, H.; Hack, G.; Konrad, R.; Kreienberg, R.; Wulff, C. Potential role of renin–angiotensin-system for tumor angiogenesis in receptor negative breast cancer. Gynecol. Oncol. 2008, 109, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.R.; Ateeq, B.; Cao, Q.; Tomlins, S.A.; Mehra, R.; Laxman, B.; Kalyana-Sundaram, S.; Lonigro, R.J.; Helgeson, B.E.; Bhojani, M.S. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc. Natl. Acad. Sci. USA 2009, 106, 10284–10289. [Google Scholar] [CrossRef]
- Arafat, H.A.; Gong, Q.; Chipitsyna, G.; Rizvi, A.; Saa, C.T.; Yeo, C.J. Antihypertensives as novel antineoplastics: Angiotensin-I-converting enzyme inhibitors and angiotensin II type 1 receptor blockers in pancreatic ductal adenocarcinoma. J. Am. Coll. Surg. 2007, 204, 996–1005. [Google Scholar] [CrossRef]
- Juillerat-Jeanneret, L.; Celerier, J.; Bernasconi, C.C.; Nguyen, G.; Wostl, W.; Maerki, H.; Janzer, R.; Corvol, P.; Gasc, J. Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br. J. Cancer 2004, 90, 1059–1068. [Google Scholar] [CrossRef]
- Suganuma, T.; Ino, K.; Shibata, K.; Kajiyama, H.; Nagasaka, T.; Mizutani, S.; Kikkawa, F. Functional expression of the angiotensin II type1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin. Cancer Res. 2005, 11, 2686–2694. [Google Scholar] [CrossRef]
- Arrieta, O.; Pineda-Olvera, B.; Guevara-Salazar, P.; Hernández-Pedro, N.; Morales-Espinosa, D.; Cerón-Lizarraga, T.; González-De la Rosa, C.; Rembao, D.; Segura-Pacheco, B.; Sotelo, J. Expression of AT1 and AT2 angiotensin receptors in astrocytomas is associated with poor prognosis. Br. J. Cancer 2008, 99, 160–166. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.; Wang, Q.; Yu, K.; Wang, R.; Sun, J. Blockade of vascular endothelial growth factor-A/receptor 2 exhibits a protective effect on angiotensin-II stimulated podocytes. Mol. Med. Rep. 2015, 12, 4340–4345. [Google Scholar] [CrossRef][Green Version]
- Ohashi, H.; Takagi, H.; Oh, H.; Suzuma, K.; Suzuma, I.; Miyamoto, N.; Uemura, A.; Watanabe, D.; Murakami, T.; Sugaya, T. Phosphatidylinositol 3-kinase/Akt regulates angiotensin II–induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ. Res. 2004, 94, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Elbaz, N.; Bedecs, K.; Masson, M.; Sutren, M.N.; Strosberg, A.D.; Nahmias, C. Functional trans-inactivation of insulin receptor kinase by growth-inhibitory angiotensin II AT2 receptor. Mol. Endocrinol. 2000, 14, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Karpe, P.A.; Gupta, J.; Marthong, R.F.; Ramarao, P.; Tikoo, K. Insulin resistance induces a segmental difference in thoracic and abdominal aorta: Differential expression of AT1 and AT2 receptors. J. Hypertens. 2012, 30, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Nouet, S.; Amzallag, N.; Li, J.-M.; Louis, S.; Seitz, I.; Cui, T.-X.; Alleaume, A.-M.; Di Benedetto, M.; Boden, C.; Masson, M. Trans-inactivation of receptor tyrosine kinases by novel angiotensin II AT2 receptor-interacting protein, ATIP. J. Biol. Chem. 2004, 279, 28989–28997. [Google Scholar] [CrossRef]
- Fujiyama, S.; Matsubara, H.; Nozawa, Y.; Maruyama, K.; Mori, Y.; Tsutsumi, Y.; Masaki, H.; Uchiyama, Y.; Koyama, Y.; Nose, A. Angiotensin AT1 and AT2 receptors differentially regulate angiopoietin-2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding–epidermal growth factor (EGF)–mediated EGF receptor transactivation. Circ. Res. 2001, 88, 22–29. [Google Scholar] [CrossRef]
- Seibold, S.; Rudroff, C.; Weber, M.; Galle, J.; Wanner, C.; Marx, M. Identification of a new tumor suppressor gene located at chromosome 8p21.3–22. FASEB J. 2003, 17, 1180–1182. [Google Scholar] [CrossRef]
- Imai, N.; Hashimoto, T.; Kihara, M.; Yoshida, S.-I.; Kawana, I.; Yazawa, T.; Kitamura, H.; Umemura, S. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab. Investig. 2007, 87, 189–198. [Google Scholar] [CrossRef]
- Lindberg, H.; Nielsen, D.; Jensen, B.V.; Eriksen, J.; Skovsgaard, T. Angiotensin converting enzyme inhibitors for cancer treatment? Acta Oncol. 2004, 43, 142–152. [Google Scholar]
- Chaudhary, S.K.; De, A.; Bhadra, S.; Mukherjee, P.K. Angiotensin-converting enzyme (ACE) inhibitory potential of standardized Mucuna pruriens seed extract. Pharmaceut. Biol. 2015, 53, 1614–1620. [Google Scholar] [CrossRef]
- Hashemzadeh, M.; Park, S.; Ju, H.; Movahed, M.R. A novel design of combining the angiotensin converting enzyme (ACE) inhibitor captopril with the angiotensin receptor blocker (ARB) losartan using homo coupling via PEG diacid linker. Recent Pat. Cardiovasc. Drug Discov. 2013, 8, 221–225. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.; Huang, X.; Lu, X. Captopril-polyethyleneimine conjugate modified gold nanoparticles for co-delivery of drug and gene in anti-angiogenesis breast cancer therapy. J. Biomater. Sci. Polym. Edit. 2015, 26, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Odović, J.; Marković, B.; Vladimirov, S.; Karljiković-Rajić, K. In Vitro modeling of angiotensin-converting enzyme inhibitor’s absorption with chromatographic retention data and selected molecular descriptors. J. Chromatogr. B 2014, 953, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Hicks, B.M.; Filion, K.B.; Yin, H.; Sakr, L.; Udell, J.A.; Azoulay, L. Angiotensin converting enzyme inhibitors and risk of lung cancer: Population based cohort study. BMJ 2018, 363, k4209. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wang, W.; Wang, C.; Wang, Y.; Zhou, J.; Ding, Y.; Wang, X.; Jin, Y. A chitosan-graft-PEI-candesartan conjugate for targeted co-delivery of drug and gene in anti-angiogenesis cancer therapy. Biomaterials 2014, 35, 8450–8466. [Google Scholar] [CrossRef]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Takahara, N.; Ito, Y.; Matsubara, S.; Uchino, R.; Yagioka, H.; Arizumi, T. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Investig. New Drugs 2013, 31, 1294–1299. [Google Scholar] [CrossRef]
- Okazaki, M.; Fushida, S.; Harada, S.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Tajima, H.; Ninomiya, I.; Fujimura, T.; Ohta, T. The angiotensin II type 1 receptor blocker candesartan suppresses proliferation and fibrosis in gastric cancer. Cancer Lett. 2014, 355, 46–53. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Sasaki, T.; Tsuchida, A.; Chayama, K. Angiotensin II type 1 receptor expression in human pancreatic cancer and growth inhibition by angiotensin II type 1 receptor antagonist. FEBS Lett. 2001, 495, 197–200. [Google Scholar] [CrossRef]
- Arrieta, O.; Guevara, P.; Escobar, E.; García-Navarrete, R.; Pineda, B.; Sotelo, J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 2005, 92, 1247–1252. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Lin, C.-L.; Lin, C.-C.; Hsu, W.-H.; Lin, C.-D.; Wang, I.; Hsu, C.-Y.; Kao, C.-H. Association between angiotensin-converting enzyme inhibitors and lung cancer—A nationwide, population-based, propensity score-matched cohort study. Cancers 2020, 12, 747. [Google Scholar] [CrossRef]
- Kim, K.W.; Paul, P.; Qiao, J.; Lee, S.; Chung, D.H. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy 2013, 9, 1579–1590. [Google Scholar] [CrossRef]
- Qiao, J.; Kang, J.; Ishola, T.A.; Rychahou, P.G.; Evers, B.M.; Chung, D.H. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proc. Natl. Acad. Sci. USA 2008, 105, 12891–12896. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Qiao, J.; Paul, P.; Chung, D.H. Integrin β1 is critical for gastrin-releasing peptide receptor-mediated neuroblastoma cell migration and invasion. Surgery 2013, 154, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Patel, O.; Shulkes, A.; Baldwin, G.S. Gastrin-releasing peptide and cancer. Biochim. Biophys. Acta Rev. Cancer 2006, 1766, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Qiao, J.; Paul, P.; O’connor, K.L.; Evers, B.M.; Chung, D.H. FAK is a critical regulator of neuroblastoma liver metastasis. Oncotarget 2012, 3, 1576. [Google Scholar] [CrossRef] [PubMed]
- Egloff, A.M.; Liu, X.; Davis, A.L.G.; Trevelline, B.K.; Vuga, M.; Siegfried, J.M.; Grandis, J.R. Elevated gastrin-releasing peptide receptor mRNA expression in buccal mucosa: Association with head and neck squamous cell carcinoma. Head Neck 2013, 35, 270–279. [Google Scholar] [CrossRef]
- Zhang, Q.; Bhola, N.E.; Lui, V.W.Y.; Siwak, D.R.; Thomas, S.M.; Gubish, C.T.; Siegfried, J.M.; Mills, G.B.; Shin, D.; Grandis, J.R. Antitumor mechanisms of combined gastrin-releasing peptide receptor and epidermal growth factor receptor targeting in head and neck cancer. Mol. Cancer Ther. 2007, 6, 1414–1424. [Google Scholar] [CrossRef]
- Liu, X.; Carlisle, D.L.; Swick, M.C.; Gaither-Davis, A.; Grandis, J.R.; Siegfried, J.M. Gastrin-releasing peptide activates Akt through the epidermal growth factor receptor pathway and abrogates the effect of gefitinib. Exp. Cell Res. 2007, 313, 1361–1372. [Google Scholar] [CrossRef]
- Li, X.; Lv, Y.; Yuan, A.; Yi, S.; Ma, Y.; Li, Z. Gastrin-releasing peptide promotes the growth of HepG2 cells via EGFR-independent ERK1/2 activation. Oncol. Rep. 2010, 24, 441–448. [Google Scholar]
- Chaudhry, A.; Carrasquillo, J.A.; Avis, I.L.; Shuke, N.; Reynolds, J.C.; Bartholomew, R.; Larson, S.M.; Cuttitta, F.; Johnson, B.E.; Mulshine, J.L. Phase I and imaging trial of a monoclonal antibody directed against gastrin-releasing peptide in patients with lung cancer. Clin. Cancer Res. 1999, 5, 3385–3393. [Google Scholar]
- Fang, J.; Lu, Y.; Ouyang, K.; Wu, G.; Zhang, H.; Liu, Y.; Chen, Y.; Lin, M.; Wang, H.; Jin, L. Specific antibodies elicited by a novel DNA vaccine targeting gastrin-releasing peptide inhibit murine melanoma growth in vivo. Clin. Vaccine Immunol. 2009, 16, 1033–1039. [Google Scholar] [CrossRef][Green Version]
- Martínez, A.; Zudaire, E.; Julian, M.; Moody, T.W.; Cuttitta, F. Gastrin-releasing peptide (GRP) induces angiogenesis and the specific GRP blocker 77427 inhibits tumor growth in vitro and in vivo. Oncogene 2005, 24, 4106–4113. [Google Scholar] [CrossRef]
- Szepeshazi, K.; Schally, A.V.; Nagy, A.; Wagner, B.W.; Bajo, A.M.; Halmos, G. Preclinical evaluation of therapeutic effects of targeted cytotoxic analogs of somatostatin and bombesin on human gastric carcinomas. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 98, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Mantey, S.A.; Pradhan, T.K.; Schumann, M.; Nakagawa, T.; Martinez, A.; Fuselier, J.; Coy, D.H.; Jensen, R.T. Development of high affinity camptothecin-bombesin conjugates that have targeted cytotoxicity for bombesin receptor-containing tumor cells. J. Biol. Chem. 2004, 279, 23580–23589. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Dev, K.K. The structure and function of the S1P1 receptor. Trends Pharmacol. Sci. 2013, 34, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Usui, S.; Sugimoto, N.; Takuwa, N.; Sakagami, S.; Takata, S.; Kaneko, S.; Takuwa, Y. Blood lipid mediator sphingosine 1-phosphate potently stimulates platelet-derived growth factor-A and-B chain expression through S1P1-Gi-Ras-MAPK-dependent induction of Krüppel-like factor 5. J. Biol. Chem. 2004, 279, 12300–12311. [Google Scholar] [CrossRef] [PubMed]
- Safarian, F.; Khallaghi, B.; Ahmadiani, A.; Dargahi, L. Activation of S1P1 receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J. Mol. Neurosci. 2015, 56, 177–187. [Google Scholar] [CrossRef]
- Fujii, K.; Machida, T.; Iizuka, K.; Hirafuji, M. Sphingosine 1-phosphate increases an intracellular Ca2+ concentration via S1P3 receptor in cultured vascular smooth muscle cells. J. Pharm. Pharmacol. 2014, 66, 802–810. [Google Scholar] [CrossRef]
- Rhee, S.A.; Zhang, P.; Hunter, K.; Mama, S.; Caraballo, R.; Holzberg, A.; Seftel, R.; Seftel, A.; Echols, K.; DiSanto, M. Pelvic organ prolapse is associated with alteration of sphingosine-1-phosphate/Rho-kinase signalling pathway in human vaginal wall. J. Obstet. Gynaecol. 2015, 35, 726–732. [Google Scholar] [CrossRef]
- Deng, L.; Zhou, J.-F.; Sellers, R.S.; Li, J.-F.; Nguyen, A.V.; Wang, Y.; Orlofsky, A.; Liu, Q.; Hume, D.A.; Pollard, J.W. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am. J. Pathol. 2010, 176, 952–967. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef]
- Urbano, M.; Guerrero, M.; Rosen, H.; Roberts, E. Modulators of the Sphingosine 1-phosphate receptor 1. Bioorganic Med. Chem. Lett. 2013, 23, 6377–6389. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.W.; Lee, D.T.W.; Ling, M.T.; Zhou, C.; Man, K.; Ho, J.; Chan, F.L.; Wang, X.; Wong, Y.C. FTY720, a fungus metabolite, inhibits in vivo growth of androgen-independent prostate cancer. Int. J. Cancer 2005, 117, 1039–1048. [Google Scholar] [CrossRef]
- Schmid, G.; Guba, M.; Papyan, A.; Ischenko, I.; Brückel, M.; Bruns, C.; Jauch, K.-W.; Graeb, C. FTY720 inhibits tumor growth and angiogenesis. Transplant. Proc. 2005, 37, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Qi, Y.; Wadham, C.; Wang, L.; Warren, A.; Di, W.; Xia, P. FTY720 induces necrotic cell death and autophagy in ovarian cancer cells: A protective role of autophagy. Autophagy 2010, 6, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Man, K.; Ho, J.W.; Sun, C.K.; Ng, K.T.; Wang, X.H.; Wong, Y.C.; Ng, I.O.; Xu, R.; Fan, S.T. FTY720 induces apoptosis of human hepatoma cell lines through PI3-K-mediated Akt dephosphorylation. Carcinogenesis 2004, 25, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.; Guerrero, M.; Urbano, M.; Rosen, H. Sphingosine 1-phosphate receptor agonists: A patent review (2010–2012). Expert Opin. Ther. Pat. 2013, 23, 817–841. [Google Scholar] [CrossRef]
- Albert, R.; Ehrhardt, C.; Ettmayer, P.; Hinterding, K.; Högenauer, K.; Nussbaumer, P. Aminopropanol Derivatives as Sphingosine-1-Phosphate Receptor Modulators. U.S. Patent 7,825,260, 2 November 2010. [Google Scholar]
- Heidelbaugh, T.M.; Nguyen, P.X. Novel Compounds as Receptor Modulators with Therapeutic Utility. U.S. Patent 4,325,121, 15 March 2017. Available online: https://patents.google.com/patent/EP2504323B1/en%20US4325121.pdf (accessed on 21 November 2021).
- Lin, X.; Ren, F.; Si, Y. 1, 2, 4-Oxadiazol Derivatives, Their Pharmaceutical Compositions and Their Use as Sphingosine 1-Phosphate 1 Receptor Agonists. U.S. Patent Application 13,379,214, 26 April 2012. [Google Scholar]
- Heidelbaugh, T.M.; Nguyen, P.X. Compounds as Receptor Modulators with Therapeutic Utility. U.S. Patent 8,653,270, 18 February 2014. Available online: https://data.epo.org/gpi/EP2643331A1-NOVEL-COMPOUNDS-AS-RECEPTOR-MODULATORS-WITH-THERAPEUTIC-UTILITY (accessed on 21 November 2021).
- Labrie, F.; Belanger, A.; Luu-The, V.; Labrie, C.; Simard, J.; Cusan, L.; Gomez, J.; Candas, B. Gonadotropin-releasing hormone agonists in the treatment of prostate cancer. Endocr. Rev. 2005, 26, 361–379. [Google Scholar] [CrossRef]
- Meyer, T.; Caplin, M.; Palmer, D.; Valle, J.W.; Larvin, M.; Waters, J.; Coxon, F.; Borbath, I.; Peeters, M.; Nagano, E. A phase Ib/IIa trial to evaluate the CCK2 receptor antagonist Z-360 in combination with gemcitabine in patients with advanced pancreatic cancer. Eur. J. Cancer 2010, 46, 526–533. [Google Scholar] [CrossRef]
- Paciaroni, N.G.; Norwood, V.M., IV; Ratnayake, R.; Luesch, H.; Huigens, R.W., III. Yohimbine as a starting point to access diverse natural product-like agents with re-programmed activities against cancer-relevant GPCR targets. Bioorg. Med. Chem. 2020, 28, 115546. [Google Scholar] [CrossRef]
- Usman, S.; Khawer, M.; Rafique, S.; Naz, Z.; Saleem, K. The current status of anti-GPCR drugs against different cancers. J. Pharm. Anal. 2020, 10, 517–521. [Google Scholar] [CrossRef]
- Soond, S.M.; Zamyatnin, A.A., Jr. Targeting G protein-coupled receptors in cancer therapy. Adv. Cancer Res. 2020, 145, 49–97. [Google Scholar] [PubMed]
- Xiang, Y.; Yao, X.; Chen, K.; Wang, X.; Zhou, J.; Gong, W.; Yoshimura, T.; Huang, J.; Wang, R.; Wu, Y. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am. J. Cancer Res. 2016, 6, 2599. [Google Scholar] [PubMed]
- Su, L.; Peng, J.; Ge, Y. Formyl peptide receptor 2 mediated chemotherapeutics drug resistance in colon cancer cells. Eur. Rev. Med. Pharmacol. Sci 2018, 22, 95–100. [Google Scholar] [PubMed]
- Schuller, H.M. Regulatory role of G protein-coupled receptors in pancreatic cancer development and progression. Curr. Med. Chem. 2018, 25, 2566–2575. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Rezmann, L.; Imamura, K.; Wang, L.; Catt, K.; Tikellis, C.; Louis, W.J.; Frauman, A.G.; Louis, S.N. Functional angiotensin II type 2 receptors inhibit growth factor signaling in LNCaP and PC3 prostate cancer cell lines. Prostate 2008, 68, 651–660. [Google Scholar] [CrossRef]
- Zhou, C.; Dai, X.; Chen, Y.; Shen, Y.; Lei, S.; Xiao, T.; Bartfai, T.; Ding, J.; Wang, M.-W. G protein-coupled receptor GPR160 is associated with apoptosis and cell cycle arrest of prostate cancer cells. Oncotarget 2016, 7, 12823. [Google Scholar] [CrossRef]
- Maussang, D.; Mujić-Delić, A.; Descamps, F.J.; Stortelers, C.; Vanlandschoot, P.; Stigter-van Walsum, M.; Vischer, H.F.; van Roy, M.; Vosjan, M.; Gonzalez-Pajuelo, M. Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 2013, 288, 29562–29572. [Google Scholar] [CrossRef]
- Voisin, T.; El Firar, A.; Fasseu, M.; Rouyer-Fessard, C.; Descatoire, V.; Walker, F.; Paradis, V.; Bedossa, P.; Henin, D.; Lehy, T. Aberrant expression of OX1 receptors for orexins in colon cancers and liver metastases: An openable gate to apoptosis. Cancer Res. 2011, 71, 3341–3351. [Google Scholar] [CrossRef]
- Insel, P.A.; Sriram, K.; Wiley, S.Z.; Wilderman, A.; Katakia, T.; McCann, T.; Yokouchi, H.; Zhang, L.; Corriden, R.; Liu, D. GPCRomics: GPCR expression in cancer cells and tumors identifies new, potential biomarkers and therapeutic targets. Front. Pharmacol. 2018, 9, 431. [Google Scholar] [CrossRef]
- Wiley, S.Z.; Sriram, K.; Liang, W.; Chang, S.E.; French, R.; McCann, T.; Sicklick, J.; Nishihara, H.; Lowy, A.M.; Insel, P.A. GPR68, a proton-sensing GPCR, mediates interaction of cancer-associated fibroblasts and cancer cells. FASEB J. 2018, 32, 1170–1183. [Google Scholar] [CrossRef]
- Feigin, M.E.; Xue, B.; Hammell, M.C.; Muthuswamy, S.K. G-protein–coupled receptor GPR161 is overexpressed in breast cancer and is a promoter of cell proliferation and invasion. Proc. Natl. Acad. Sci. USA 2014, 111, 4191–4196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Telonis, A.; Jing, Y.; Xia, N.; Biederman, L.; Jimbo, M.; Blanco, F.; Londin, E.; Brody, J.; Rigoutsos, I. GPRC5A is a potential oncogene in pancreatic ductal adenocarcinoma cells that is upregulated by gemcitabine with help from HuR. Cell Death Dis. 2016, 7, e2294. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 6220. [Google Scholar] [CrossRef] [PubMed]
| Receptor/s | Ligand/s | Pathway/s | Cancer Type |
|---|---|---|---|
| Lysophosphatidic acid receptors LPA1-6) | LPA | Rho-dependent pathways [26,27] | |
| β-cantenin stabilization [33,34] | |||
| Kruppel-like factor 5 [35] | |||
| Protease-activated receptors (PAR1&2) LPA | Thrombin, trypsin, or TFLLRN (PAR1) or SLIGKV (PAR2) Lysophosphatidic acid (Gαq) | Hippo/YAP pathways via activation of Gα12/13-coupled receptors or Gαq. Inhibition of Hippo pathway (via the inhibition of Lats1/2 kinases) [36] | |
| Frizzled (Fz) PAR1 Parathyroid receptor1 (PTHR1) | Wnt 3A (canonical pathway) | Canonical Wnt signaling stabilization of β-catenin and its transcription activity [40] |
|
| Thrombin or TFLLRN | |||
| PTH | |||
| Chemokine receptor (CXCR4) | CXCL12, SDF-1 | PI3K, Akt, Src, PIP2, IP3, Ras, Raf, ERK1/2, PLC, JNK [46] |
|
| Endothelin receptors (ETAR and ETBR) | Endothelin 1–3 (ET-1, ET-2, ET-3) | c-Src/cross-talk with EGFR | |
| β-arrestin1 or 2 PDZRhoGEF and Rho A, C | |||
| β-catenin stabilization [49,50] | |||
| Prostaglandin receptors (PE2, PE4) | PGE2 | Cyclooxygenase pathway, PI3K (coupling to Gαi) [51,52,53] | |
| Bradykinin receptor Type 1 and 2 (B1R, B2R) | Kinins | Gαq and cross-talk with EGFR Ras, Raf, ERK |
|
| Sphingosine 1- phosphate receptor (S1PR) | S1P | Ras-ERK, PI3K/-Akt/-Rac, Rho, STAT3 (coupling to Gαi) [55,56] |
|
| Angiotensin II type 1 receptor | Angiotensin II | TNF-α, ERK1/2, NF-κB, STAT [60,61] | |
| Gastrin-releasing peptide receptor | Gastrin-releasing peptide | NF-κB, p38MAPK, PI3K/-Akt [63,64] |
|
| Drugs | Receptor | Cancer Types | Year of Approval |
|---|---|---|---|
| Cabergoline | Dopamine receptor D1 (DRD1) | Neuroendocrine tumors, pituitary tumors | 1996 |
| Lanreotide | Somatostatin receptor (SSTR) | Pancreatic cancer | 2007 |
| Degarelix | GnRH | Prostate cancer | 2008 |
| Vismodegib (Erivedge) | SMO | Locally advanced and metastatic basal cell carcinoma | 2012 |
| Sonidegib (Odomzo) | SMO | Locally advanced and metastatic basal cell carcinoma | 2015 |
| Mogamulizumab | CCR4 | T-cell lymphoma | 2018 |
| Cancer | Inhibitor | Type of Molecule | Receptor | Phase | Sponsor/s |
|---|---|---|---|---|---|
| Head and neck cancer | GDC-0449 (Vismodegib) | Small molecule | SMO | Phase II | Sue Yom in collaboration with Genentech, Inc. |
| Ovarian cancer | GDC-0449 (Vismodegib) | Small molecule | SMO | Phase II | Genentech, Inc. |
| Propranolol (beta-blockers) | Small molecule | Beta-adrenergic receptor | Phase I | Washington University School of Medicine | |
| Pancreatic cancer | CCX872 (OMP-18R5) | Small molecule | CCR2 | Phase I | ChemoCentryx |
| Vantictumab | Antibodies | Frizzled receptor FZD7 | Phase I (combine with nab-paclitaxel and gemcitabine) | OncoMed Pharmaceuticals, Inc. | |
| G17DT | Immunogen | Cholescystokinin-2 receptor | Phase III | Cancer Advances Inc. | |
| Multiple myeloma | BMS-936564 | Antibodies | CXCR4 | Phase I | Bristol-Myers Squibb |
| Melanoma | Plozalizumab | Humanized monoclonal antibody | CCR2 | Phase I | Millennium Pharmaceuticals, Inc |
| Adult T-cell leukemia and lymphoma | KW-0761 (Mogamulizumab) | Antibodies | CCR4 | Phase II | Kyowa Kakko Kirin |
| Metastatic breast cancer | OMP-18R5 (Vantictumab) | Antibodies | Frizzled receptors (FZD1, 2, 5, 7, 8) | Phase I (combined with paclitaxel) | OncoMed Pharmaceuticals, Inc. |
| Beta-blockers | Small molecule | Beta-adrenergic receptor | Phase II | Columbia University | |
| Non-small cell lung carcinoma | OMP-18R5 (Vantictumab) | Antibodies | Frizzled receptors (FZD1, 2, 5, 7, 8) | Phase I (combined with docetaxel) | OncoMed Pharmaceuticals, Inc. |
| Advanced solid tumors | AAT-007 | Small molecule | Prostagladin E2 receptor (EP4) | Phase II | University of Maryland |
| Advanced or metastatic cancer | LY-2624587 | Antibodies | CXCR4 | Phase I | Eli Lilly and company |
| Cancer type | Receptor | Ligand | Experiment Model/s | Results | References |
|---|---|---|---|---|---|
| Colon cancer | Formylpeptide receptor-2 (FPR2) | F2L | Human colon cancer cell lines | Knockdown of FPR2 from colon cancer lines resulted in reduced tumorigenicity. | [381,382] |
| Pancreatic cancer | Gα-coupled beta-adrenergic receptor | Beta-blocker | Hamsters, transgenic mice | Blockage of beta-adrenergic signaling by beta-blocker prevented pancreatic cancer in mice. | [383] |
| Prostate cancer | AT1R | Ang II | LNCap and PC3 cells | Inhibition of growth factor signaling was observed in LNCaP and PC3 cell lines. | [384] |
| GPR160 | Instead of cognate ligands, lentivirus-mediated shRNA system was used to suppress GPR160 transcription. | PC3, LNCaP, DU145, and 22Rv1 cells | Treatment of PC3 cells with GPR160-targeting shRNA lentiviruses resulted in cell apoptosis and growth arrest. | [385] | |
| Head and neck cancer | CXCR7, an atypical chemokine receptor also referred to as ACKR3 | Single variable domains of a highly selective immunoglobulin were used. | HNSCC cells | Immunoglobin therapy inhibited CXCR7-expressing head and neck cancer xenografted cells in nude mice. | [386] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. https://doi.org/10.3390/cells10123288
Chaudhary PK, Kim S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells. 2021; 10(12):3288. https://doi.org/10.3390/cells10123288
Chicago/Turabian StyleChaudhary, Preeti Kumari, and Soochong Kim. 2021. "An Insight into GPCR and G-Proteins as Cancer Drivers" Cells 10, no. 12: 3288. https://doi.org/10.3390/cells10123288
APA StyleChaudhary, P. K., & Kim, S. (2021). An Insight into GPCR and G-Proteins as Cancer Drivers. Cells, 10(12), 3288. https://doi.org/10.3390/cells10123288