
Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites

 ,
, 
 , and
, and
Abstract
:1. Introduction
Study Outcomes
2. Methods
2.1. Patients
2.2. Laboratory Procedures
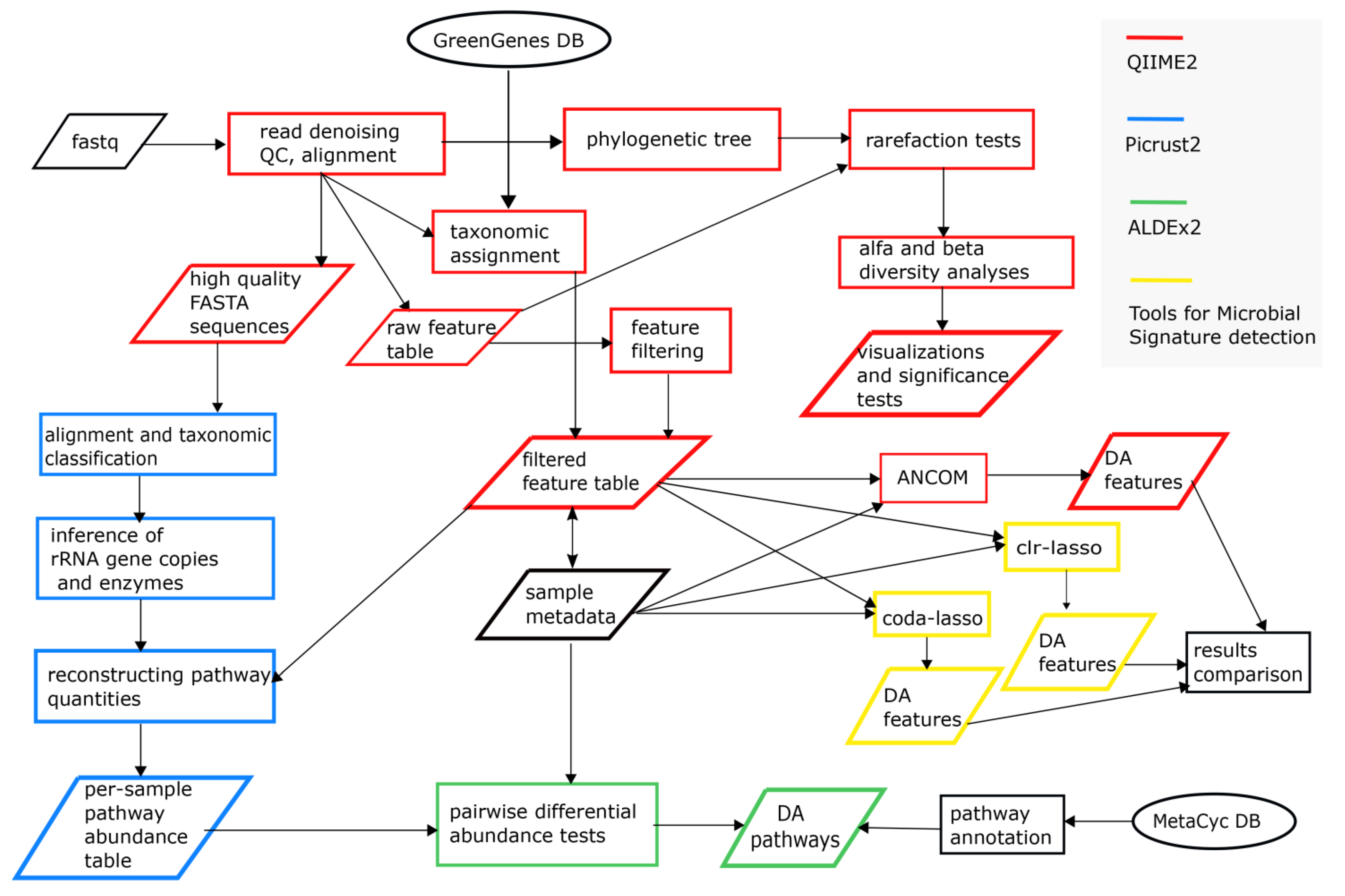
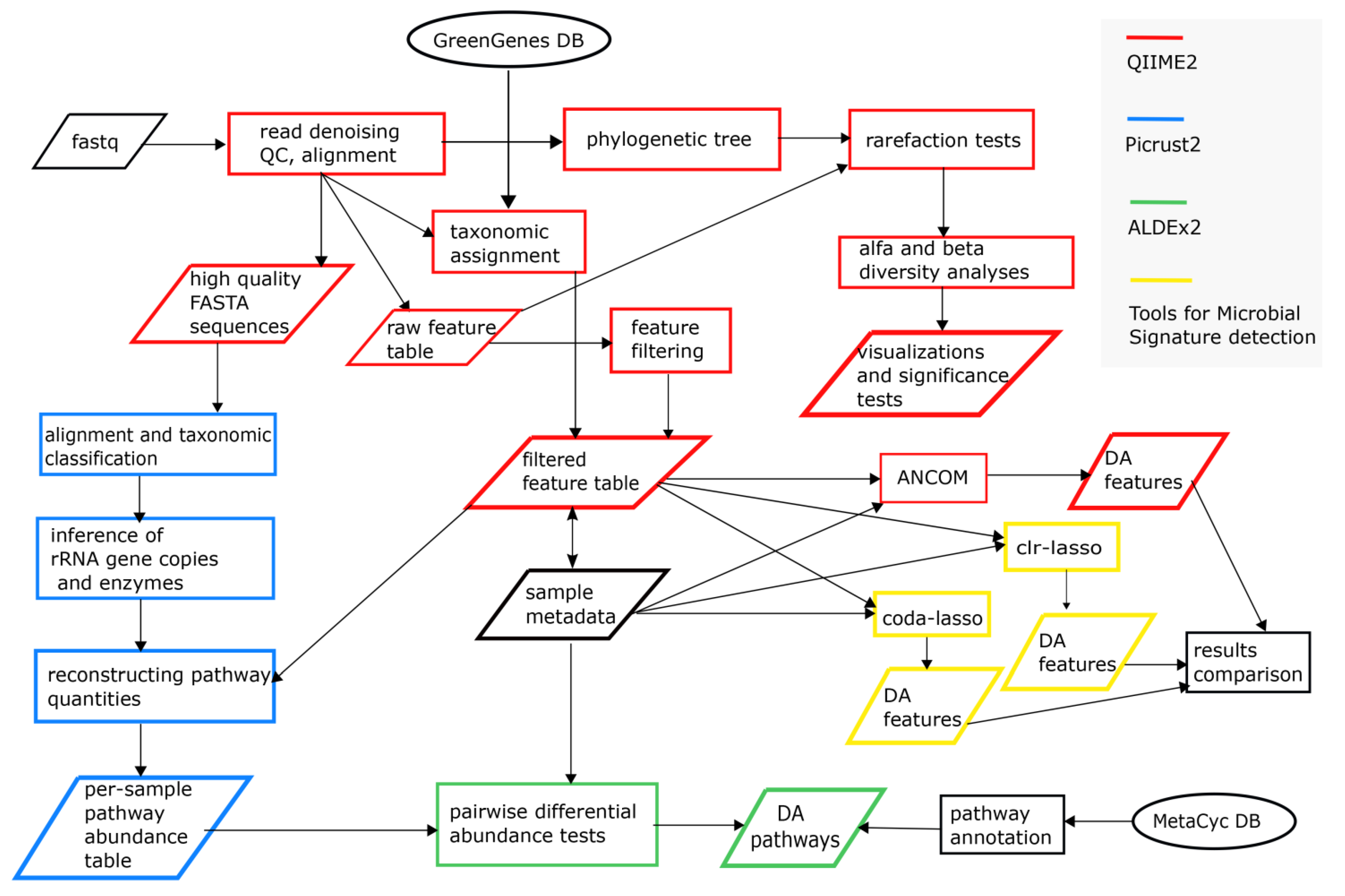
2.3. Bioinformatics and Statistical Sata Analyses
3. Results
3.1. Clinicopathologic Characteristics
3.2. 16S rRNA V3-V4 Region Sequencing Results
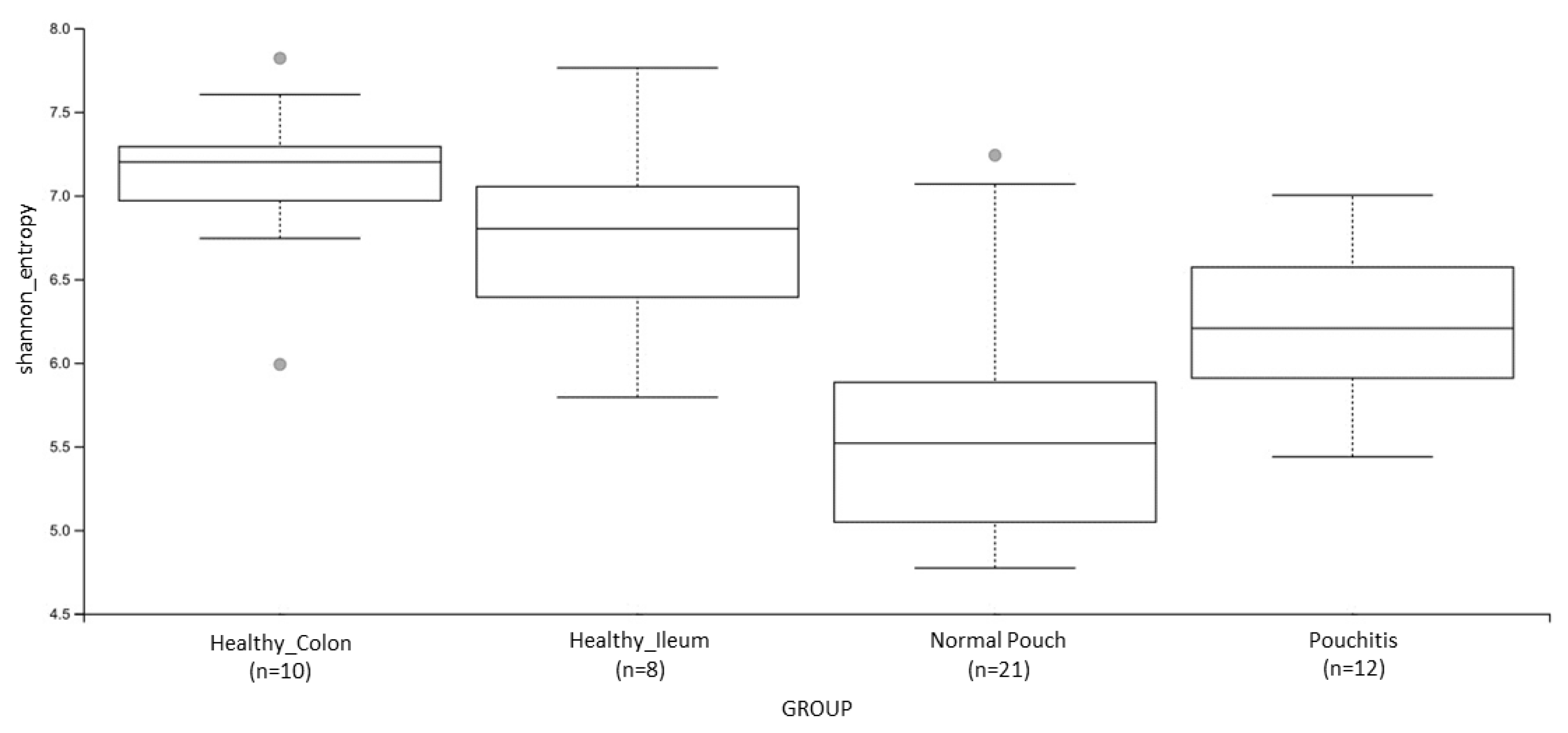
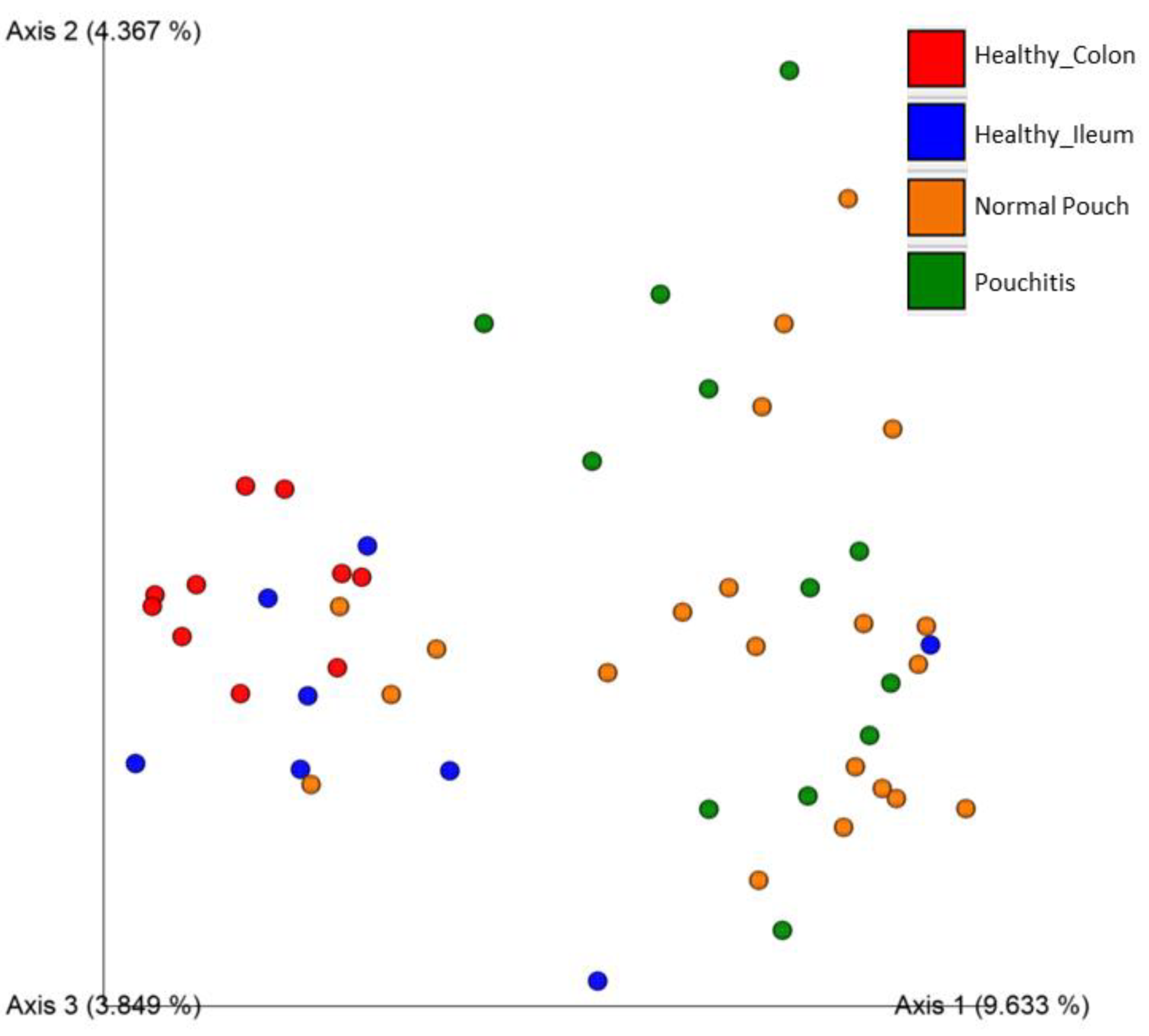
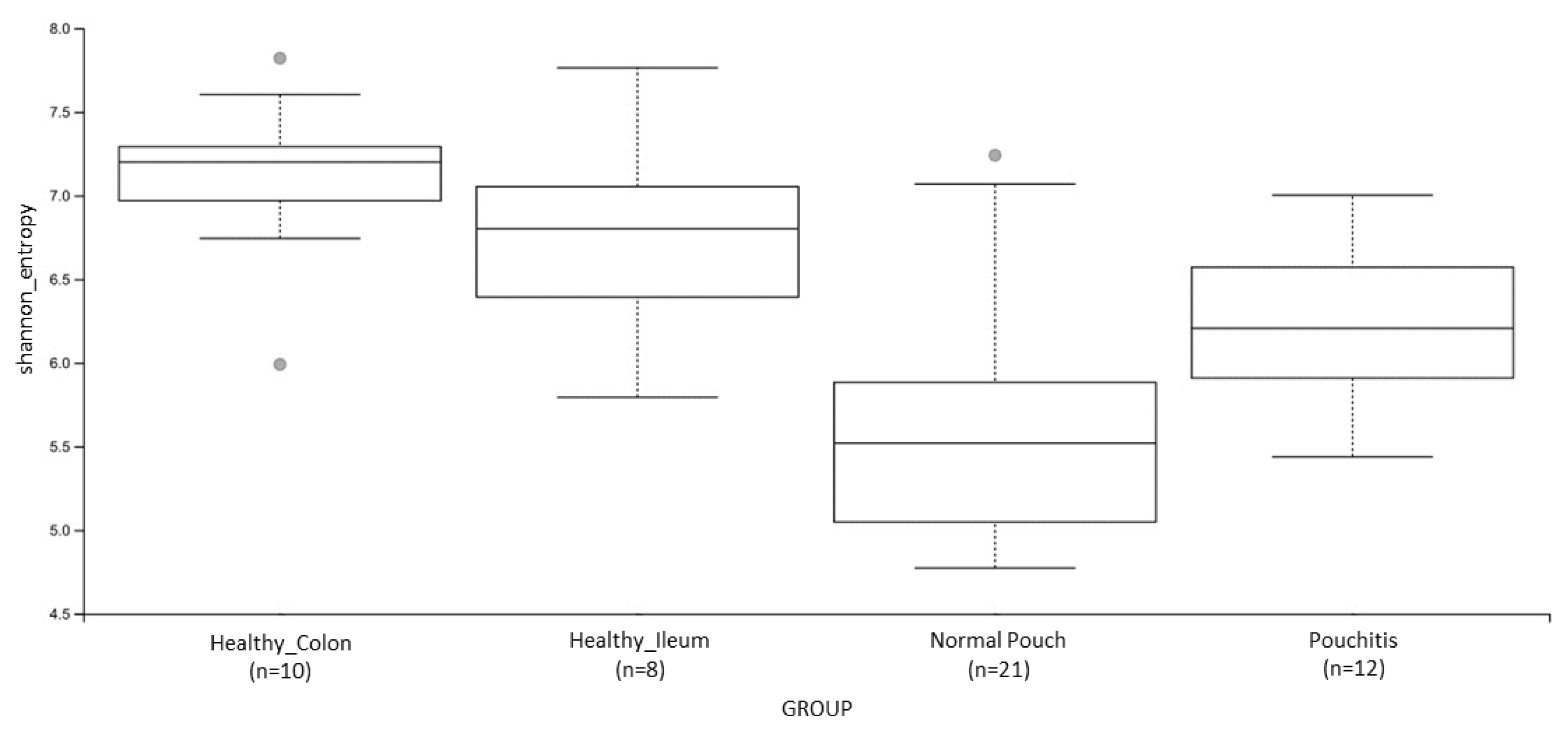
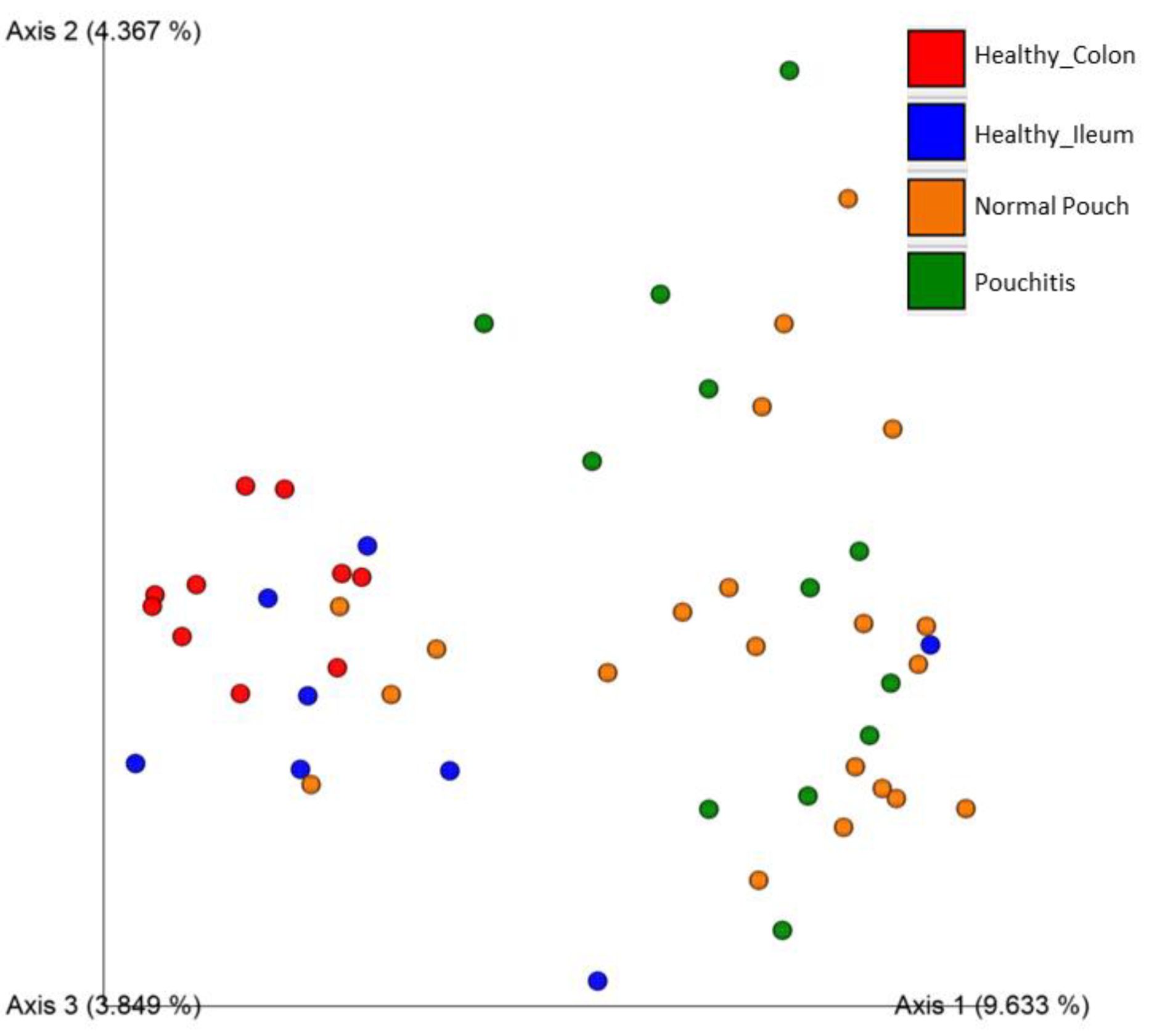
3.3. Microbial Diversity and Community Analyses
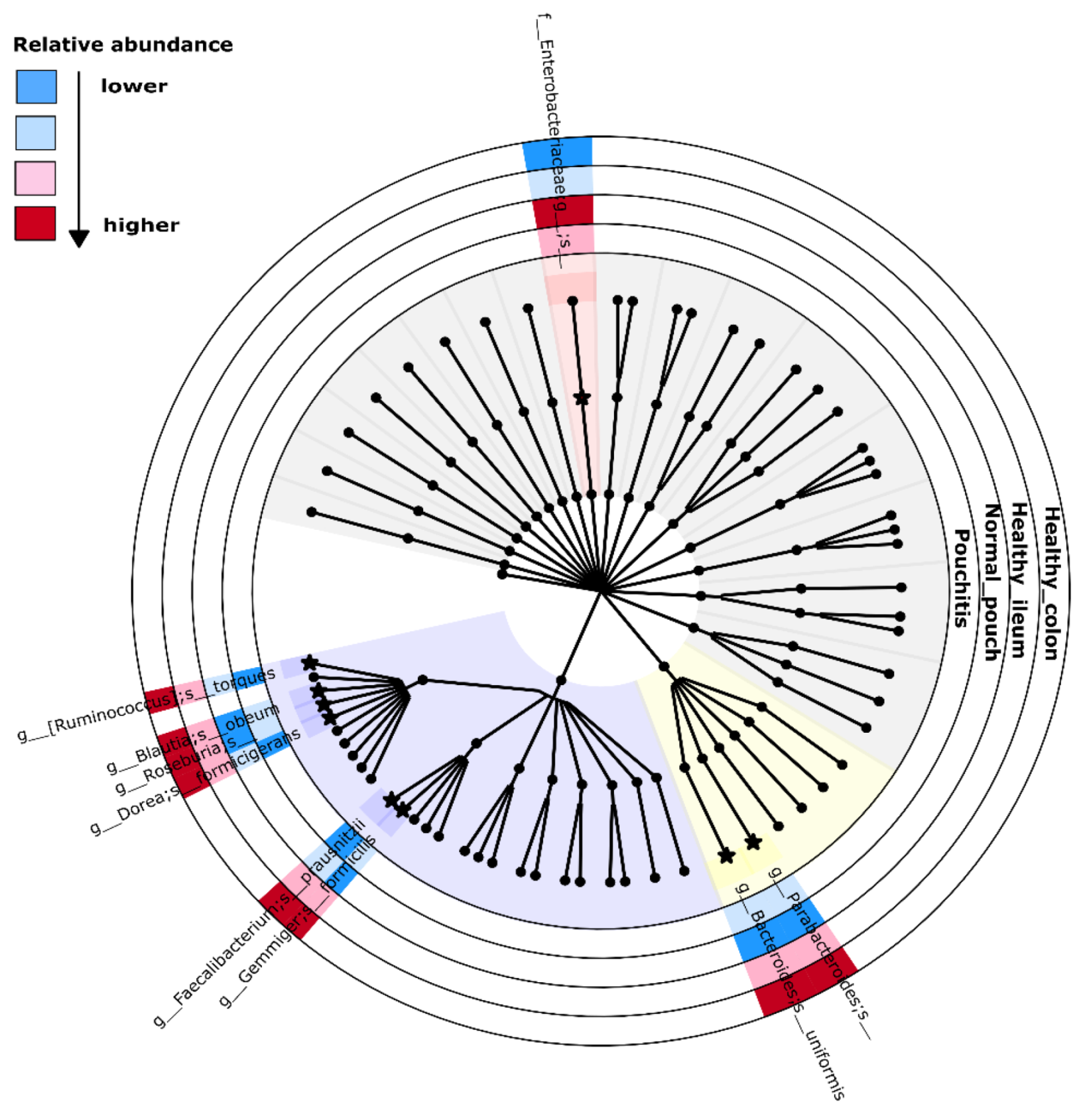
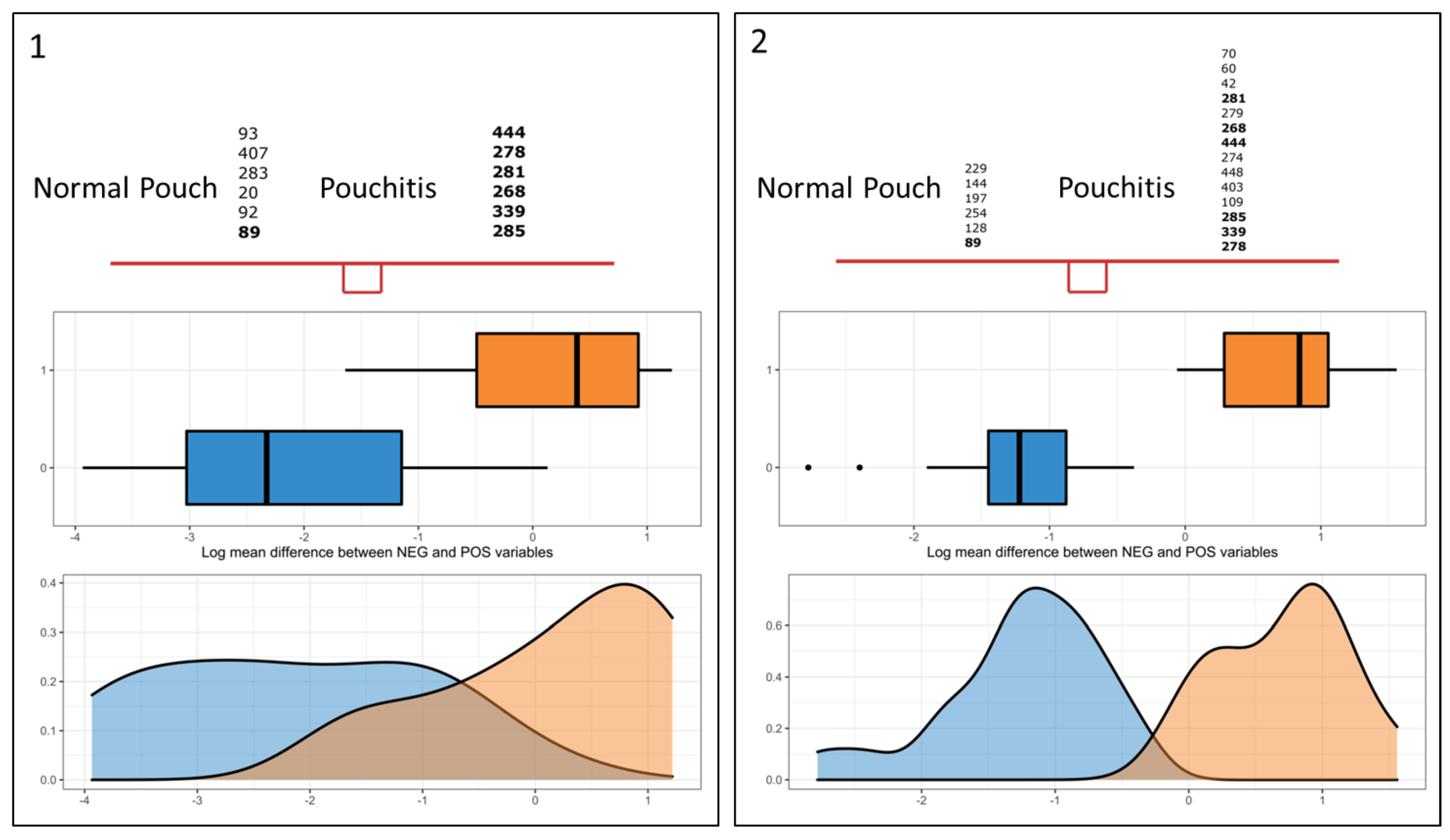
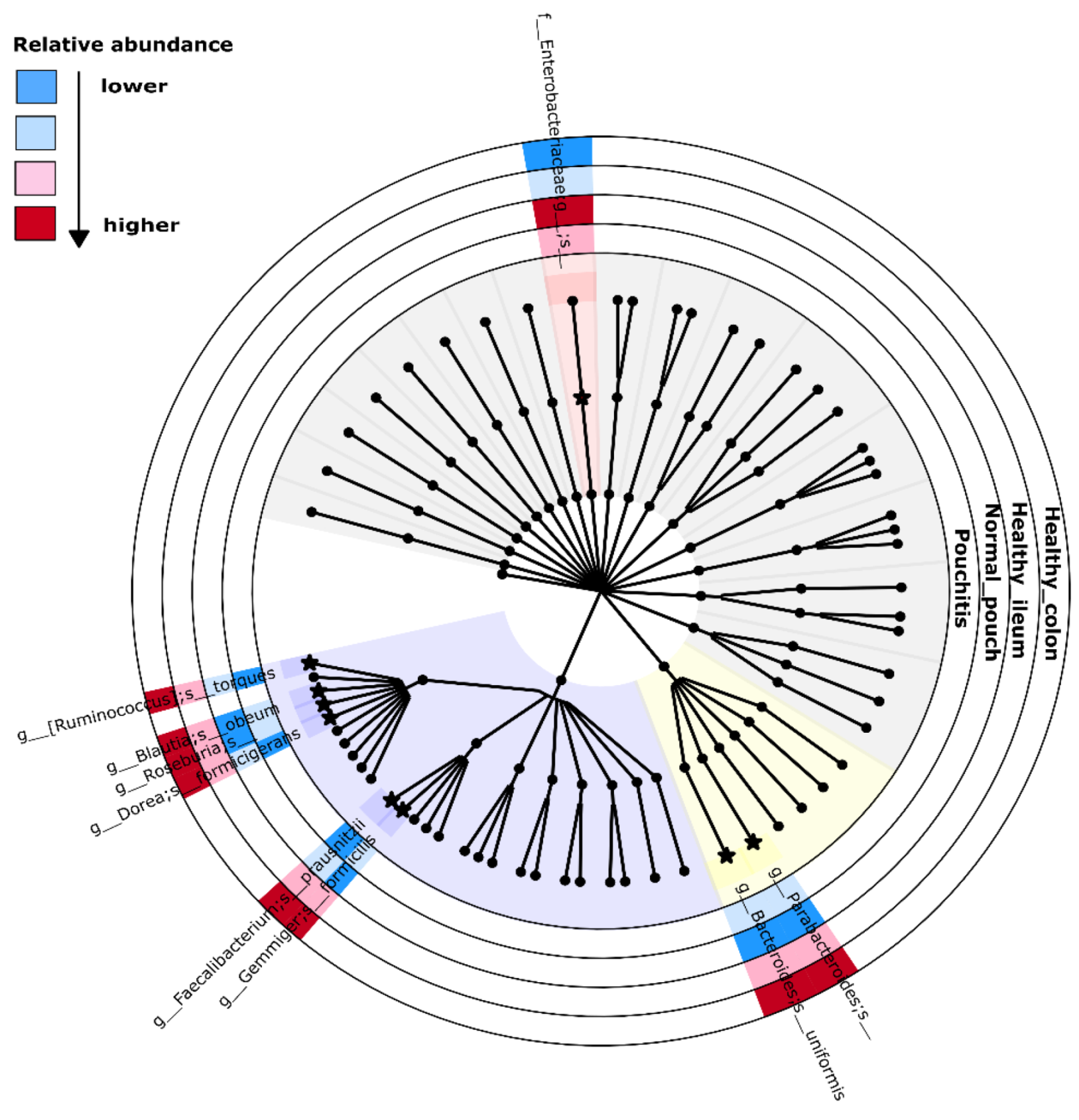
3.4. Microbial Signature from Pairwise Group Comparisons
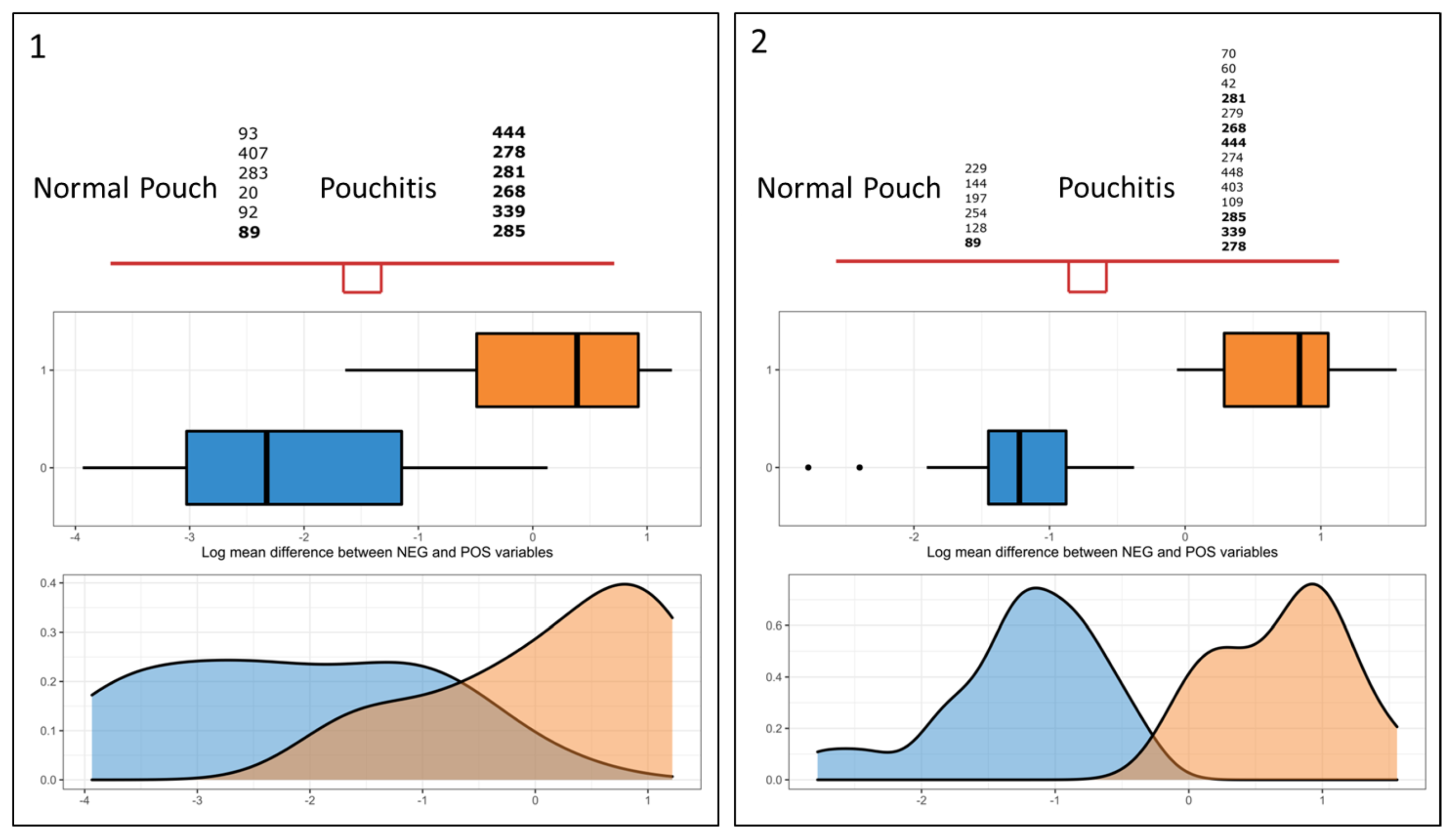
3.5. Differential Analysis of Inferred Microbial Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Prim. 2020, 6, 1–20. [Google Scholar] [CrossRef]
- Landy, J.; Al-Hassi, H.O.; McLaughlin, S.D.; Knight, S.C.; Ciclitira, P.J.; Nicholls, R.J.; Clark, S.K.; Hart, A.L. Etiology of pouchitis *. Inflamm. Bowel Dis. 2012, 18, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Kirat, H.; Remzi, F. Technical Aspects of Ileoanal Pouch Surgery in Patients with Ulcerative Colitis. Clin. Colon Rectal Surg. 2010, 23, 239–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinicke, J.A. Ileal Pouch Complications. Surg. Clin. N. Am. 2019, 99, 1185–1196. [Google Scholar] [CrossRef]
- Hoda, K.M.; Collins, J.F.; Knigge, K.L.; DeVeney, K.E. Predictors of Pouchitis after Ileal Pouch-Anal Anastomosis: A Retrospective Review. Dis. Colon Rectum 2008, 51, 554–560. [Google Scholar] [CrossRef]
- Navaneethan, U.; Shen, B. Laboratory Tests for Patients with Ileal Pouch–Anal Anastomosis: Clinical Utility in Predicting, Diagnosing, and Monitoring Pouch Disorders. Am. J. Gastroenterol. 2009, 104, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.J.; Di Giovine, F.S.; Cox, A.; Goodfellow, P.; Jones, S.; Shorthouse, A.J.; Duff, G.W.; Lobo, A.J. The interleukin 1 receptor antagonist gene allele 2 as a predictor of pouchitis following colectomy and IPAA in ulcerative colitis. Gastroenterol. 2001, 121, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Lammers, S.O.K. Combined carriership ofTLR9-1237C and CD14 -260T alleles enhances the risk of developing chronic relapsing pouchitis. World J. Gastroenterol. 2005, 11, 7323–7329. [Google Scholar] [CrossRef]
- Meier, C.B.; Hegazi, R.A.; Aisenberg, J.; Legnani, P.E.; Nilubol, N.; Cobrin, G.M.; Duerr, R.H.; Gorfine, S.R.; Bauer, J.J.; Sachar, D.B.; et al. Innate Immune Receptor Genetic Polymorphisms in Pouchitis: Is CARD15 a Susceptibility Factor? Inflamm. Bowel Dis. 2005, 11, 965–971. [Google Scholar] [CrossRef]
- Pardi, D.S.; Dʼhaens, G.; Shen, B.; Campbell, S.; Gionchetti, P. Clinical guidelines for the management of pouchitis. Inflamm. Bowel Dis. 2009, 15, 1424–1431. [Google Scholar] [CrossRef]
- Gionchetti, P.; Calafiore, A.; Riso, D.; Liguori, G.; Calabrese, C.; Vitali, G.; Laureti, S.; Poggioli, G.; Campieri, M.; Rizzello, F. The role of antibiotics and prebiotics in pouchitis. Ann. Gastroenterol. 2012, 25, 100–105. [Google Scholar]
- Segal, J.P.; Oke, S.; Hold, G.L.; Clark, S.K.; Faiz, O.D.; Hart, A.L. Systematic review: Ileoanal pouch microbiota in health and disease. Aliment. Pharmacol. Ther. 2018, 47, 466–477. [Google Scholar] [CrossRef] [Green Version]
- Reshef, L.; Kovacs, A.; Ofer, A.; Yahav, L.; Maharshak, N.; Keren, N.; Konikoff, F.M.; Tulchinsky, H.; Gophna, U.; Dotan, I. Pouch Inflammation Is Associated With a Decrease in Specific Bacterial Taxa. Gastroenterol. 2015, 149, 718–727. [Google Scholar] [CrossRef]
- Hinata, M.; Kohyama, A.; Ogawa, H.; Haneda, S.; Watanabe, K.; Suzuki, H.; Shibata, C.; Funayama, Y.; Takahashi, K.-I.; Sasaki, I.; et al. A Shift from Colon- to Ileum-Predominant Bacteria in Ileal-Pouch Feces Following Total Proctocolectomy. Dig. Dis. Sci. 2012, 57, 2965–2974. [Google Scholar] [CrossRef]
- Young, V.B.; Raffals, L.H.; Huse, S.M.; Vital, M.; Dai, D.; Schloss, P.D.; Brulc, J.M.; Antonopoulos, D.A.; Arrieta, R.L.; Kwon, J.H.; et al. Multiphasic analysis of the temporal development of the distal gut microbiota in patients following ileal pouch anal anastomosis. Microbiome 2013, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandborn, W.J.; Tremaine, W.J.; Batts, K.P.; Pemberton, J.H.; Phillips, S.F. Pouchitis After Ileal Pouch-Anal Anastomosis: A Pouchitis Disease Activity Index. Mayo Clin. Proc. 1994, 69, 409–415. [Google Scholar] [CrossRef]
- 16S Metagenomic Sequencing Library Preparation”. 2013. Available online: https://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 18 October 2021).
- R Core Team. The R Project for Statistical Computing. 2021. Available online: https://www.R-project.org/ (accessed on 18 October 2021).
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Briefings Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susin, A.; Wang, Y.; Cao, K.-A.L.; Calle, M.L. Variable selection in microbiome compositional data analysis. NAR Genom. Bioinform. 2020, 2, lqaa029. [Google Scholar] [CrossRef]
- Lin, H.; Das Peddada, S. Analysis of microbial compositions: A review of normalization and differential abundance analysis. npj Biofilms Microbiomes 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Quinn, T.P.; Erb, I.; Gloor, G.; Notredame, C.; Richardson, M.F.; Crowley, T.M. A field guide for the compositional analysis of any-omics data. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W.; Wiener, N. The Mathematical Theory of Communication. Phys. Today 1950, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 1908, 44, 223–270. [Google Scholar] [CrossRef]
- Sørensen, Thorvald Julius. A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content and Its Application to Analyses of the Vegetation on Danish Commons; I kommission hos E. Munksgaard: Copenhagen, Denmark, 1948. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnicar, F.; Weingart, G.; Tickle, T.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shi, P.; Li, H. Generalized linear models with linear constraints for microbiome compositional data. Biometrics 2018, 75, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.D.; Reid, J.N.; Macklaim, J.M.; McMurrough, T.A.; Edgell, D.R.; Gloor, G.B. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2014, 2, 15. [Google Scholar] [CrossRef] [Green Version]
- Strober, W.; Fuss, I.J.; Blumberg, R.S. The Immunology of Mucosal Models of Inflammation. Annu. Rev. Immunol. 2002, 20, 495–549. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670.e5. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Duncan, S.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium prausnitzii: From microbiology to diagnostics and prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, K.; Wright, J.R.; Harris, L.R.; Deiling, S.; Yang, Z.; Lamendella, R.; Yochum, G.S.; Koltun, W.A. NOD2 Genetic Variants Predispose One of Two Familial Adenomatous Polyposis Siblings to Pouchitis Through Microbiome Dysbiosis. J. Crohn’s Coliti 2017, 11, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Sabino, J.; Vandermosten, L.; Joossens, M.; Arijs, I.; De Bruyn, M.; Eeckhaut, V.; Van Assche, G.; Ferrante, M.; Verhaegen, J.; et al. Specific members of the predominant gut microbiota predict pouchitis following colectomy and IPAA in UC. Gut 2017, 66, 79–88. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Mena-Vázquez, N.; Ruiz-Limón, P.; Moreno-Indias, I.; Manrique-Arija, S.; Tinahones, F.J.; Fernández-Nebro, A. Expansion of Rare and Harmful Lineages is Associated with Established Rheumatoid Arthritis. J. Clin. Med. 2020, 9, 1044. [Google Scholar] [CrossRef] [Green Version]
- Mondot, S.; Lepage, P.; Seksik, P.; Allez, M.; Treton, X.; Bouhnik, Y.; Colombel, J.F.; Leclerc, M.; Pochart, P.; Dore, J.; et al. Structural robustness of the gut mucosal microbiota is associated with Crohn’s disease remission after surgery. Gut 2015, 65, 954–962. [Google Scholar] [CrossRef]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Andoh, A.; Sugimoto, M. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef]
- Tyler, A.D.; Knox, N.; Kabakchiev, B.; Milgrom, R.; Kirsch, R.; Cohen, Z.; McLeod, R.S.; Guttman, D.S.; Krause, D.O.; Silverberg, M.S. Characterization of the Gut-Associated Microbiome in Inflammatory Pouch Complications Following Ileal Pouch-Anal Anastomosis. PLoS ONE 2013, 8, e66934. [Google Scholar] [CrossRef]
- Landy, J.; Walker, A.; Li, J.; Al-Hassi, H.O.; Ronde, E.; English, N.R.; Mann, E.R.; Bernardo, D.; McLaughlin, S.D.; Parkhill, J.; et al. Variable alterations of the microbiota, without metabolic or immunological change, following faecal microbiota transplantation in patients with chronic pouchitis. Sci. Rep. 2015, 5, 12955. [Google Scholar] [CrossRef] [Green Version]
- Kousgaard, S.J.; Michaelsen, T.Y.; Nielsen, H.L.; Kirk, K.F.; Albertsen, M.; Thorlacius-Ussing, O. The Microbiota Profile in Inflamed and Non-Inflamed Ileal Pouch–Anal Anastomosis. Microorganisms 2020, 8, 1611. [Google Scholar] [CrossRef] [PubMed]
- Gerner, R.R.; Nuccio, S.-P.; Raffatellu, M. Iron at the host-microbe interface. Mol. Asp. Med. 2020, 75, 100895. [Google Scholar] [CrossRef] [PubMed]
- Constante, M.; Fragoso, G.; Calvé, A.; Samba-Mondonga, M.; Santos, M.M. Dietary Heme Induces Gut Dysbiosis, Aggravates Colitis, and Potentiates the Development of Adenomas in Mice. Front. Microbiol. 2017, 8, 1809. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Byun, E.J.; Kim, H.S. Potential Role of the Microbiome in Acne: A Comprehensive Review. J. Clin. Med. 2019, 8, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosavljevic, M.N.; Kostic, M.; Milovanovic, J.; Zaric, R.Z.; Stojadinovic, M.; Jankovic, S.M.; Stefanovic, S.M. Antimicrobial treatment of Erysipelatoclostridium ramosum invasive infections: A systematic review. Rev. Inst. Med. Trop. São Paulo 2021, 63, e30. [Google Scholar] [CrossRef]
- Nagayama, M.; Yano, T.; Atarashi, K.; Tanoue, T.; Sekiya, M.; Kobayashi, Y.; Sakamoto, H.; Miura, K.; Sunada, K.; Kawaguchi, T.; et al. TH1 cell-inducing Escherichia coli strain identified from the small intestinal mucosa of patients with Crohn’s disease. Gut Microbes 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-K.; Kim, H.-N.; Choi, C.H.; Im, J.P.; Cha, J.M.; Eun, C.S.; Kim, T.-O.; Kang, S.-B.; Bang, K.B.; Kim, H.G.; et al. Differentially Abundant Bacterial Taxa Associated with Prognostic Variables of Crohn’s Disease: Results from the IMPACT Study. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Gulick, A.M.; Hubbard, B.K.; Gerlt, J.A.; Rayment, I. Evolution of Enzymatic Activities in the Enolase Superfamily: Identification of the General Acid Catalyst in theActive Site of d-Glucarate Dehydratase from Escherichia coli. Biochemistry 2001, 40, 10054–10062. [Google Scholar] [CrossRef]
- Aghaie, A.; Lechaplais, C.; Sirven, P.; Tricot, S.; Besnard-Gonnet, M.; Muselet, D.; de Berardinis, V.; Kreimeyer, A.; Gyapay, G.; Salanoubat, M.; et al. New Insights into the Alternative d-Glucarate Degradation Pathway. J. Biol. Chem. 2008, 283, 15638–15646. [Google Scholar] [CrossRef] [Green Version]
- Pradel, E.; Boquet, P. Utilization of exogenous glucose-1-phosphate as a source of carbon or phosphate byEscherichia coli K12: Respective roles of acid glucose-1-phosphatase, hexose-phosphate permease, phosphoglucomutase and alkaline phosphatase. Res. Microbiol. 1991, 142, 37–45. [Google Scholar] [CrossRef]
- Campbell, J.W.; Morgan-Kiss, R.M.; Cronan, J.E. A new Escherichia coli metabolic competency: Growth on fatty acids by a novel anaerobic β-oxidation pathway. Mol. Microbiol. 2003, 47, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Stefely, J.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci. 2017, 42, 824–843. [Google Scholar] [CrossRef] [PubMed]
- Kulp, A.J.; Sun, B.; Ai, T.; Manning, A.J.; Orench-Rivera, N.; Schmid, A.K.; Kuehn, M. Genome-Wide Assessment of Outer Membrane Vesicle Production in Escherichia coli. PLoS ONE 2015, 10, e0139200. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Nakamura, A.; Kurihara, S.; Takahashi, D.; Ohashi, W.; Nakamura, Y.; Kimura, S.; Onuki, M.; Kume, A.; Sasazawa, Y.; Furusawa, Y.; et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nat. Cell Biol. 2016, 535, 75–84. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Michaudel, C.; Sokol, H. The Gut Microbiota at the Service of Immunometabolism. Cell Metab. 2020, 32, 514–523. [Google Scholar] [CrossRef]
- McDonald, D.; Kaehler, B.; Gonzalez, A.; DeReus, J.; Ackermann, G.; Marotz, C.; Huttley, G.; Knight, R. redbiom: A Rapid Sample Discovery and Feature Characterization System. mSystems 2019, 4, e00215-19. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; et al. MGnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2019, 48, D570–D578. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Pouch | Pouchitis | |
|---|---|---|
| (n = 21) | (n = 13) | |
| Gender (female %) | 12 (57%) | 4 (31%) |
| Mean age at recruitment (years SE) | 48.2 (3.1) | 49.3 (3.9) |
| Mean age at diagnosis (years SE) | 33.9 (2.9) | 34.4 (4.3) |
| Time from surgery to sample collection (years SE) | 7.8 (1.2) | 8.1 (1.7) |
| Mean age at surgery | 40.8 (3.1) | 41.6 (4.2) |
| Antibiotic previous month (% using) | 0 (0%) | 3 (25%) |
| Antibiotics ever (% used) | 1 (4.8%) | 3 (23%) |
| Comparison | Normal Pouch | Healthy Colon | Healthy Ileum | Healthy Ileum | ||
|---|---|---|---|---|---|---|
| vs. | vs. | vs. | vs. | |||
| Measure | Pouchitis | Normal Pouch | Pouchitis | Normal Pouch | Pouchitis | Healthy Colon |
| Shannon’s Entropy | 0.009 | 0.0004 | 0.004 | 0.005 | ns | ns |
| Pielou’s evenness | 0.013 | 0.003 | 0.035 | 0.001 | 0.011 | ns |
| Number of Observed Features | ns | 0.0005 | 0.049 | ns | ns | 0.013 |
| Faith’s Phylogenetic Distance | ns | 0.064 | ns | 0.022 | 0.022 | 0.008 |
| Comparison | Normal Pouch | Healthy Colon | Healthy Ileum | Healthy Ileum | ||
|---|---|---|---|---|---|---|
| vs. | vs. | vs. | vs. | |||
| Measure | Pouchitis | Normal Pouch | Pouchitis | Normal Pouch | Pouchitis | Healthy Colon |
| Jaccard similarity | ns | 0.002 | 0.002 | 0.002 | 0.002 | 0.02 |
| Bray–Curtis dissimilarity | ns | 0.002 | 0.002 | 0.005 | 0.002 | ns |
| Unweighted UniFrac dissimilarity | ns | 0.008 | 0.063 | 0.004 | 0.004 | 0.002 |
| Weighted UniFrac dissimilarity | 0.048 | 0.04 | 0.04 | 0.05 | 0.04 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palmieri, O.; Castellana, S.; Biscaglia, G.; Panza, A.; Latiano, A.; Fontana, R.; Guerra, M.; Corritore, G.; Latiano, T.; Martino, G.; et al. Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites. Cells 2021, 10, 3243. https://doi.org/10.3390/cells10113243
Palmieri O, Castellana S, Biscaglia G, Panza A, Latiano A, Fontana R, Guerra M, Corritore G, Latiano T, Martino G, et al. Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites. Cells. 2021; 10(11):3243. https://doi.org/10.3390/cells10113243
Chicago/Turabian StylePalmieri, Orazio, Stefano Castellana, Giuseppe Biscaglia, Anna Panza, Anna Latiano, Rosanna Fontana, Maria Guerra, Giuseppe Corritore, Tiziana Latiano, Giuseppina Martino, and et al. 2021. "Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites" Cells 10, no. 11: 3243. https://doi.org/10.3390/cells10113243
APA StylePalmieri, O., Castellana, S., Biscaglia, G., Panza, A., Latiano, A., Fontana, R., Guerra, M., Corritore, G., Latiano, T., Martino, G., Mazza, T., Andriulli, A., Perri, F., & Bossa, F. (2021). Microbiome Analysis of Mucosal Ileoanal Pouch in Ulcerative Colitis Patients Revealed Impairment of the Pouches Immunometabolites. Cells, 10(11), 3243. https://doi.org/10.3390/cells10113243