
High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review

 , , ,
, , , 

Abstract
1. Introduction

{kind=link}
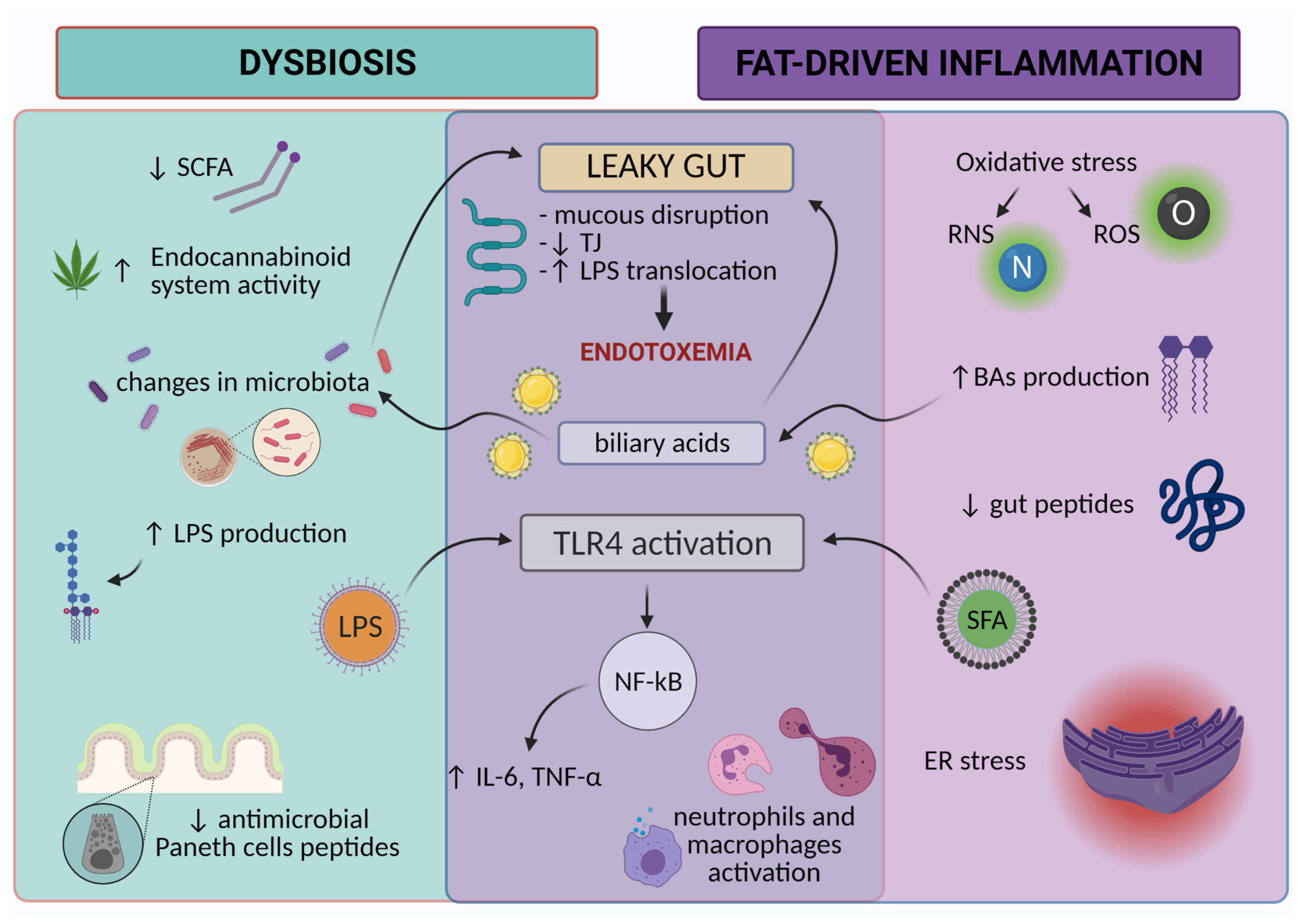{kind=link}
| Reference | Study Models | Fat (%kcal) |
|---|---|---|
| Cani et al., 2008 [37] | C57bl6/J mice and ob/ob mice C57bl6 background | 72% |
| Garidou et al., 2015 [38] | Wildtype (WT) C57Bl6/J mice, (RORγt−/−) mice, Rag1-deficient (Rag1−/−) mice, OVA-specific TCR transgenic (OTII) mice | 72% |
| Tomas et al., 2016 [39] | mice (C57BL/6JRj, Janvier, France) | 71% |
| Agus et al., 2016 [29] | C57BL/6 mice | 60% |
| Amar et al., 2011 [40] | C57bl6, ob/ob, CD14/, ob/obxCD14/, Myd88/, Nod1/or Nod2/mice | 60% |
| Brandsma et al., 2019 [41] | Female Casp1−/− mice (B6N.129S2-Casp1tm1Flv/J)) and Ldlr−/− mice (B6.129S7-Ldlrtm1Her/J) | 60% |
| Chelakkot et al., 2018 [42] | C57BL/6 mice | 60% |
| Crawford et al., 2019 [30] | Sprague-Dawley rats | 60% |
| Guo et al., 2017 [43] | C57BL/6 mice | 60% |
| Hu, Zhang, 2016 [44] | Toll-like receptor 4 knockout (TLR4−/−) and C57BL/6J (WT) mice | 60% |
| Jeong et al., 2019 [45] | Male C57BL/6 J mice | 60% |
| Kawano et al., 2016 [46] | (M-Ccr2KO) and (Vil-Ccl2KO) mice | 60% |
| Kim et al., 2012 [47] | C57BL/6J and TLR4-deficient C57BL/10ScNJ mice | 60% |
| In Kim et al., 2019 [48] | C57BL/6 mice | 60% |
| Li et al., 2019 [49] | C57BL/6 mice | 60% |
| Perez et al., 2019 [50] | C57BL/6 mice (IL-17RA−/−) | 60% |
| Schmid et al., 2015 [51] | Healthy human | 60% |
| Talukdar et al., 2012 [52] | NE KO, JAX labs B6.129X1–Elanetm1Sds/J mice and WT C57BL/6J mice | 60% |
| Wang et al., 2020 [53] | C57BL/6J mice | 60% |
| Gulhane et al., 2016 [54] | Wild type (WT) C57BL/6 mice | 46% |
| de la Serre et al., 2010 [55] | Male Sprague Dawley rats | 45% |
| Kim et al., 2019 [31] | male C57BL/6 J mice | 45% |
| Park et al., 2016 [56] | ApcMin/+ mice | 45% |
| Sen et al., 2017 [57] | Male Sprague Dawley rats | 45% |
| Napier et al., 2019 [58] | BALB/c mice, C57BL/6 mice | 42% |
| Murakami et al., 2016 [59] | C57/BL6 mice | 40% |
| Wan et al., 2019 [60] | Healthy adults | 40% |
| Laugerette et al., 2011 [61] | C57Bl6/J mice | 37.7% |
| Guo et al., 2016 [62] | C57BL/6J ApoE−/− mice | 37% |
2. Methodology
3. Microbiota
3.1. Healthy Microbiota Composition and Dysbiosis
3.2. Anti-Inflammatory and Proinflammatory Microbiota
3.3. High-Fat Diet-Driven Dysbiosis
3.4. Disruption of Spatial Microbiota Distribution
3.5. Increased Gut Permeability
3.6. Decreased SCFA
3.7. Endocannabinoid System
3.8. Endotoxemia
3.9. Bile Acids
3.10. Inflammation-Related Gene Expression
3.11. Neutrophils and Macrophage Activation
3.12. Decreased Production of Antimicrobial Peptides by Paneth Cells
4. Non-Dysbiosis Related Inflammation
4.1. Saturated Fatty Acids
4.2. Oxidative Stress
4.3. Endoplasmic Reticulum Stress
4.4. TLR4 and NF-κB Pathway
4.5. TNF-α and IL-6
4.6. Increased Gut Permeability and Decreased Tight Junctions
4.7. Gut Microbiota—An Innocent Passerby
4.8. Bile Acids
4.9. Macrophages and Neutrophils Activation
4.10. The Decrease in Gut Peptides
5. Summary
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| Bas | bile acids |
| Ccl2 | chemokine (C-C motif) ligand 2 |
| Ccr2 | encoding a chemokine receptor for Ccl2 |
| CD14KO | CD14 knockout mice |
| CDCA | chenodeoxycholic acid |
| Cftr | cystic fibrosis transmembrane conductance regulator |
| DCA | deoxycholic acid |
| EC | endocannabinoid system |
| EGFR | epidermal growth factor receptor |
| ER | endoplasmic reticulum |
| FAK | focal adhesion kinase |
| FISH | fluorescent in situ hybridization |
| FXR | farnesoid X receptor |
| GLP-1 | glucagon-like peptide 1 |
| HFD | High-fat diet |
| IFN-γ | interferon-γ |
| IKKβ | IκB kinase |
| iNOS | inducible NO synthase |
| JNK | NH2-terminal kinase |
| LBP | LPS-binding protein |
| LCA | lithocholic acid |
| LFD | low-fat diet |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MCP1 | monocyte chemoattractant protein 1 |
| MPO | neutrophil myeloperoxidase |
| MUFA | monounsaturated fatty acid |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| OCLN | occludin |
| OxS | oxidative stress |
| PBMC | peripheral blood mononuclear cells |
| PPAR-γ | peroxisome proliferator-activated receptor gamma |
| PUFA | polyunsaturated fatty acids |
| qPCR | quantitative polymerase chain reaction |
| RAS | renin-angiotensin system |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SCFA | short-chain fatty acids |
| SFA | saturated fatty acids |
| TGR5 | Takeda G protein-coupled BA receptor-1 |
| Th17 | T helper 17 cells |
| TJ | tight junctions |
| TJP | junction protein |
| TNF-α | tumor necrosis factor-α |
| UDCA | ursodeoxycholic acid |
| UPR | unfolded protein response |
| WD | Western diet |
References
- Varlamov, O. Western-Style Diet, Sex Steroids and Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1147–1155. [Google Scholar] [CrossRef]
- Carrera-Bastos, P.; Fontes-Villalba, M.; O’Keefe, J.H.; Lindeberg, S.; Cordain, L. The Western Diet and Lifestyle and Diseases of Civilization. Res. Rep. Clin. Cardiol. 2011, 2, 15–35. [Google Scholar] [CrossRef]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef] [PubMed]
- CHAPTER 2 Profiling Food Consumption in America. pp. 1–10. Available online: https://fliphtml5.com/nzsh/vvna/basic (accessed on 20 September 2021).
- Mozaffarian, D. Dietary and Policy Priorities for Cardiovascular Disease, Diabetes, and Obesity: A Comprehensive Review. Circulation 2016, 133, 187–225. [Google Scholar] [CrossRef] [PubMed]
- Halton, T.L.; Willett, W.C.; Liu, S.; Manson, J.E.; Stampfer, M.J.; Hu, F.B. Potato and French Fry Consumption and Risk of Type 2 Diabetes in Women. Am. J. Clin. Nutr. 2006, 83, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and Evolution of the Western Diet: Health Implications for the 21st Century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Rehm, C.D.; Rogers, G.; Ruan, M.; Wang, D.D.; Hu, F.B.; Mozaffarian, D.; Zhang, F.F.; Bhupathiraju, S.N. Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 2019, 322, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Carbohydrates in Your Diet: It’s the Quality That Counts. Not All Carbohydrates-Rich Foods Are Created Equal. Here’s How to Identify Healthy Carbs and Add Get More of Them. Available online: https://pubmed.ncbi.nlm.nih.gov/24818288/ (accessed on 25 February 2021).
- National Research Council (US) Committee on Diet and Health. Diet and Health: Implications for Reducing Chronic Disease Risk; National Academies Press (US): Washington, DC, USA, 1989; ISBN 978-0-309-03994-9. [Google Scholar]
- Jain, A.P.; Aggarwal, K.K.; Zhang, P.-Y. Omega-3 Fatty Acids and Cardiovascular Disease. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 441–445. [Google Scholar] [PubMed]
- Shahidi, F.; Ambigaipalan, P. Omega-3 Polyunsaturated Fatty Acids and Their Health Benefits. Annu. Rev. Food Sci. Technol. 2018, 9, 345–381. [Google Scholar] [CrossRef]
- Cani, P.D. Metabolism: Dietary Emulsifiers--Sweepers of the Gut Lining? Nat. Rev. Endocrinol. 2015, 11, 319–320. [Google Scholar] [CrossRef]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary Emulsifiers Impact the Mouse Gut Microbiota Promoting Colitis and Metabolic Syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Laster, J.; Bonnes, S.L.; Rocha, J. Increased Use of Emulsifiers in Processed Foods and the Links to Obesity. Curr. Gastroenterol. Rep. 2019, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E. Banting Lecture 2011: Hyperinsulinemia: Cause or Consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia Drives Diet-Induced Obesity Independently of Brain Insulin Production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef]
- Kopp, W. High-Insulinogenic Nutrition--an Etiologic Factor for Obesity and the Metabolic Syndrome? Metabolism. 2003, 52, 840–844. [Google Scholar] [CrossRef]
- Nolan, C.J.; Prentki, M. Insulin Resistance and Insulin Hypersecretion in the Metabolic Syndrome and Type 2 Diabetes: Time for a Conceptual Framework Shift. Diab. Vasc. Dis. Res. 2019, 16, 118–127. [Google Scholar] [CrossRef]
- Erion, K.A.; Corkey, B.E. Hyperinsulinemia: A Cause of Obesity? Curr. Obes. Rep. 2017, 6, 178–186. [Google Scholar] [CrossRef]
- Templeman, N.M.; Skovsø, S.; Page, M.M.; Lim, G.E.; Johnson, J.D. A Causal Role for Hyperinsulinemia in Obesity. J. Endocrinol. 2017, 232, R173–R183. [Google Scholar] [CrossRef]
- Rowe, J.W.; Young, J.B.; Minaker, K.L.; Stevens, A.L.; Pallotta, J.; Landsberg, L. Effect of Insulin and Glucose Infusions on Sympathetic Nervous System Activity in Normal Man. Diabetes 1981, 30, 219–225. [Google Scholar] [CrossRef]
- Rooney, D.P.; Edgar, J.D.; Sheridan, B.; Atkinson, A.B.; Bell, P.M. The Effects of Low Dose Insulin Infusions on the Renin Angiotensin and Sympathetic Nervous Systems in Normal Man. Eur. J. Clin. Investig. 1991, 21, 430–435. [Google Scholar] [CrossRef]
- Haenni, A.; Reneland, R.; Lind, L.; Lithell, H. Serum Aldosterone Changes during Hyperinsulinemia Are Correlated to Body Mass Index and Insulin Sensitivity in Patients with Essential Hypertension. J. Hypertens. 2001, 19, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.B.; Leone, V.; Chang, E.B. Western Diets, Gut Dysbiosis, and Metabolic Diseases: Are They Linked? Gut Microbes 2017, 8, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative Stress and Beta-Cell Dysfunction. Pflugers Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef]
- Park, K.-H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef]
- Cohen, D.H.; LeRoith, D. Obesity, Type 2 Diabetes, and Cancer: The Insulin and IGF Connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western Diet Induces a Shift in Microbiota Composition Enhancing Susceptibility to Adherent-Invasive E. coli Infection and Intestinal Inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef]
- Crawford, M.; Whisner, C.; Al-Nakkash, L.; Sweazea, K.L. Six-Week High-Fat Diet Alters the Gut Microbiome and Promotes Cecal Inflammation, Endotoxin Production, and Simple Steatosis without Obesity in Male Rats. Lipids 2019, 54, 119–131. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, S.-E.; Kim, A.-R.; Kang, S.; Park, M.-Y.; Sung, M.-K. Dietary Fat Intake and Age Modulate the Composition of the Gut Microbiota and Colonic Inflammation in C57BL/6J Mice. BMC Microbiol. 2019, 19, 193. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia Muciniphila Gen. Nov., Sp. Nov., a Human Intestinal Mucin-Degrading Bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic Endotoxemia Initiates Obesity and Insulin Resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.J.; Magness, S.; Jobin, C.; Lund, P.K. High-Fat Diet: Bacteria Interactions Promote Intestinal Inflammation Which Precedes and Correlates with Obesity and Insulin Resistance in Mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef]
- Wu, W.; Lv, L.; Shi, D.; Ye, J.; Fang, D.; Guo, F.; Li, Y.; He, X.; Li, L. Protective Effect of Akkermansia Muciniphila against Immune-Mediated Liver Injury in a Mouse Model. Front. Microbiol. 2017, 8, 1804. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health Relevance of the Modification of Low Grade Inflammation in Ageing (Inflammageing) and the Role of Nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Garidou, L.; Pomié, C.; Klopp, P.; Waget, A.; Charpentier, J.; Aloulou, M.; Giry, A.; Serino, M.; Stenman, L.; Lahtinen, S.; et al. The Gut Microbiota Regulates Intestinal CD4 T Cells Expressing RORγt and Controls Metabolic Disease. Cell Metab. 2015, 22, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Tomas, J.; Mulet, C.; Saffarian, A.; Cavin, J.-B.; Ducroc, R.; Regnault, B.; Kun Tan, C.; Duszka, K.; Burcelin, R.; Wahli, W.; et al. High-Fat Diet Modifies the PPAR-γ Pathway Leading to Disruption of Microbial and Physiological Ecosystem in Murine Small Intestine. Proc. Natl. Acad. Sci. USA 2016, 113, E5934–E5943. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal Mucosal Adherence and Translocation of Commensal Bacteria at the Early Onset of Type 2 Diabetes: Molecular Mechanisms and Probiotic Treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Ríos-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Choi, Y.; Kim, D.-K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.-S.; Jee, Y.-K.; Gho, Y.S.; et al. Akkermansia Muciniphila-Derived Extracellular Vesicles Influence Gut Permeability through the Regulation of Tight Junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef]
- Guo, X.; Li, J.; Tang, R.; Zhang, G.; Zeng, H.; Wood, R.J.; Liu, Z. High Fat Diet Alters Gut Microbiota and the Expression of Paneth Cell-Antimicrobial Peptides Preceding Changes of Circulating Inflammatory Cytokines. Mediat. Inflamm. 2017, 2017, 9474896. [Google Scholar] [CrossRef]
- Hu, N.; Zhang, Y. TLR4 Knockout Attenuated High Fat Diet-Induced Cardiac Dysfunction via NF-ΚB/JNK-Dependent Activation of Autophagy. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.-Y.; Jang, H.-M.; Kim, D.-H. High-Fat Diet Causes Psychiatric Disorders in Mice by Increasing Proteobacteria Population. Neurosci. Lett. 2019, 698, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Nakae, J.; Watanabe, N.; Kikuchi, T.; Tateya, S.; Tamori, Y.; Kaneko, M.; Abe, T.; Onodera, M.; Itoh, H. Colonic Pro-Inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab. 2016, 24, 295–310. [Google Scholar] [CrossRef]
- Kim, K.-A.; Gu, W.; Lee, I.-A.; Joh, E.-H.; Kim, D.-H. High Fat Diet-Induced Gut Microbiota Exacerbates Inflammation and Obesity in Mice via the TLR4 Signaling Pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef] [PubMed]
- In Kim, H.; Kim, J.-K.; Kim, J.-Y.; Jang, S.-E.; Han, M.J.; Kim, D.-H. Lactobacillus Plantarum LC27 and Bifidobacterium Longum LC67 Simultaneously Alleviate High-Fat Diet-Induced Colitis, Endotoxemia, Liver Steatosis, and Obesity in Mice. Nutr. Res. N. Y. 2019, 67, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-Fat Diet Promotes Experimental Colitis by Inducing Oxidative Stress in the Colon. Am. J. Physiol.-Gastrointest. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.M.; Martins, L.M.S.; Dias, M.S.; Pereira, C.A.; Leite, J.A.; Gonçalves, E.C.S.; de Almeida, P.Z.; de Freitas, E.N.; Tostes, R.C.; Ramos, S.G.; et al. Interleukin-17/Interleukin-17 Receptor Axis Elicits Intestinal Neutrophil Migration, Restrains Gut Dysbiosis and Lipopolysaccharide Translocation in High-Fat Diet-Induced Metabolic Syndrome Model. Immunology 2019, 156, 339–355. [Google Scholar] [CrossRef]
- Schmid, A.; Petry, N.; Walther, B.; Bütikofer, U.; Luginbühl, W.; Gille, D.; Chollet, M.; McTernan, P.G.; Gijs, M.A.M.; Vionnet, N.; et al. Inflammatory and Metabolic Responses to High-Fat Meals with and without Dairy Products in Men. Br. J. Nutr. 2015, 113, 1853–1861. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils Mediate Insulin Resistance in Mice Fed a High-Fat Diet through Secreted Elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef]
- Wang, B.; Kong, Q.; Li, X.; Zhao, J.; Zhang, H.; Chen, W.; Wang, G. A High-Fat Diet Increases Gut Microbiota Biodiversity and Energy Expenditure Due to Nutrient Difference. Nutrients 2020, 12, 3197. [Google Scholar] [CrossRef]
- Gulhane, M.; Murray, L.; Lourie, R.; Tong, H.; Sheng, Y.H.; Wang, R.; Kang, A.; Schreiber, V.; Wong, K.Y.; Magor, G.; et al. High Fat Diets Induce Colonic Epithelial Cell Stress and Inflammation That Is Reversed by IL-22. Sci. Rep. 2016, 6, 28990. [Google Scholar] [CrossRef]
- De La Serre, C.B.; Ellis, C.L.; Lee, J.; Hartman, A.L.; Rutledge, J.C.; Raybould, H.E. Propensity to High-Fat Diet-Induced Obesity in Rats Is Associated with Changes in the Gut Microbiota and Gut Inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G440–G448. [Google Scholar] [CrossRef]
- Park, M.-Y.; Kim, M.Y.; Seo, Y.R.; Kim, J.-S.; Sung, M.-K. High-Fat Diet Accelerates Intestinal Tumorigenesis Through Disrupting Intestinal Cell Membrane Integrity. J. Cancer Prev. 2016, 21, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Cawthon, C.R.; Ihde, B.T.; Hajnal, A.; DiLorenzo, P.M.; de La Serre, C.B.; Czaja, K. Diet-Driven Microbiota Dysbiosis Is Associated with Vagal Remodeling and Obesity. Physiol. Behav. 2017, 173, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Napier, B.A.; Andres-Terre, M.; Massis, L.M.; Hryckowian, A.J.; Higginbottom, S.K.; Cumnock, K.; Casey, K.M.; Haileselassie, B.; Lugo, K.A.; Schneider, D.S.; et al. Western Diet Regulates Immune Status and the Response to LPS-Driven Sepsis Independent of Diet-Associated Microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Tanabe, S.; Suzuki, T. High-Fat Diet-Induced Intestinal Hyperpermeability Is Associated with Increased Bile Acids in the Large Intestine of Mice. J. Food Sci. 2016, 81, H216–H222. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, F.; Yuan, J.; Li, J.; Jiang, D.; Zhang, J.; Li, H.; Wang, R.; Tang, J.; Huang, T.; et al. Effects of Dietary Fat on Gut Microbiota and Faecal Metabolites, and Their Relationship with Cardiometabolic Risk Factors: A 6-Month Randomised Controlled-Feeding Trial. Gut 2019, 68, 1417–1429. [Google Scholar] [CrossRef]
- Laugerette, F.; Furet, J.-P.; Debard, C.; Daira, P.; Loizon, E.; Géloën, A.; Soulage, C.O.; Simonet, C.; Lefils-Lacourtablaise, J.; Bernoud-Hubac, N.; et al. Oil Composition of High-Fat Diet Affects Metabolic Inflammation Differently in Connection with Endotoxin Receptors in Mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E374–E386. [Google Scholar] [CrossRef]
- Guo, H.; Diao, N.; Yuan, R.; Chen, K.; Geng, S.; Li, M.; Li, L. Subclinical Dose Endotoxin Sustains Low-Grade Inflammation and Exacerbates Steatohepatitis in High-Fat Diet Fed Mice. J. Immunol. Baltim. 2016, 196, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Allam-Ndoul, B.; Castonguay-Paradis, S.; Veilleux, A. Gut Microbiota and Intestinal Trans-Epithelial Permeability. Int. J. Mol. Sci. 2020, 21, 6402. [Google Scholar] [CrossRef]
- Salazar, J.; Angarita, L.; Morillo, V.; Navarro, C.; Martínez, M.S.; Chacín, M.; Torres, W.; Rajotia, A.; Rojas, M.; Cano, C.; et al. Microbiota and Diabetes Mellitus: Role of Lipid Mediators. Nutrients 2020, 12, 3039. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Graf, D.; Di Cagno, R.; Fåk, F.; Flint, H.J.; Nyman, M.; Saarela, M.; Watzl, B. Contribution of Diet to the Composition of the Human Gut Microbiota. Microb. Ecol. Health Dis. 2015, 26, 26164. [Google Scholar] [CrossRef]
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.S.; Koller, K.R.; Ramaboli, M.C.; Nesengani, L.T.; Ocvirk, S.; Chen, C.; Flanagan, C.A.; Sapp, F.R.; Merritt, Z.T.; Bhatti, F.; et al. Diet and the Human Gut Microbiome: An International Review. Dig. Dis. Sci. 2020, 65, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Martí, J.M.; Martínez-Martínez, D.; Rubio, T.; Gracia, C.; Peña, M.; Latorre, A.; Moya, A.; Garay, C.P. Health and Disease Imprinted in the Time Variability of the Human Microbiome. mSystems 2017, 2, e00144-16. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Sonnenburg, J.L.; Manchester, J.K.; Hansen, E.E.; Chiang, H.C.; Gordon, J.I. A Hybrid Two-Component System Protein of a Prominent Human Gut Symbiont Couples Glycan Sensing in Vivo to Carbohydrate Metabolism. Proc. Natl. Acad. Sci. USA 2006, 103, 8834–8839. [Google Scholar] [CrossRef]
- Morowitz, M.J.; Carlisle, E.; Alverdy, J.C. Contributions of Intestinal Bacteria to Nutrition and Metabolism in the Critically Ill. Surg. Clin. N. Am. 2011, 91, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J. Intestinal Flora and Endogenous Vitamin Synthesis. Eur. J. Cancer Prev. Off. J. Eur. Cancer Prev. Organ. ECP 1997, 6 (Suppl. 1), S43–S45. [Google Scholar] [CrossRef]
- Buffie, C.G.; Pamer, E.G. Microbiota-Mediated Colonization Resistance against Intestinal Pathogens. Nat. Rev. Immunol. 2013, 13, 790–801. [Google Scholar] [CrossRef]
- Alakomi, H.L.; Skyttä, E.; Saarela, M.; Mattila-Sandholm, T.; Latva-Kala, K.; Helander, I.M. Lactic Acid Permeabilizes Gram-Negative Bacteria by Disrupting the Outer Membrane. Appl. Environ. Microbiol. 2000, 66, 2001–2005. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.-U.; Chen, G.Y.; Núñez, G. Role of the Gut Microbiota in Immunity and Inflammatory Disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Diaz Heijtz, R.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal Gut Microbiota Modulates Brain Development and Behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal Microbial Colonization Programs the Hypothalamic-Pituitary-Adrenal System for Stress Response in Mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Wilson, I.D. Gut Microorganisms, Mammalian Metabolism and Personalized Health Care. Nat. Rev. Microbiol. 2005, 3, 431–438. [Google Scholar] [CrossRef]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology Consensus Guidelines on the Management of Inflammatory Bowel Disease in Adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium Difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2018, 66, e1–e48. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Totino, V.; Gagliardi, A.; Santangelo, F.; Cacciotti, F.; Trancassini, M.; Mancini, C.; Cicerone, C.; Corazziari, E.; Pantanella, F.; et al. Eubiosis and Dysbiosis: The Two Sides of the Microbiota. New Microbiol. 2016, 39, 1–12. [Google Scholar]
- Scarmozzino, F.; Poli, A.; Visioli, F. Microbiota and Cardiovascular Disease Risk: A Scoping Review. Pharmacol. Res. 2020, 159, 104952. [Google Scholar] [CrossRef]
- Silverman, E.K.; Schmidt, H.H.H.W.; Anastasiadou, E.; Altucci, L.; Angelini, M.; Badimon, L.; Balligand, J.-L.; Benincasa, G.; Capasso, G.; Conte, F.; et al. Molecular Networks in Network Medicine: Development and Applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1489. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional Interactions between the Gut Microbiota and Host Metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and Evolutionary Forces Shaping Microbial Diversity in the Human Intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Darzi, Y.; Lima-Mendez, G.; Garcia Yunta, R.; Okuda, S.; Vandeputte, D.; Valles-Colomer, M.; Hildebrand, F.; Chaffron, S.; et al. Species-Function Relationships Shape Ecological Properties of the Human Gut Microbiome. Nat. Microbiol. 2016, 1, 16088. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through Ageing, and beyond: Gut Microbiota and Inflammatory Status in Seniors and Centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Hemalatha, R. Diet and Gut Microbiota in Human Health. Proc. Indian Natl. Sci. Acad. 2016, 82, 1437–1447. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Costello, E.K.; Berg-Lyons, D.; Gonzalez, A.; Stombaugh, J.; Knights, D.; Gajer, P.; Ravel, J.; Fierer, N.; et al. Moving Pictures of the Human Microbiome. Genome Biol. 2011, 12, R50. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Pushpanathan, P.; Mathew, G.S.; Selvarajan, S.; Seshadri, K.G.; Srikanth, P. Gut Microbiota and Its Mysteries. Indian J. Med. Microbiol. 2019, 37, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Gut Microbiota, Obesity and Diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Bawden, E. Gut Microbiome: What We Do and Don’t Know. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2015, 30, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut Microbiota and IBD: Causation or Correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Scarpellini, E.; Colica, C.; Boccuto, L.; Salehi, B.; Sharifi-Rad, J.; Aiello, V.; Romano, B.; De Lorenzo, A.; Izzo, A.A.; et al. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients 2019, 11, 2690. [Google Scholar] [CrossRef]
- Ottman, N.; Reunanen, J.; Meijerink, M.; Pietilä, T.E.; Kainulainen, V.; Klievink, J.; Huuskonen, L.; Aalvink, S.; Skurnik, M.; Boeren, S.; et al. Pili-like Proteins of Akkermansia Muciniphila Modulate Host Immune Responses and Gut Barrier Function. PLoS ONE 2017, 12, e0173004. [Google Scholar] [CrossRef]
- Macchione, I.G.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia Muciniphila: Key Player in Metabolic and Gastrointestinal Disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar] [CrossRef]
- Belzer, C.; de Vos, W.M. Microbes Inside--from Diversity to Function: The Case of Akkermansia. ISME J. 2012, 6, 1449–1458. [Google Scholar] [CrossRef]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A Purified Membrane Protein from Akkermansia Muciniphila or the Pasteurized Bacterium Improves Metabolism in Obese and Diabetic Mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef]
- Everard, A.; Lazarevic, V.; Derrien, M.; Girard, M.; Muccioli, G.G.; Muccioli, G.M.; Neyrinck, A.M.; Possemiers, S.; Van Holle, A.; François, P.; et al. Responses of Gut Microbiota and Glucose and Lipid Metabolism to Prebiotics in Genetic Obese and Diet-Induced Leptin-Resistant Mice. Diabetes 2011, 60, 2775–2786. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-Talk between Akkermansia Muciniphila and Intestinal Epithelium Controls Diet-Induced Obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Isolauri, E.; Laitinen, K.; Salminen, S. Distinct Composition of Gut Microbiota during Pregnancy in Overweight and Normal-Weight Women. Am. J. Clin. Nutr. 2008, 88, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.L.J.; Onnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The Microbiota of the Gut in Preschool Children with Normal and Excessive Body Weight. Obes. Silver Spring Md 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides Vulgatus and Bacteroides Dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef]
- Drissi, F.; Merhej, V.; Angelakis, E.; El Kaoutari, A.; Carrière, F.; Henrissat, B.; Raoult, D. Comparative Genomics Analysis of Lactobacillus Species Associated with Weight Gain or Weight Protection. Nutr. Diabetes 2014, 4, e109. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, W.; Rył, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczyńska, M. Immunomodulatory Potential of Gut Microbiome-Derived Short-Chain Fatty Acids (SCFAs). Acta Biochim. Pol. 2019, 66, 1–12. [Google Scholar] [CrossRef]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-Chain Fatty Acids Stimulate Angiopoietin-like 4 Synthesis in Human Colon Adenocarcinoma Cells by Activating Peroxisome Proliferator-Activated Receptor γ. Mol. Cell. Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef]
- Myhrstad, M.C.W.; Tunsjø, H.; Charnock, C.; Telle-Hansen, V.H. Dietary Fiber, Gut Microbiota, and Metabolic Regulation-Current Status in Human Randomized Trials. Nutrients 2020, 12, 859. [Google Scholar] [CrossRef]
- Morales, P.; Fujio, S.; Navarrete, P.; Ugalde, J.A.; Magne, F.; Carrasco-Pozo, C.; Tralma, K.; Quezada, M.; Hurtado, C.; Covarrubias, N.; et al. Impact of Dietary Lipids on Colonic Function and Microbiota: An Experimental Approach Involving Orlistat-Induced Fat Malabsorption in Human Volunteers. Clin. Transl. Gastroenterol. 2016, 7, e161. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Delgado, S.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Molecular Communication with the Immune System. Front. Microbiol. 2017, 8, 2345. [Google Scholar] [CrossRef]
- Xu, J.; Lian, F.; Zhao, L.; Zhao, Y.; Chen, X.; Zhang, X.; Guo, Y.; Zhang, C.; Zhou, Q.; Xue, Z.; et al. Structural Modulation of Gut Microbiota during Alleviation of Type 2 Diabetes with a Chinese Herbal Formula. ISME J. 2015, 9, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Yanagibashi, T.; Hosono, A.; Oyama, A.; Tsuda, M.; Suzuki, A.; Hachimura, S.; Takahashi, Y.; Momose, Y.; Itoh, K.; Hirayama, K.; et al. IgA Production in the Large Intestine Is Modulated by a Different Mechanism than in the Small Intestine: Bacteroides Acidifaciens Promotes IgA Production in the Large Intestine by Inducing Germinal Center Formation and Increasing the Number of IgA+ B Cells. Immunobiology 2013, 218, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Madhulika, A.; Deepika, G.; Rao, G.V.; Reddy, D.N.; Subramanyam, C.; Sasikala, M.; Talukdar, R. Altered Intestinal Microbiota in Patients with Chronic Pancreatitis: Implications in Diabetes and Metabolic Abnormalities. Sci. Rep. 2017, 7, 43640. [Google Scholar] [CrossRef]
- Salguero, M.V.; Al-Obaide, M.A.I.; Singh, R.; Siepmann, T.; Vasylyeva, T.L. Dysbiosis of Gram-Negative Gut Microbiota and the Associated Serum Lipopolysaccharide Exacerbates Inflammation in Type 2 Diabetic Patients with Chronic Kidney Disease. Exp. Ther. Med. 2019, 18, 3461–3469. [Google Scholar] [CrossRef]
- d’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems 2017, 2, e00046-17. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial Ecology: Human Gut Microbes Associated with Obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The Gut Microbiota as an Environmental Factor That Regulates Fat Storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in Plasma Endotoxin Concentrations and the Expression of Toll-like Receptors and Suppressor of Cytokine Signaling-3 in Mononuclear Cells after a High-Fat, High-Carbohydrate Meal: Implications for Insulin Resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Neff, L.M.; Suárez-Fariñas, M.; Holt, P.R. Diet-Induced Weight Loss Reduces Colorectal Inflammation: Implications for Colorectal Carcinogenesis. Am. J. Clin. Nutr. 2011, 93, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Reigstad, C.S.; Bäckhed, H.K.; Reinhardt, C.; Ketonen, M.; Lundén, G.Ö.; Cani, P.D.; Bäckhed, F. Gut-Derived Lipopolysaccharide Augments Adipose Macrophage Accumulation but Is Not Essential for Impaired Glucose or Insulin Tolerance in Mice. Gut 2012, 61, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Hildebrandt, M.A.; Hoffmann, C.; Sherrill-Mix, S.A.; Keilbaugh, S.A.; Hamady, M.; Chen, Y.-Y.; Knight, R.; Ahima, R.S.; Bushman, F.; Wu, G.D. High-Fat Diet Determines the Composition of the Murine Gut Microbiome Independently of Obesity. Gastroenterology 2009, 137, 1716–1724. [Google Scholar] [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes Ratio of the Human Microbiota Changes with Age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged High-Fat-Diet Feeding Promotes Non-Alcoholic Fatty Liver Disease and Alters Gut Microbiota in Mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Velasquez, M.T. Altered Gut Microbiota: A Link Between Diet and the Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2018, 16, 321–328. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Cai, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. Gut Microbiome May Contribute to Insulin Resistance and Systemic Inflammation in Obese Rodents: A Meta-Analysis. Physiol. Genom. 2018, 50, 244–254. [Google Scholar] [CrossRef]
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Mujico, J.R.; Baccan, G.C.; Gheorghe, A.; Díaz, L.E.; Marcos, A. Changes in Gut Microbiota Due to Supplemented Fatty Acids in Diet-Induced Obese Mice. Br. J. Nutr. 2013, 110, 711–720. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; de La Serre, C.B.; et al. Energy-Dense Diet Triggers Changes in Gut Microbiota, Reorganization of Gut-brain Vagal Communication and Increases Body Fat Accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Delzenne, N.M. The Role of the Gut Microbiota in Energy Metabolism and Metabolic Disease. Curr. Pharm. Des. 2009, 15, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [PubMed]
- Netto Candido, T.L.; Bressan, J.; de Alfenas, R.C.G. Dysbiosis and Metabolic Endotoxemia Induced by High-Fat Diet. Nutr. Hosp. 2018, 35, 1432–1440. [Google Scholar] [CrossRef]
- Patterson, E.; O’ Doherty, R.M.; Murphy, E.F.; Wall, R.; O’ Sullivan, O.; Nilaweera, K.; Fitzgerald, G.F.; Cotter, P.D.; Ross, R.P.; Stanton, C. Impact of Dietary Fatty Acids on Metabolic Activity and Host Intestinal Microbiota Composition in C57BL/6J Mice. Br. J. Nutr. 2014, 111, 1905–1917. [Google Scholar] [CrossRef]
- Simões, C.D.; Maukonen, J.; Kaprio, J.; Rissanen, A.; Pietiläinen, K.H.; Saarela, M. Habitual Dietary Intake Is Associated with Stool Microbiota Composition in Monozygotic Twins. J. Nutr. 2013, 143, 417–423. [Google Scholar] [CrossRef]
- Wolters, M.; Ahrens, J.; Romaní-Pérez, M.; Watkins, C.; Sanz, Y.; Benítez-Páez, A.; Stanton, C.; Günther, K. Dietary Fat, the Gut Microbiota, and Metabolic Health—A Systematic Review Conducted within the MyNewGut Project. Clin. Nutr. Edinb. Scotl. 2019, 38, 2504–2520. [Google Scholar] [CrossRef]
- Blædel, T.; Holm, J.B.; Sundekilde, U.K.; Schmedes, M.S.; Hess, A.L.; Lorenzen, J.K.; Kristiansen, K.; Dalsgaard, T.K.; Astrup, A.; Larsen, L.H. A Randomised, Controlled, Crossover Study of the Effect of Diet on Angiopoietin-like Protein 4 (ANGPTL4) through Modification of the Gut Microbiome. J. Nutr. Sci. 2016, 5, e45. [Google Scholar] [CrossRef]
- Pu, S.; Khazanehei, H.R.; Krause, D.O.; West, S.G.; Kris-Etherton, P.M.; Jenkins, D.J.; Lamarche, B.; Jones, P.J.; Khafipour, E. Effects of Unsaturated Fatty Acids (USFA) on Human Gut Microbiome Profile in a Subset of Canola Oil Multicenter Intervention Trial (COMIT). FASEB J. 2013, 27, 1056.7. [Google Scholar] [CrossRef]
- Rajkumar, H.; Mahmood, N.; Kumar, M.; Varikuti, S.R.; Challa, H.R.; Myakala, S.P. Effect of Probiotic (VSL#3) and Omega-3 on Lipid Profile, Insulin Sensitivity, Inflammatory Markers, and Gut Colonization in Overweight Adults: A Randomized, Controlled Trial. Mediat. Inflamm. 2014, 2014, 348959. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Odenwald, M.A.; Turner, J.R. The Intestinal Epithelial Barrier: A Therapeutic Target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective Increases of Bifidobacteria in Gut Microflora Improve High-Fat-Diet-Induced Diabetes in Mice through a Mechanism Associated with Endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef]
- Heisel, T.; Montassier, E.; Johnson, A.; Al-Ghalith, G.; Lin, Y.-W.; Wei, L.-N.; Knights, D.; Gale, C.A. High-Fat Diet Changes Fungal Microbiomes and Interkingdom Relationships in the Murine Gut. mSphere 2017, 2, e00351-17. [Google Scholar] [CrossRef]
- Lin, H.; An, Y.; Hao, F.; Wang, Y.; Tang, H. Correlations of Fecal Metabonomic and Microbiomic Changes Induced by High-Fat Diet in the Pre-Obesity State. Sci. Rep. 2016, 6, 21618. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Nugent, C.A.; Tsompana, M.; Guan, L.; Wang, Y.; Buck, M.J.; Genco, R.J.; Baker, R.D.; Zhu, R.; et al. High-Fat Diet Increases Clostridium Clusters XIVa in Obese Rodents. FASEB J. 2017, 31, 965.9. [Google Scholar] [CrossRef]
- Sun, J.; Qiao, Y.; Qi, C.; Jiang, W.; Xiao, H.; Shi, Y.; Le, G.-W. High-Fat-Diet-Induced Obesity Is Associated with Decreased Antiinflammatory Lactobacillus Reuteri Sensitive to Oxidative Stress in Mouse Peyer’s Patches. Nutrition 2016, 32, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.Y.; Hoffmann, J.M.A.; Oscarsson, J.; Dinudom, A.; Mather, T.J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Effects of Dietary Fat Profile on Gut Permeability and Microbiota and Their Relationships with Metabolic Changes in Mice. Obes. Silver Spring Md 2015, 23, 1429–1439. [Google Scholar] [CrossRef]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-Fat-Induced Taurocholic Acid Promotes Pathobiont Expansion and Colitis in Il10−/− Mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide Causes an Increase in Intestinal Tight Junction Permeability in Vitro and in Vivo by Inducing Enterocyte Membrane Expression and Localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. Baltim. Md 1950 2015, 195, 4999–5010. [Google Scholar] [CrossRef]
- Derrien, M.; van Passel, M.W.; van de Bovenkamp, J.H.; Schipper, R.G.; de Vos, W.M.; Dekker, J. Mucin-Bacterial Interactions in the Human Oral Cavity and Digestive Tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Capaldo, C.T.; Powell, D.N.; Kalman, D. Layered Defense: How Mucus and Tight Junctions Seal the Intestinal Barrier. J. Mol. Med. Berl. Ger. 2017, 95, 927–934. [Google Scholar] [CrossRef]
- Anderson, R.C.; Cookson, A.L.; McNabb, W.C.; Park, Z.; McCann, M.J.; Kelly, W.J.; Roy, N.C. Lactobacillus Plantarum MB452 Enhances the Function of the Intestinal Barrier by Increasing the Expression Levels of Genes Involved in Tight Junction Formation. BMC Microbiol. 2010, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, E.; Morita, H.; Tanabe, S. Lactobacillus Rhamnosus Alleviates Intestinal Barrier Dysfunction in Part by Increasing Expression of Zonula Occludens-1 and Myosin Light-Chain Kinase in Vivo. J. Dairy Sci. 2009, 92, 2400–2408. [Google Scholar] [CrossRef]
- Patel, R.M.; Myers, L.S.; Kurundkar, A.R.; Maheshwari, A.; Nusrat, A.; Lin, P.W. Probiotic Bacteria Induce Maturation of Intestinal Claudin 3 Expression and Barrier Function. Am. J. Pathol. 2012, 180, 626–635. [Google Scholar] [CrossRef]
- Hsieh, C.-Y.; Osaka, T.; Moriyama, E.; Date, Y.; Kikuchi, J.; Tsuneda, S. Strengthening of the Intestinal Epithelial Tight Junction by Bifidobacterium Bifidum. Physiol. Rep. 2015, 3, e12327. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.Y.; Campbell, C.R.; Mitchell, A.J.; Dinudom, A.; Oscarsson, J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Increased Gut Permeability and Microbiota Change Associate with Mesenteric Fat Inflammation and Metabolic Dysfunction in Diet-Induced Obese Mice. PLoS ONE 2012, 7, e34233. [Google Scholar] [CrossRef]
- Cavallari, J.F.; Denou, E.; Foley, K.P.; Khan, W.I.; Schertzer, J.D. Different Th17 Immunity in Gut, Liver, and Adipose Tissues during Obesity: The Role of Diet, Genetics, and Microbes. Gut Microbes 2016, 7, 82–89. [Google Scholar] [CrossRef]
- Kim, C.H. Microbiota or Short-Chain Fatty Acids: Which Regulates Diabetes? Cell. Mol. Immunol. 2018, 15, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Evidence for the Gut Microbiota Short-Chain Fatty Acids as Key Pathophysiological Molecules Improving Diabetes. Mediators Inflamm. 2014, 2014, 162021. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Elce, A.; Amato, F.; Zarrilli, F.; Calignano, A.; Troncone, R.; Castaldo, G.; Canani, R.B. Butyrate Modulating Effects on Pro-Inflammatory Pathways in Human Intestinal Epithelial Cells. Benef. Microbes 2017, 8, 841–847. [Google Scholar] [CrossRef]
- Andoh, A.; Bamba, T.; Sasaki, M. Physiological and Anti-Inflammatory Roles of Dietary Fiber and Butyrate in Intestinal Functions. JPEN J. Parenter. Enteral Nutr. 1999, 23, S70–S73. [Google Scholar] [CrossRef]
- Segain, J.P.; Raingeard de la Blétière, D.; Bourreille, A.; Leray, V.; Gervois, N.; Rosales, C.; Ferrier, L.; Bonnet, C.; Blottière, H.M.; Galmiche, J.P. Butyrate Inhibits Inflammatory Responses through NFkappaB Inhibition: Implications for Crohn’s Disease. Gut 2000, 47, 397–403. [Google Scholar] [CrossRef]
- Song, M.; Xia, B.; Li, J. Effects of Topical Treatment of Sodium Butyrate and 5-Aminosalicylic Acid on Expression of Trefoil Factor 3, Interleukin 1beta, and Nuclear Factor KappaB in Trinitrobenzene Sulphonic Acid Induced Colitis in Rats. Postgrad. Med. J. 2006, 82, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.E.M.; Koetsier, M.A.; van Deventer, S.J.H.; van Tol, E.A.F. Short Chain Fatty Acids Stimulate Epithelial Mucin 2 Expression through Differential Effects on Prostaglandin E(1) and E(2) Production by Intestinal Myofibroblasts. Gut 2003, 52, 1442–1447. [Google Scholar] [CrossRef]
- Miao, W.; Wu, X.; Wang, K.; Wang, W.; Wang, Y.; Li, Z.; Liu, J.; Li, L.; Peng, L. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. Int. J. Mol. Sci. 2016, 17, 1696. [Google Scholar] [CrossRef]
- Ma, X.; Fan, P.X.; Li, L.S.; Qiao, S.Y.; Zhang, G.L.; Li, D.F. Butyrate Promotes the Recovering of Intestinal Wound Healing through Its Positive Effect on the Tight Junctions. J. Anim. Sci. 2012, 90 (Suppl. 4), 266–268. [Google Scholar] [CrossRef] [PubMed]
- Cândido, F.G.; Valente, F.X.; Grześkowiak, Ł.M.; Moreira, A.P.B.; Rocha, D.M.U.P.; de Alfenas, R.C.G. Impact of Dietary Fat on Gut Microbiota and Low-Grade Systemic Inflammation: Mechanisms and Clinical Implications on Obesity. Int. J. Food Sci. Nutr. 2018, 69, 125–143. [Google Scholar] [CrossRef]
- Díaz-Rúa, A.; Chivite, M.; Comesaña, S.; Velasco, C.; Valente, L.M.P.; Soengas, J.L.; Conde-Sieira, M. The Endocannabinoid System Is Affected by a High-Fat-Diet in Rainbow Trout. Horm. Behav. 2020, 125, 104825. [Google Scholar] [CrossRef]
- Cani, P.D.; Geurts, L.; Matamoros, S.; Plovier, H.; Duparc, T. Glucose Metabolism: Focus on Gut Microbiota, the Endocannabinoid System and Beyond. Diabetes Metab. 2014, 40, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The Endocannabinoid System Links Gut Microbiota to Adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.M.; Duckworth, C.A.; Watson, A.J.M.; Frey, M.R.; Miguel, J.C.; Burkitt, M.D.; Sutton, R.; Hughes, K.R.; Hall, L.J.; Caamaño, J.H.; et al. A Mouse Model of Pathological Small Intestinal Epithelial Cell Apoptosis and Shedding Induced by Systemic Administration of Lipopolysaccharide. Dis. Model. Mech. 2013, 6, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhang, Q.; Wang, C.; Wu, H.; Jiao, L.; Hong, Q.; Hu, C. LPS Challenge Increased Intestinal Permeability, Disrupted Mitochondrial Function and Triggered Mitophagy of Piglets. Innate Immun. 2018, 24, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Rohr, M.W.; Narasimhulu, C.A.; Rudeski-Rohr, T.A.; Parthasarathy, S. Negative Effects of a High-Fat Diet on Intestinal Permeability: A Review. Adv. Nutr. Bethesda Md 2020, 11, 77–91. [Google Scholar] [CrossRef]
- Yu, S.; Gao, N. Compartmentalizing Intestinal Epithelial Cell Toll-like Receptors for Immune Surveillance. Cell. Mol. Life Sci. CMLS 2015, 72, 3343–3353. [Google Scholar] [CrossRef]
- Moreira, A.P.B.; Texeira, T.F.S.; Ferreira, A.B.; Peluzio, M.d.C.G.; Alfenas, R.d.C.G. Influence of a High-Fat Diet on Gut Microbiota, Intestinal Permeability and Metabolic Endotoxaemia. Br. J. Nutr. 2012, 108, 801–809. [Google Scholar] [CrossRef]
- Manco, M.; Putignani, L.; Bottazzo, G.F. Gut Microbiota, Lipopolysaccharides, and Innate Immunity in the Pathogenesis of Obesity and Cardiovascular Risk. Endocr. Rev. 2010, 31, 817–844. [Google Scholar] [CrossRef]
- Cani, P.D.; Delzenne, N.M. The Gut Microbiome as Therapeutic Target. Pharmacol. Ther. 2011, 130, 202–212. [Google Scholar] [CrossRef]
- Elena, G.; Giovanna, D.; Brunella, P.; De Anna, F.; Alessandro, M.; Antonietta, T.M. Proinflammatory Signal Transduction Pathway Induced by Shigella Flexneri Porins in Caco-2 Cells. Braz. J. Microbiol. 2009, 40, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like Receptor 4 Is Associated with Insulin Resistance in Adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef]
- Islam, K.B.M.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile Acid Is a Host Factor That Regulates the Composition of the Cecal Microbiota in Rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural Resilience of the Gut Microbiota in Adult Mice under High-Fat Dietary Perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial Pathways in Colonic Sulfur Metabolism and Links with Health and Disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Bleau, C.; Karelis, A.D.; St-Pierre, D.H.; Lamontagne, L. Crosstalk between Intestinal Microbiota, Adipose Tissue and Skeletal Muscle as an Early Event in Systemic Low-Grade Inflammation and the Development of Obesity and Diabetes. Diabetes Metab. Res. Rev. 2015, 31, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Margioris, A.N. Fatty Acids and Postprandial Inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Al-Mass, A.; Atizado, V.; Al-Hubail, A.; Al-Ghimlas, F.; Al-Arouj, M.; Bennakhi, A.; Dermime, S.; Behbehani, K. Elevated Expression of the Toll like Receptors 2 and 4 in Obese Individuals: Its Significance for Obesity-Induced Inflammation. J. Inflamm. Lond. Engl. 2012, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Pillon, N.J.; Bilan, P.J.; Fink, L.N.; Klip, A. Cross-Talk between Skeletal Muscle and Immune Cells: Muscle-Derived Mediators and Metabolic Implications. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E453–E465. [Google Scholar] [CrossRef] [PubMed]
- Soeters, M.R.; Soeters, P.B.; Schooneman, M.G.; Houten, S.M.; Romijn, J.A. Adaptive Reciprocity of Lipid and Glucose Metabolism in Human Short-Term Starvation. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1397–E1407. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Annuzzi, G.; Ragucci, M.; Di Donato, O.; Della Pepa, G.; Della Corte, G.; Griffo, E.; Anniballi, G.; Giacco, A.; Mancini, M.; et al. Insulin Resistance, Postprandial GLP-1 and Adaptive Immunity Are the Main Predictors of NAFLD in a Homogeneous Population at High Cardiovascular Risk. Nutr. Metab. Cardiovasc. Dis. NMCD 2016, 26, 623–629. [Google Scholar] [CrossRef]
- Tang, M.W.; Koopman, F.A.; Visscher, J.P.M.; de Hair, M.J.; Gerlag, D.M.; Tak, P.P. Hormone, Metabolic Peptide, and Nutrient Levels in the Earliest Phases of Rheumatoid Arthritis-Contribution of Free Fatty Acids to an Increased Cardiovascular Risk during Very Early Disease. Clin. Rheumatol. 2017, 36, 269–278. [Google Scholar] [CrossRef]
- Zilversmit, D.B. Atherogenesis: A Postprandial Phenomenon. Circulation 1979, 60, 473–485. [Google Scholar] [CrossRef]
- Ohashi, K.; Shibata, R.; Murohara, T.; Ouchi, N. Role of Anti-Inflammatory Adipokines in Obesity-Related Diseases. Trends Endocrinol. Metab. TEM 2014, 25, 348–355. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 Links Innate Immunity and Fatty Acid-Induced Insulin Resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Luu, N.-T.; Madden, J.; Calder, P.C.; Grimble, R.F.; Shearman, C.P.; Chan, T.; Dastur, N.; Howell, W.M.; Rainger, G.E.; Nash, G.B. Dietary Supplementation with Fish Oil Modifies the Ability of Human Monocytes to Induce an Inflammatory Response. J. Nutr. 2007, 137, 2769–2774. [Google Scholar] [CrossRef]
- Calder, P.C. Polyunsaturated Fatty Acids and Inflammation. Biochem. Soc. Trans. 2005, 33, 423–427. [Google Scholar] [CrossRef]
- Bogani, P.; Galli, C.; Villa, M.; Visioli, F. Postprandial Anti-Inflammatory and Antioxidant Effects of Extra Virgin Olive Oil. Atherosclerosis 2007, 190, 181–186. [Google Scholar] [CrossRef]
- Pacheco, Y.M.; López, S.; Bermúdez, B.; Abia, R.; Villar, J.; Muriana, F.J.G. A Meal Rich in Oleic Acid Beneficially Modulates Postprandial SICAM-1 and SVCAM-1 in Normotensive and Hypertensive Hypertriglyceridemic Subjects. J. Nutr. Biochem. 2008, 19, 200–205. [Google Scholar] [CrossRef]
- Calder, P.C. Immunomodulation by Omega-3 Fatty Acids. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 327–335. [Google Scholar] [CrossRef]
- Calder, P.C.; Deckelbaum, R.J. Omega-3 Fatty Acids: Time to Get the Messages Right! Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Ghanim, H.; Aljada, A.; Browne, R.; Hamouda, W.; Prabhala, A.; Afzal, A.; Garg, R. The Suppressive Effect of Dietary Restriction and Weight Loss in the Obese on the Generation of Reactive Oxygen Species by Leukocytes, Lipid Peroxidation, and Protein Carbonylation. J. Clin. Endocrinol. Metab. 2001, 86, 355–362. [Google Scholar] [CrossRef]
- Kobayasi, R.; Akamine, E.H.; Davel, A.P.; Rodrigues, M.A.M.; Carvalho, C.R.O.; Rossoni, L.V. Oxidative Stress and Inflammatory Mediators Contribute to Endothelial Dysfunction in High-Fat Diet-Induced Obesity in Mice. J. Hypertens. 2010, 28, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.W.; Adams, L.A. Nonalcoholic Fatty Liver Disease and Diabetes Mellitus: Pathogenesis and Treatment. Nat. Rev. Endocrinol. 2011, 7, 456–465. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High Fat Induces Acute and Chronic Inflammation in the Hypothalamus: Effect of High-Fat Diet, Palmitate and TNF-α on Appetite-Regulating NPY Neurons. Int. J. Obes. 2005 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Tapias, V.; Hu, X.; Luk, K.C.; Sanders, L.H.; Lee, V.M.; Greenamyre, J.T. Synthetic Alpha-Synuclein Fibrils Cause Mitochondrial Impairment and Selective Dopamine Neurodegeneration in Part via INOS-Mediated Nitric Oxide Production. Cell. Mol. Life Sci. CMLS 2017, 74, 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.G.; Gotoh, Y.; Jennings, M.H.; Rhoads, C.A.; Aw, T.Y. Lipid Hydroperoxide-Induced Apoptosis in Human Colonic CaCo-2 Cells Is Associated with an Early Loss of Cellular Redox Balance. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2000, 14, 1567–1576. [Google Scholar] [CrossRef]
- Kruidenier, L.; Verspaget, H.W. Review Article: Oxidative Stress as a Pathogenic Factor in Inflammatory Bowel Disease--Radicals or Ridiculous? Aliment. Pharmacol. Ther. 2002, 16, 1997–2015. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Graß, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β Activation and Cell Death as a Consequence of TLR-MyD88-Induced CIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef]
- Yabal, M.; Müller, N.; Adler, H.; Knies, N.; Groß, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwälder, M.; et al. XIAP Restricts TNF- and RIP3-Dependent Cell Death and Inflammasome Activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 Promotes Cell Death and NLRP3 Inflammasome Activation in the Absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef]
- Argüelles, S.; Camandola, S.; Cutler, R.G.; Ayala, A.; Mattson, M.P. Elongation Factor 2 Diphthamide Is Critical for Translation of Two IRES-Dependent Protein Targets, XIAP and FGF2, under Oxidative Stress Conditions. Free Radic. Biol. Med. 2014, 67, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Rahmani, M.; Dent, P.; Grant, S. Blockade of Histone Deacetylase Inhibitor-Induced RelA/P65 Acetylation and NF-KappaB Activation Potentiates Apoptosis in Leukemia Cells through a Process Mediated by Oxidative Damage, XIAP Downregulation, and c-Jun N-Terminal Kinase 1 Activation. Mol. Cell. Biol. 2005, 25, 5429–5444. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Lu, P.D.; Zhang, Y.; Scheuner, D.; Kaufman, R.J.; Sonenberg, N.; Harding, H.P.; Ron, D. Translational Repression Mediates Activation of Nuclear Factor Kappa B by Phosphorylated Translation Initiation Factor 2. Mol. Cell. Biol. 2004, 24, 10161–10168. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway through IRE1alpha-Mediated NF-KappaB Activation and down-Regulation of TRAF2 Expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress Activates Cleavage of CREBH to Induce a Systemic Inflammatory Response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and Oxidative Stress Signaling: The PERK/Nrf2 Signaling Pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Xue, X.; Piao, J.-H.; Nakajima, A.; Sakon-Komazawa, S.; Kojima, Y.; Mori, K.; Yagita, H.; Okumura, K.; Harding, H.; Nakano, H. Tumor Necrosis Factor Alpha (TNFalpha) Induces the Unfolded Protein Response (UPR) in a Reactive Oxygen Species (ROS)-Dependent Fashion, and the UPR Counteracts ROS Accumulation by TNFalpha. J. Biol. Chem. 2005, 280, 33917–33925. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Thematic Review Series: Adipocyte Biology. Adipocyte Stress: The Endoplasmic Reticulum and Metabolic Disease. J. Lipid Res. 2007, 48, 1905–1914. [Google Scholar] [CrossRef]
- Schaeffler, A.; Gross, P.; Buettner, R.; Bollheimer, C.; Buechler, C.; Neumeier, M.; Kopp, A.; Schoelmerich, J.; Falk, W. Fatty Acid-Induced Induction of Toll-like Receptor-4/Nuclear Factor-KappaB Pathway in Adipocytes Links Nutritional Signalling with Innate Immunity. Immunology 2009, 126, 233–245. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ye, J.; Gao, Z.; Youn, H.S.; Lee, W.H.; Zhao, L.; Sizemore, N.; Hwang, D.H. Reciprocal Modulation of Toll-like Receptor-4 Signaling Pathways Involving MyD88 and Phosphatidylinositol 3-Kinase/AKT by Saturated and Polyunsaturated Fatty Acids. J. Biol. Chem. 2003, 278, 37041–37051. [Google Scholar] [CrossRef]
- Wong, S.W.; Kwon, M.-J.; Choi, A.M.K.; Kim, H.-P.; Nakahira, K.; Hwang, D.H. Fatty Acids Modulate Toll-like Receptor 4 Activation through Regulation of Receptor Dimerization and Recruitment into Lipid Rafts in a Reactive Oxygen Species-Dependent Manner. J. Biol. Chem. 2009, 284, 27384–27392. [Google Scholar] [CrossRef]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated Fatty Acids, but Not Unsaturated Fatty Acids, Induce the Expression of Cyclooxygenase-2 Mediated through Toll-like Receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef]
- Fritsche, K.L. The Science of Fatty Acids and Inflammation. Adv. Nutr. Bethesda Md 2015, 6, 293S–301S. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, H.; Yao, H.; Wei, Q.; Mao, X.-M.; Jiang, T.; Xiang, J.; Dila, N. Expression and Activity of the TLR4/NF-ΚB Signaling Pathway in Mouse Intestine Following Administration of a Short-Term High-Fat Diet. Exp. Ther. Med. 2013, 6, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/P62--More than Just a Scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T.; Wooten, M.W. Signal Integration and Diversification through the P62 Scaffold Protein. Trends Biochem. Sci. 2007, 32, 95–100. [Google Scholar] [CrossRef]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial Endothelial Activation in Healthy Subjects and in Type 2 Diabetic Patients: Role of Fat and Carbohydrate Meals. J. Am. Coll. Cardiol. 2002, 39, 1145–1150. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Li, Y.-H.; Lin, C.-C.; Chao, T.-H.; Chen, J.-H. Effects of Oxidative Stress on Endothelial Function after a High-Fat Meal. Clin. Sci. Lond. Engl. 1979 2004, 106, 315–319. [Google Scholar] [CrossRef]
- Aljada, A.; Mohanty, P.; Ghanim, H.; Abdo, T.; Tripathy, D.; Chaudhuri, A.; Dandona, P. Increase in Intranuclear Nuclear Factor KappaB and Decrease in Inhibitor KappaB in Mononuclear Cells after a Mixed Meal: Evidence for a Proinflammatory Effect. Am. J. Clin. Nutr. 2004, 79, 682–690. [Google Scholar] [CrossRef]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the Tight Junction Protein ZO-1 in Dextran Sulfate Sodium Induced Colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Feng, W.; Wang, Y.; Liu, Y.; Barker, D.F.; Barve, S.S.; McClain, C.J. The Type of Dietary Fat Modulates Intestinal Tight Junction Integrity, Gut Permeability, and Hepatic Toll-like Receptor Expression in a Mouse Model of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2012, 36, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hara, H. Dietary Fat and Bile Juice, but Not Obesity, Are Responsible for the Increase in Small Intestinal Permeability Induced through the Suppression of Tight Junction Protein Expression in LETO and OLETF Rats. Nutr. Metab. 2010, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.Y.; Söderholm, J.D. Importance of Disrupted Intestinal Barrier in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef]
- Shen, L.; Su, L.; Turner, J.R. Mechanisms and Functional Implications of Intestinal Barrier Defects. Dig. Dis. Basel Switz. 2009, 27, 443–449. [Google Scholar] [CrossRef]
- Tso, P.; Balint, J.A. Formation and Transport of Chylomicrons by Enterocytes to the Lymphatics. Am. J. Physiol. 1986, 250, G715–G726. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Sakata, Y.; Tso, P. Nutrient-Induced Inflammation in the Intestine. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 315–321. [Google Scholar] [CrossRef]
- Rietschel, E.T.; Kirikae, T.; Schade, F.U.; Mamat, U.; Schmidt, G.; Loppnow, H.; Ulmer, A.J.; Zähringer, U.; Seydel, U.; Di Padova, F. Bacterial Endotoxin: Molecular Relationships of Structure to Activity and Function. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1994, 8, 217–225. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons Promote Intestinal Absorption of Lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef]
- Mani, V.; Hollis, J.H.; Gabler, N.K. Dietary Oil Composition Differentially Modulates Intestinal Endotoxin Transport and Postprandial Endotoxemia. Nutr. Metab. 2013, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A High-Fat Meal Induces Low-Grade Endotoxemia: Evidence of a Novel Mechanism of Postprandial Inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Lyte, J.M.; Gabler, N.K.; Hollis, J.H. Postprandial Serum Endotoxin in Healthy Humans Is Modulated by Dietary Fat in a Randomized, Controlled, Cross-over Study. Lipids Health Dis. 2016, 15, 186. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Bile Acids: Regulation of Synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef]
- Araki, Y.; Katoh, T.; Ogawa, A.; Bamba, S.; Andoh, A.; Koyama, S.; Fujiyama, Y.; Bamba, T. Bile Acid Modulates Transepithelial Permeability via the Generation of Reactive Oxygen Species in the Caco-2 Cell Line. Free Radic. Biol. Med. 2005, 39, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Santoro, P.; Barone, M.V.; Pappacoda, S.; Barretta, M.L.; Nanayakkara, M.; Apicella, C.; Capasso, L.; Paludetto, R. Bile Acids Modulate Tight Junction Structure and Barrier Function of Caco-2 Monolayers via EGFR Activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G906–G913. [Google Scholar] [CrossRef]
- Stenman, L.K.; Holma, R.; Korpela, R. High-Fat-Induced Intestinal Permeability Dysfunction Associated with Altered Fecal Bile Acids. World J. Gastroenterol. 2012, 18, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.K.; Basuroy, S.; Rao, V.U.; Karnaky, K.J.; Gupta, A. Tyrosine Phosphorylation and Dissociation of Occludin-ZO-1 and E-Cadherin-Beta-Catenin Complexes from the Cytoskeleton by Oxidative Stress. Biochem. J. 2002, 368, 471–481. [Google Scholar] [CrossRef]
- Sheth, P.; Seth, A.; Atkinson, K.J.; Gheyi, T.; Kale, G.; Giorgianni, F.; Desiderio, D.M.; Li, C.; Naren, A.; Rao, R. Acetaldehyde Dissociates the PTP1B-E-Cadherin-Beta-Catenin Complex in Caco-2 Cell Monolayers by a Phosphorylation-Dependent Mechanism. Biochem. J. 2007, 402, 291–300. [Google Scholar] [CrossRef]
- Penumetcha, M.; Khan-Merchant, N.; Parthasarathy, S. Enhanced Solubilization and Intestinal Absorption of Cholesterol by Oxidized Linoleic Acid. J. Lipid Res. 2002, 43, 895–903. [Google Scholar] [CrossRef]
- Kullmann, F.; Arndt, H.; Gross, V.; Rüschoff, J.; Schölmerich, J. Beneficial Effect of Ursodeoxycholic Acid on Mucosal Damage in Trinitrobenzene Sulphonic Acid-Induced Colitis. Eur. J. Gastroenterol. Hepatol. 1997, 9, 1205–1211. [Google Scholar] [PubMed]
- Rodrigues, C.M.; Fan, G.; Wong, P.Y.; Kren, B.T.; Steer, C.J. Ursodeoxycholic Acid May Inhibit Deoxycholic Acid-Induced Apoptosis by Modulating Mitochondrial Transmembrane Potential and Reactive Oxygen Species Production. Mol. Med. Camb. Mass 1998, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Bernardes-Silva, C.F.; Damião, A.O.M.C.; Sipahi, A.M.; Laurindo, F.R.M.; Iriya, K.; Lopasso, F.P.; Buchpiguel, C.A.; Lordello, M.L.L.; Agostinho, C.L.O.; Laudanna, A.A. Ursodeoxycholic Acid Ameliorates Experimental Ileitis Counteracting Intestinal Barrier Dysfunction and Oxidative Stress. Dig. Dis. Sci. 2004, 49, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Perrone, E.E.; Chen, C.; Longshore, S.W.; Okezie, O.; Warner, B.W.; Sun, C.-C.; Alaish, S.M.; Strauch, E.D. Dietary Bile Acid Supplementation Improves Intestinal Integrity and Survival in a Murine Model. J. Pediatr. Surg. 2010, 45, 1256–1265. [Google Scholar] [CrossRef][Green Version]
- Margheritis, E.; Castellani, B.; Magotti, P.; Peruzzi, S.; Romeo, E.; Natali, F.; Mostarda, S.; Gioiello, A.; Piomelli, D.; Garau, G. Bile Acid Recognition by NAPE-PLD. ACS Chem. Biol. 2016, 11, 2908–2914. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.L.; Renooij, W.; Murzilli, S.; Klomp, L.W.J.; Siersema, P.D.; Schipper, M.E.I.; Danese, S.; et al. Farnesoid X Receptor Activation Inhibits Inflammation and Preserves the Intestinal Barrier in Inflammatory Bowel Disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Oldenburg, B.; Willemsen, E.C.L.; Spit, M.; Murzilli, S.; Salvatore, L.; Klomp, L.W.J.; Siersema, P.D.; van Erpecum, K.J.; van Mil, S.W.C. Activation of Bile Salt Nuclear Receptor FXR Is Repressed by Pro-Inflammatory Cytokines Activating NF-ΚB Signaling in the Intestine. Biochim. Biophys. Acta 2011, 1812, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Mil, S.W.C.; Oldenburg, B.; Siersema, P.D.; Klomp, L.W.J.; van Erpecum, K.J. Bile Acids and Their Nuclear Receptor FXR: Relevance for Hepatobiliary and Gastrointestinal Disease. Biochim. Biophys. Acta 2010, 1801, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Everard, A.; Van Hul, M.; Essaghir, A.; Duparc, T.; Matamoros, S.; Plovier, H.; Castel, J.; Denis, R.G.P.; Bergiers, M.; et al. Adipose Tissue NAPE-PLD Controls Fat Mass Development by Altering the Browning Process and Gut Microbiota. Nat. Commun. 2015, 6, 6495. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Tian, W.; Hong, J.; Li, D.; Tavares, R.; Noble, L.; Moss, S.F.; Resnick, M.B. Expression of Bile Acid Receptor TGR5 in Gastric Adenocarcinoma. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G322–G327. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile Acid-Microbiota Crosstalk in Gastrointestinal Inflammation and Carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Ní Gabhann, J.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic Acid and Lithocholic Acid Exert Anti-Inflammatory Actions in the Colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef]
- Ajouz, H.; Mukherji, D.; Shamseddine, A. Secondary Bile Acids: An Underrecognized Cause of Colon Cancer. World J. Surg. Oncol. 2014, 12, 164. [Google Scholar] [CrossRef] [PubMed]
- Payne, C.M.; Bernstein, C.; Dvorak, K.; Bernstein, H. Hydrophobic Bile Acids, Genomic Instability, Darwinian Selection, and Colon Carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of Deoxycholate, a Secondary Bile Acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorakova, K.; Garewal, H. Bile Acids as Carcinogens in Human Gastrointestinal Cancers. Mutat. Res. 2005, 589, 47–65. [Google Scholar] [CrossRef]
- Rao, Y.P.; Stravitz, R.T.; Vlahcevic, Z.R.; Gurley, E.C.; Sando, J.J.; Hylemon, P.B. Activation of Protein Kinase C Alpha and Delta by Bile Acids: Correlation with Bile Acid Structure and Diacylglycerol Formation. J. Lipid Res. 1997, 38, 2446–2454. [Google Scholar] [CrossRef]
- Mencarelli, A.; Renga, B.; Migliorati, M.; Cipriani, S.; Distrutti, E.; Santucci, L.; Fiorucci, S. The Bile Acid Sensor Farnesoid X Receptor Is a Modulator of Liver Immunity in a Rodent Model of Acute Hepatitis. J. Immunol. Baltim. Md 1950 2009, 183, 6657–6666. [Google Scholar] [CrossRef] [PubMed]
- Calmus, Y.; Poupon, R. Shaping Macrophages Function and Innate Immunity by Bile Acids: Mechanisms and Implication in Cholestatic Liver Diseases. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 550–556. [Google Scholar] [CrossRef]
- Charo, I.F.; Ransohoff, R.M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354, 610–621. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 Contributes to Macrophage Infiltration into Adipose Tissue, Insulin Resistance, and Hepatic Steatosis in Obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity Is Associated with Macrophage Accumulation in Adipose Tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic Inflammation in Fat Plays a Crucial Role in the Development of Obesity-Related Insulin Resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Aljada, A.; Hofmeyer, D.; Syed, T.; Mohanty, P.; Dandona, P. Circulating Mononuclear Cells in the Obese Are in a Proinflammatory State. Circulation 2004, 110, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- RostamiRad, A.; Ebrahimi, S.S.S.; Sadeghi, A.; Taghikhani, M.; Meshkani, R. Palmitate-Induced Impairment of Autophagy Turnover Leads to Increased Apoptosis and Inflammation in Peripheral Blood Mononuclear Cells. Immunobiology 2018, 223, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Lowry, J.E.; Tumurbaatar, B.; D’Agostino, C.; Main, E.; Wright, T.J.; Dillon, E.L.; Saito, T.B.; Porter, C.; Andersen, C.R.; Brining, D.L.; et al. Effect of High-Fat Diet on Peripheral Blood Mononuclear Cells and Adipose Tissue in Early Stages of Diet-Induced Weight Gain. Br. J. Nutr. 2019, 122, 1359–1367. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired Macrophage Autophagy Increases the Immune Response in Obese Mice by Promoting Proinflammatory Macrophage Polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Istrate, N.; Wang, L.; Nichols, A.J.; Tozzo, E.; Stricker-Krongrad, A. Resistance to the Orexigenic Effect of Ghrelin in Dietary-Induced Obesity in Mice: Reversal upon Weight Loss. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2004, 28, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Hyvarinen, N.; Lilly, N.; Kay, K.; Dossat, A.; Parise, E.; Torregrossa, A.-M. Maintenance on a High-Fat Diet Impairs the Anorexic Response to Glucagon-like-Peptide-1 Receptor Activation. Physiol. Behav. 2011, 103, 557–564. [Google Scholar] [CrossRef] [PubMed]


| Bacteria | HFD Impact |
|---|---|
| Phylum: Firmicutes | ↑ |
| Order: Erysipelotrichales | ↑ |
| Class: Bacili | ↑ |
| Genus: Lactobacillus | ↑ |
| Order: Clostridiales | ↑ |
| Genus: Oscillibacter | ↑ |
| Genus: Dorea | ↑ |
| Genus: Ruminococcus | ↑ |
| Phylum: Bacteroidetes | ↓ |
| Family: Prevotellaceae | ↓ |
| Family: Rikenellaceae | ↓ |
| Phylum: Actinobacteria | ↑/↓ |
| Genus: Bifidobacterium | ↑/↓ |
| Phylum: Tenericutes | ↓ |
| Phylum: Proteobacteria | ↑ |
| Order: Enterobacteriales | ↑ |
| Family: Enterobacteriaceae | ↑ |
| Order: Desulfovibrionales | ↑ |
| Genus: Desulfovibrio | ↑ |
| Species: Bilophila wadsworthia | ↑ |
| Phylum: Verrucomicrobia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. https://doi.org/10.3390/cells10113164
Malesza IJ, Malesza M, Walkowiak J, Mussin N, Walkowiak D, Aringazina R, Bartkowiak-Wieczorek J, Mądry E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells. 2021; 10(11):3164. https://doi.org/10.3390/cells10113164
Chicago/Turabian StyleMalesza, Ida Judyta, Michał Malesza, Jarosław Walkowiak, Nadiar Mussin, Dariusz Walkowiak, Raisa Aringazina, Joanna Bartkowiak-Wieczorek, and Edyta Mądry. 2021. "High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review" Cells 10, no. 11: 3164. https://doi.org/10.3390/cells10113164
APA StyleMalesza, I. J., Malesza, M., Walkowiak, J., Mussin, N., Walkowiak, D., Aringazina, R., Bartkowiak-Wieczorek, J., & Mądry, E. (2021). High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells, 10(11), 3164. https://doi.org/10.3390/cells10113164