
Signaling through the S1P−S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment


Abstract
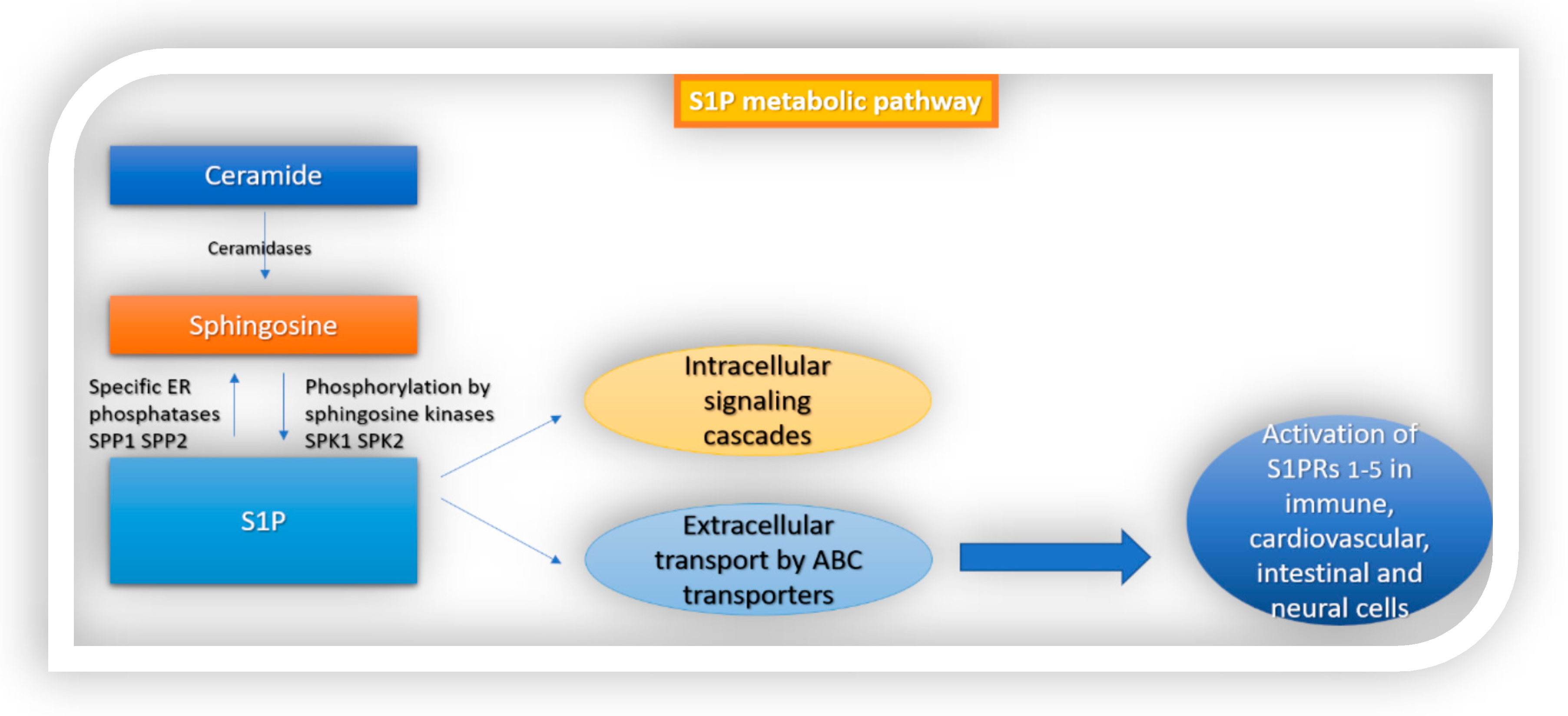
1. Introduction
2. S1P Signaling in the Immune System
2.1. Lymphocyte Trafficking and Regulation of Adaptive Immune Functions
2.2. Regulation of Innate Immune Cell Trafficking
3. S1P Signaling in the Intestine
Implication in Intestinal Barrier Homeostasis
4. S1P Signaling in the CNS
Implication in BBB Homeostasis
5. Implication for the Role of S1P in Multiple Sclerosis
6. The Role of the Gut–Brain Axis in MS Course and Progression
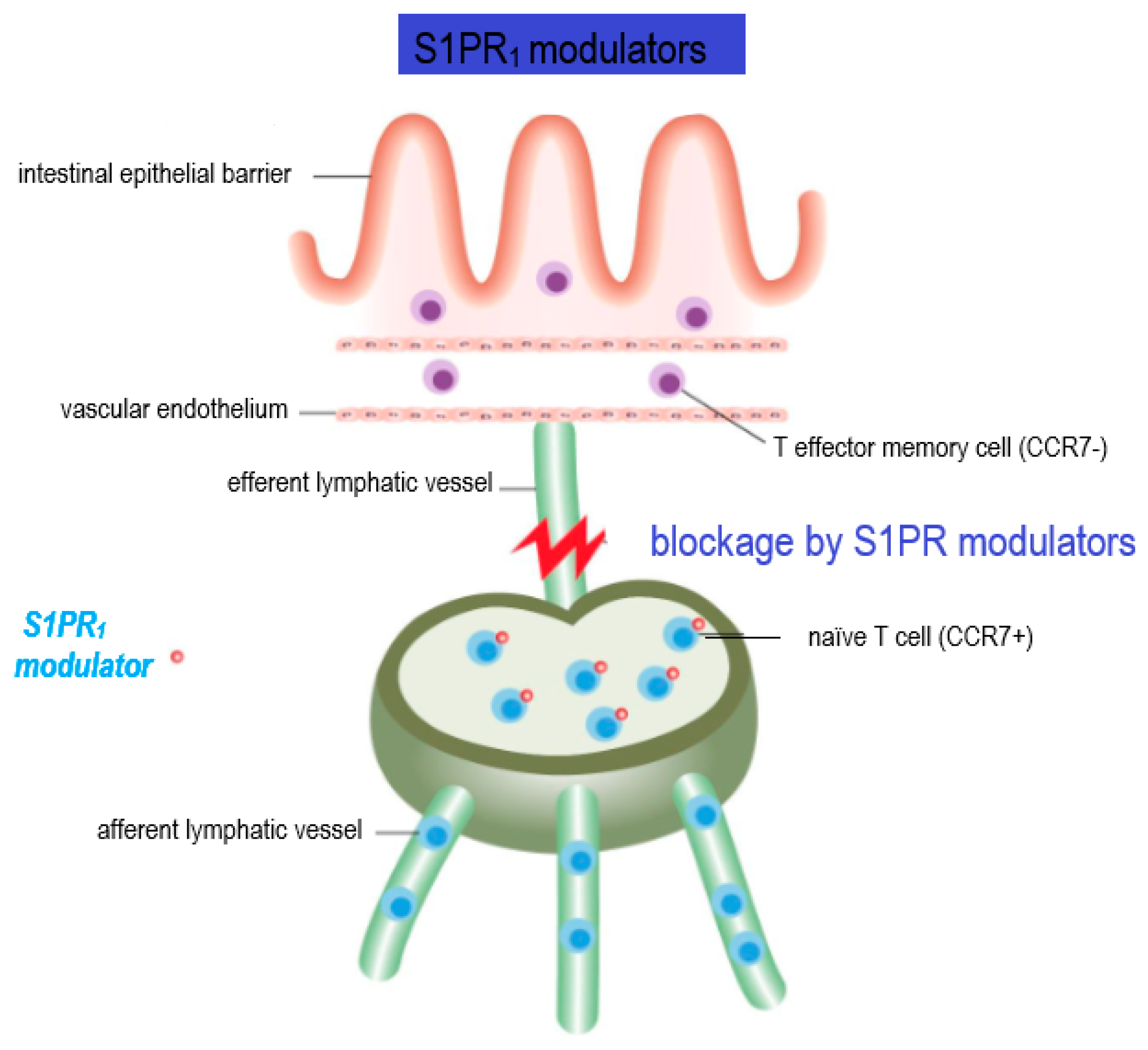
7. S1PR Modulators in MS Therapy
7.1. Immunomodulatory Effects
7.2. Potential Neuroprotective Effects
7.3. Additional Effects under Investigation
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Thudichum, J.L.W. Treatise on the Chemical Constitution of the Brain: Based Throughout upon Original Researches. Glasgow Med. J. 1884, 22, 363–364. [Google Scholar]
- An, S.; Bleu, T.; Huang, W.; Hallmark, O.G.; Coughlin, S.R.; Goetzl, E.J. Identification of cDNAs encoding two G protein-coupled receptors for lysosphingolipids. FEBS Lett. 1997, 417, 279–282. [Google Scholar] [CrossRef]
- Lee, M.J.; van Brocklyn, J.R.; Thangada, S.; Liu, C.H.; Hand, A.R.; Menzeleev, R.; Spiegel, S.; Hla, T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 1998, 279, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology 2011, 76, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.; Gonzalez-Cabrera, P.J.; Sanna, M.G.; Brown, S. Sphingosine 1-phosphate receptor signalling. Annu. Rev. Biochem. 2009, 78, 743–768. [Google Scholar] [CrossRef]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef]
- Aoki, M.; Aoki, H.; Ramanathan, R.; Hait, N.C.; Takabe, K. Sphingosine-1-Phosphate Signaling in Immune Cells and Inflammation: Roles and Therapeutic Potential. Mediators Inflamm. 2016, 2016, 8606878. [Google Scholar]
- Marsolais, D.; Rosen, H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307. [Google Scholar] [CrossRef]
- Mendoza, A.; Bréart, B.; Ramos-Perez, W.R.; Pitt, L.A.; Gobert, M.; Sunkara, M.; Lafaille, J.; Morris, A.J.; Schwab, S.R. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1- phosphate. Cell Rep. 2012, 2, 1104–1110. [Google Scholar] [CrossRef]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 23, 617–625. [Google Scholar] [CrossRef]
- Galvani, S.; Sanson, M.; Blaho, V.A.; Swendeman, S.L.; Obinata, H.; Conger, H.; Dahlbäck, B.; Kono, M.; Proia, R.L.; Smith, J.D.; et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 2015, 8, ra79. [Google Scholar] [CrossRef]
- Dash, R.P.; Rais, R.; Srinivas, N.R. Ponesimod, a selective sphingosine 1-phosphate (S1P 1) receptor modulator for autoimmune diseases: Review of clinical pharmacokinetics and drug disposition. Xenobiotica 2018, 48, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Arish, M.; Alaidarous, M.; Ali, R.; Akhter, Y.; Rub, A. Implication of sphingosine-1-phosphate signaling in diseases: Molecular mechanism and therapeutic strategies. J. Recept. Signal Transduct. Res. 2017, 37, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Schwab, S.R. Sphingosine-1-Phosphate and Lymphocyte Egress from Lymphoid Organs. Annu. Rev. Immunol. 2012, 30, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Bryan, A.M.; del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef]
- Mao-Draayer, Y.; Sarazin, J.; Fox, D.; Schiopu, E. The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin. Immunol. 2017, 175, 10–15. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Han, M.H. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway: Therapeutic Targets in Autoimmunity and Inflammation. Drugs 2016, 76, 1067–1079. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355. [Google Scholar] [CrossRef]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Allende, M.L.; Bektas, M.; Lee, B.G.; Bonifacino, E.; Kang, J.; Tuymetova, G.; Chen, W.; Saba, J.D.; Proia, R.L. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 2011, 286, 7348–7358. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Seki, N.; Kataoka, H.; Takemoto, K.; Utsumi, H.; Fukunari, A.; Sugahara, K.; Chiba, K. IL-17-producing Vγ4+ γδ T cells require sphingosine 1-phosphate receptor 1 for their egress from the lymph nodes under homeostatic and inflammatory conditions. J. Immunol. 2015, 195, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yang, K.; Burns, S.; Shrestha, S.; Chi, H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat. Immunol. 2010, 11, 1047–1056. [Google Scholar] [CrossRef]
- Czeloth, N.; Bernhardt, G.; Hofmann, F.; Genth, H.; Förster, R. Sphingosine-1-phosphate mediates migration of mature dendritic cells. J. Immunol. 2005, 175, 2960. [Google Scholar] [CrossRef]
- Walzer, T.; Chiossone, L.; Chaix, J.; Calver, A.; Carozzo, C.; Garrigue-Antar, L.; Jacques, Y.; Baratin, M.; Tomasello, E.; Vivier, E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 2007, 8, 1337. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Enders, A.; Rivera, R.; Watson, S.R.; Bankovich, A.; Pereira, J.; Xu, Y.; Roots, C.M.; Beilke, J.N.; Banerjee, A.; et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J. Exp. Med. 2009, 206, 2469–2481. [Google Scholar] [CrossRef]
- Massberg, S.; Schaerli, P.; Knezevic-Maramica, I.; Köllnberger, M.; Tubo, N.; Moseman, E.A.; Huff, I.V.; Junt, T.; Wagers, A.J.; Mazo, I.B.; et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 2007, 131, 994. [Google Scholar] [CrossRef]
- Olesch, C.; Ringel, C.; Brüne, B.; Weigert, A. Beyond Immune Cell Migration: The Emerging Role of the Sphingosine-1-phosphate Receptor S1PR4 as a Modulator of Innate Immune Cell Activation. Mediators Inflamm. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Finley, A.; Chen, Z.; Esposito, E.; Cuzzocrea, S.; Sabbadini, R.; Salvemini, D. Sphingosine 1-Phosphate Mediates Hyperalgesia via a Neutrophil-Dependent Mechanism. PLoS ONE 2013, 8, e55255. [Google Scholar]
- Miyabe, C.; Miyabe, Y.; Komiya, T.; Shioya, H.; Miura, N.N.; Takahashi, K.; Ohno, N.; Tsuboi, R.; Luster, A.D.; Kawai, S.; et al. A sphingosine 1-phosphate receptor agonist ameliorates animal model of vasculitis. Inflamm. Res. 2017, 66, 335–340. [Google Scholar] [CrossRef]
- Gonzalez, L.; Qian, A.S.; Tahir, U.; Yu, P.; Trigatti, B.L. Sphingosine-1-Phosphate Receptor 1, Expressed in Myeloid Cells, Slows Diet-Induced Atherosclerosis and Protects against Macrophage Apoptosis in Ldlr KO Mice. Int. J. Mol. Sci. 2017, 18, 2721. [Google Scholar] [CrossRef]
- McQuiston, T.; Luberto, C.; del Poeta, M. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology 2011, 157, 1416–1427. [Google Scholar] [CrossRef]
- Hou, J.; Chen, Q.; Zhang, K.; Cheng, B.; Xie, G.; Wu, X.; Luo, C.; Chen, L.; Liu, H.; Zhao, B.; et al. Sphingosine 1-phosphate Receptor 2 Signaling Suppresses Macrophage Phagocytosis and Impairs Host Defense against Sepsis. Anesthesiology 2015, 123, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.; Canals, D.; Hannun, Y.A.; Obeid, L.M. Sphingosine-1-phosphate receptor 2. FEBS J. 2013, 280, 6354–6366. [Google Scholar] [CrossRef] [PubMed]
- Keul, P.; Lucke, S.; Lipinski, K.V.W.; Bode, C.; Gräler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2011, 108, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, C.; Mora, J.; Olesch, C.; Brüne, B.; Weigert, A. S1PR4 is required for plasmacytoid dendritic cell differentiation. Biol. Chem. 2015, 396, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; von Bernstorff, W.; Heidecke, C.-D.; Schulze, T. Differential S1P Receptor Profiles on M1- and M2-Polarized Macrophages Affect Macrophage Cytokine Production and Migration. BioMed Res. Int. 2017, 2017, 1–10. [Google Scholar]
- Debien, E.; Mayol, K.; Biajoux, V.; Daussy, C.; de Agüero, M.G.; Taillardet, M.; Dagany, N.; Brinza, L.; Henry, T.; Dubois, T.; et al. S1PR5 is pivotal for the homeostasis of patrolling monocytes. Eur. J. Immunol. 2013, 43, 1667–1675. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Nguyen, K.; Hashemi, E.; Engleman, E.; Hla, T.; Han, M.H. Myeloid sphingosine-1-phosphate receptor 1 is important for CNS autoimmunity and neuroinflammation. J. Autoimmun. 2019, 105, 102290. [Google Scholar] [CrossRef]
- Cohen, J.; Bar-Or, A.; Cree, B.A.C.; Mao-Draayer, Y.; Han, M.H.; Singer, B.; Jannu, A.; Kolodny, S.; Meng, X.; Winger, R.C. The FLUENT study design: Investigating immune cell subset and neurofilament changes in patients with relapsing multiple sclerosis treated with fingolimod. Mult. Scler. J. Exp. Transl. Clin. 2019, 5, 2055217318819245. [Google Scholar] [CrossRef]
- Salvo Romero, E.; Alonso Cotoner, C.; Pardo Camacho, C.; Casado Bedmar, M.; Vicario, M. The intestinal barrier function and its involvement in digestive disease. Rev. Esp. Enferm. Dig. 2015, 107, 686–696. [Google Scholar] [CrossRef]
- Klunder, L.J.; Faber, K.N.; Dijkstra, G.; van Ijzendoorn, S.C.D. Mechanisms of Cell Polarity-Controlled Epithelial Homeostasis and Immunity in the Intestine. Cold Spring Harb. Perspect Biol. 2017, 9, a027888. [Google Scholar] [CrossRef]
- Chen, T.; Huang, Z.; Liu, R.; Yang, J.; Hylemon, P.B.; Zhou, H. Sphingosine-1 phosphate promotes intestinal epithelial cell proliferation via S1PR2. Front. Biosci. 2017, 22, 596–608. [Google Scholar]
- Smith, A.D.; Rao, J.N.; Turner, D.J. Sphingosine-1-Phosphate and the Intestine. Surgery 2012, 2, 1. [Google Scholar] [CrossRef]
- Kunisawa, J.; Kiyono, H. Immunological Function of Sphingosine 1-Phosphate in the Intestine. Nutrients 2012, 4, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Maines, L.W.; Fitzpatrick, L.R.; French, K.J.; Zhuang, Y.; Xia, Z.; Keller, S.N.; Upson, J.J.; Smith, C.D. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig. Dis. Sci. 2008, 53, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, J.C.; Zhou, H.; Brayfield, B.P.; Hontecillas, R.; Bassaganya-Riera, J.; Schmelz, E.M. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: Importance of peroxisome proliferator-activated receptor gamma expression. J. Nutr. Biochem. 2011, 22, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Fischbeck, A.; Leucht, K.; Frey-Wagner, I.; Bentz, S.; Pesch, T.; Kellermeier, S.; Krebs, M.; Fried, M.; Rogler, G.; Hausman, M.; et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 2011, 60, 55–65. [Google Scholar] [CrossRef]
- Nyberg, L.; Duan, R.D.; Nilsson, A. A mutual inhibitory effect on absorption of sphingomyelin and cholesterol. J. Nutr. Biochem. 2000, 11, 244–249. [Google Scholar] [CrossRef]
- Suh, J.H.; Saba, J.D. Sphingosine-1-phosphate in inflammatory bowel disease and colitis-associated colon cancer: The fat’s in the fire. Transl. Cancer Res. 2015, 4, 15. [Google Scholar]
- The Human Protein Atlas, Knut & Alice Wallenberg Foundation. Available online: http://www.proteinatlas.org (accessed on 8 August 2021).
- Greenspon, J.; Li, R.; Xiao, L.; Rao, J.N.; Sun, R.; Strauch, E.D.; Shea-Donohue, T.; Wang, J.; Turner, D.J. Sphingosine-1-phosphate regulates the expression of adherens junction protein E-cadherin and enhances intestinal epithelial cell barrier function. Dig. Dis. Sci. 2011, 56, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Lin, R.; Jin, S.; Chen, R.; Xue, H.; Ye, H.; Huang, Z. The Sphingosine-1-Phosphate/Sphingosine-1-Phosphate Receptor 2 Axis in Intestinal Epithelial Cells Regulates Intestinal Barrier Function During Intestinal Epithelial Cells–CD4+T-Cell Interactions. Cell Physiol. Biochem. 2018, 48, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Anbazhagan, A.N.; Priyamvada, S.; Alakkam, A.; Kumar, A.; Borthakur, A.; Saksena, S. Transcriptional Modulation of SLC26A3 (DRA) by Sphingosine-1-Phosphate. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1028–G1035. [Google Scholar] [CrossRef] [PubMed]
- Greenspon, J.; Li, R.; Xiao, L.; Rao, J.N.; Marasa, B.S.; Strauch, E.D.; Wang, J.; Turner, D.J. Sphingosine-1-phosphate protects intestinal epithelial cells from apoptosis through the Akt signaling pathway. Dig. Dis. Sci. 2009, 54, 499–510. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bavaria, M.N.; Jin, S.; Ray, R.M.; Johnson, L.R. The mechanism by which MEK/ERK regulates JNK and p38 activity in polyamine depleted IEC-6 cells during apoptosis. Apoptosis 2014, 19, 467–479. [Google Scholar] [CrossRef]
- Bordet, R.; Camu, W.; de Seze, J.; Laplaud, D.; Ouallet, J.-C.; Thouvenot, E. Mechanism of action of s1p receptor modulators in multiple sclerosis: The double requirement. Rev. Neurol. 2020, 176, 100–112. [Google Scholar] [CrossRef]
- Martin, R.; Sospedra, M. Sphingosine- 1 phosphate and central nervous system. Curr. Top. Microbiol. Immunol. 2014, 378, 149–170. [Google Scholar]
- O’Sullivan, S.; Dev, K.K. Sphingosine-1-phosphate receptor therapies: Advances in clinical trials for CNS-related diseases. Neuropharmacology 2017, 113, 597–607. [Google Scholar] [CrossRef]
- Soliven, B.; Miron, V.; Chun, J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology 2011, 76, S9–S14. [Google Scholar] [CrossRef]
- Ng, M.L.; Yarla, N.S.; Menschikowski, M.; Sukocheva, O.A. Regulatory role of sphingosine kinase and sphingosine-1-phosphate receptor signaling in progenitor/stem cells. World J. Stem Cells 2018, 10, 119–133. [Google Scholar] [CrossRef]
- Toman, R.E.; Payne, S.G.; Watterson, K.R.; Maceyka, M.; Lee, N.H.; Milstien, S.; Bigbee, J.W.; Spiegel, S. Differential transactivation of sphingosine-1-phosphate receptors modulates NGF-induced neurite extension. J. Cell Biol. 2004, 166, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Blanc, C.A.; Grist, J.J.; Rosen, H.; Sears-Kraxberger, I.; Steward, O.; Lane, T.E. Sphingosine-1-phosphate receptor antagonism enhances proliferation and migration of engrafted neural progenitor cells in a model of viral-induced demyelination. Am. J. Pathol. 2015, 185, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E. There is more to a lipid than just being a fat: Sphingolipid-guided differentiation of oligodendroglial lineage from embryonic stem cells. Neurochem. Res. 2011, 36, 1601–1611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kanno, T.; Nishizaki, T.; Proia, R.L.; Kajimoto, T.; Jahangeer, S.; Okada, T.; Nakamura, S. Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience 2010, 171, 973–980. [Google Scholar] [CrossRef]
- Norman, E.; Cutler, R.G.; Flannery, R.; Wang, Y.; Mattson, M.P. Plasma membrane sphingomyelin hydrolysis increases hippocampal neuron excitability by sphingosine-1-phosphate mediated mechanisms. J. Neurochem. 2010, 114, 430–439. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, A.J.; Carney, P.R.; Zhu, W.J.; Chaves, H.; Garcia, J.; Grimes, J.R.; Anderson, K.J.; Roper, S.N.; Lee, N. An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur. J. Neurosci. 2001, 14, 203–209. [Google Scholar] [CrossRef]
- Ishii, I.; Ye, X.; Friedman, B.; Kawamura, S.; Contos, J.J.A.; Kingsbury, M.A.; Yang, A.H.; Zhang, G.; Brown, J.H.; Chun, J. Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P/LP(B2)/EDG-5 and S1P/LP(B3)/EDG-3. J. Biol. Chem. 2002, 277, 25152–25159. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Lariosa-Willingham, K.D.; Lin, F.F.; Webb, M.; Rao, T.S. Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia 2004, 45, 17–27. [Google Scholar] [CrossRef]
- Miron, V.E.; Durafourt, B.A.; Antel, J.P.; Kennedy, T.E. Assessment of sphingosine-1-phosphate receptor expression and associated intracellular signaling cascades in primary cells of the human central nervous system. Methods Mol. Biol. 2012, 874, 141–154. [Google Scholar]
- Novgorodov, A.S.; El-Alwani, M.; Bielawski, J.; Obeid, L.M.; Gudz, T.I. Activation of sphingosine-1-phosphate receptor S1P5 inhibits oligodendrocyte progenitor migration. FASEB J. 2007, 21, 1503–1514. [Google Scholar] [CrossRef]
- Miron, V.E.; Jung, C.G.; Kim, H.J.; Kennedy, T.E.; Soliven, B.; Antel, J.P. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann. Neurol. 2008, 63, 61–71. [Google Scholar] [CrossRef]
- Jaillard, C.; Harrison, S.; Stankoff, B.; Aigrot, M.S.; Calver, A.R.; Duddy, G.; Walsh, F.S.; Pangalos, M.N.; Arimura, N.; Kaibuchi, K.; et al. Edg8/S1P5: An oligodendroglial receptor with dual function on process retraction and cell survival. J. Neurosci. 2005, 25, 1459–1469. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Miron, V.E.; Dukala, D.; Proia, R.L.; Ludwin, S.K.; Traka, M.; Antel, J.P.; Soliven, B. Neurobiological effects of sphingosine 1-phosphate receptor modulation in the cuprizone model. FASEB J. 2011, 25, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.G.; Kim, H.J.; Miron, V.E.; Cook, S.; Kennedy, T.E.; Foster, C.A.; Antel, J.P.; Soliven, B. Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 2007, 55, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.S.; Lariosa-Willingham, K.D.; Lin, F.; Yu, N.; Tham, C.; Chun, J.; Webb, M. Growth factor pretreatment differentially regulates phosphoinositide turnover downstream of lysophospholipid receptor and metabotropic glutamate receptors in cultured rat cerebrocortical astrocytes. Int. J. Dev. Neurosci. 2004, 22, 131–135. [Google Scholar] [CrossRef]
- Osinde, M.; Mullershausen, F.; Dev, K.K. Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology 2007, 52, 1210–1218. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar] [CrossRef]
- Wu, Y.P.; Mizugishi, K.; Bektas, M.; Sandhoff, R.; Proia, R.L. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Hum. Mol. Genet. 2008, 17, 2257–2264. [Google Scholar] [CrossRef]
- O’Sullivan, S.A.; O’Sullivan, C.; Healy, L.M.; Dev, K.K.; Sheridan, G.K. Sphingosine 1-phosphate receptors regulate TLR4-induced CXCL5 release from astrocytes and microglia. J. Neurochem. 2018, 144, 736–747. [Google Scholar] [CrossRef]
- Tham, C.; Lin, F.; Rao, T.; Yu, N.; Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 2003, 21, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Investig. 2009, 119, 1871–1879. [Google Scholar] [CrossRef]
- Christensen, P.M.; Liu, C.H.; Swendeman, S.L.; Obinata, H.; Qvortrup, K.; Nielsen, L.B.; Hla, T.; Di Lorenzo, A.; Christoffersen, C. Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J. 2016, 30, 2351. [Google Scholar] [CrossRef]
- Yanagida, K.; Liu, C.H.; Faraco, G.; Galvani, S.; Smith, H.K.; Burg, N.; Anrather, J.; Sanchez, T.; Iadecola, C.; Hla, T. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. Proc. Natl. Acad. Sci. USA 2017, 114, 4531–4536. [Google Scholar] [CrossRef]
- Lee, M.-J.; Thangada, S.; Claffey, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha’afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar] [CrossRef]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Investig. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef]
- Heliopoulos, I.; Patousi, A. Therapeutic Monoclonal Antibodies and Multiple Sclerosis: The Essentials. Med. Chem. 2018, 14, 144–154. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Dargahi, N.; Katsara, M.; Tselios, T.; Androutsou, M.E.; de Courten, M.; Matsoukas, J.; Apostolopoulos, V. Multiple Sclerosis: Immunopathology and treatment update. Brain Sci. 2017, 7, 78. [Google Scholar] [CrossRef]
- Bsibsi, M.; Peferoen, L.; Holtman, I.; Nacken, P.; Gerritsen, W.; Witte, M.; van Horssen, J.; Eggen, B.J.; van der Valk, P.; Amor, S.; et al. Demyelination during multiple sclerosis is associated with combined activation of microglia/macrophages by IFN-gamma and alpha B-crystallin. Acta Neuropathol. 2014, 128, 215–229. [Google Scholar] [CrossRef]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M. B cells in multiple sclerosis. Curr. Opin. Neurol. 2018, 31, 256–262. [Google Scholar] [CrossRef]
- Stadelmann, C.; Wegner, C.; Bruck, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. N. Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Ochoareparaz, J.; Mielcarz, D.W.; Wang, Y.; Begumhaque, S.; Dasgupta, S.; Kasper, D.L.; Kasper, L.H. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal. Immunol. 2010, 3, 487–495. [Google Scholar] [CrossRef]
- Jin, J.; Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Huang, Y. LPS and palmitate synergistically stimulate sphingosine kinase 1 and increase sphingosine 1 phosphate in RAW264.7 macrophages. J. Leukoc. Biol. 2018, 104, 843–853. [Google Scholar] [CrossRef]
- Custódio, R.; McLean, C.J.; Scott, A.E.; Lowther, J.; Kennedy, A.; Clarke, D.; Campopiano, D.J.; Sarkar-Tyson, M.; Brown, A.R. Characterization of secreted sphingosine-1-phosphate lyases required for virulence and intracellular survival of Burkholderia pseudomallei. Mol. Microbiol. 2016, 102, 1004–1019. [Google Scholar] [CrossRef]
- Ochoa-Repáraz, J.; Magori, K.; Kasper, L.H. The chicken or the egg dilemma: Intestinal dysbiosis in multiple sclerosis. Ann. Transl. Med. 2017, 5, 145. [Google Scholar] [CrossRef]
- Mirza, A.; Forbes, J.D.; Zhu, F.; Bernstein, C.N.; van Domselaar, G.; Graham, M.; Waubant, E.; Tremlett, H. The multiple sclerosis gut microbiota: A systematic review. Mult. Scler. Relat. Disord. 2020, 37, 101427. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef]
- Ganesh, B.P.; Klopfleisch, R.; Loh, G.; Blaut, M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PLoS ONE 2013, 8, e74963. [Google Scholar] [CrossRef]
- Mangalam, A.; Shahi, S.K.; Luckey, D.; Karau, M.; Marietta, E.; Luo, N.; Choung, R.S.; Ju, J.; Sompallae, R.; Gibson-Corley, K.; et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep. 2017, 20, 1269–1277. [Google Scholar] [CrossRef]
- Buscarinu, M.C.; Cerasoli, B.; Annibali, V.; Policano, C.; Lionetto, L.; Capi, M.; Mechelli, R.; Romano, S.; Fornasiero, A.; Mattei, G.; et al. Altered intestinal permeability in patients with relapsing-remitting multiple sclerosis: A pilot study. Mult. Scler. J. 2016, 23, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Bredberg, A.; Weström, B.; Lavasani, S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE 2014, 9, e106335. [Google Scholar]
- Secher, T.; Kassem, S.; Benamar, M.; Bernard, I.; Boury, M.; Barreau, F.; Oswald, E.; Saoudi, A. Oral Administration of the Probiotic Strain Escherichia coli Nissle 1917 Reduces Susceptibility to Neuroinflammation and Repairs Experimental Autoimmune Encephalomyelitis-Induced Intestinal Barrier Dysfunction. Front. Immunol. 2017, 8, 1096. [Google Scholar] [CrossRef]
- Camara-Lemarroy, C.R.; Metz, L.; Meddings, J.B.; Sharkey, K.A.; Wee Yong, V. The intestinal barrier in multiple sclerosis: Implications for pathophysiology and therapeutics. Brain 2018, 141, 1900–1916. [Google Scholar] [CrossRef] [PubMed]
- Boziki, M.K.; Kesidou, E.; Theotokis, P.; Mentis, A.A.; Karafoulidou, E.; Melnikov, M.; Sviridova, A.; Rogovski, V.; Boyko, A.; Grigoriadis, N. Microbiome in Multiple Sclerosis; Where Are We, What We Know and Do Not Know. Brain Sci. 2020, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Radue, E.W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Fujita, T.; Inoue, K.; Yamamoto, S.; Ikumoto, T.; Sasaki, S.; Toyama, R.; Chiba, K.; Hoshino, Y.; Okumoto, T. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J. Antibiot. 1994, 47, 208. [Google Scholar] [CrossRef]
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J. Immunol. 1998, 160, 5037. [Google Scholar]
- Kharel, Y.; Lee, S.; Snyder, A.H.; Sheasley-O’Neill, S.L.; Morris, M.A.; Setiady, Y.; Zhu, R.; Zigler, M.A.; Burcin, T.L.; Ley, K.; et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J. Biol. Chem. 2005, 280, 36865. [Google Scholar] [CrossRef]
- Thomas, K.; Sehr, T.; Proschmann, U.; Rodriguez-Leal, F.A.; Haase, R.; Ziemssen, T. Fingolimod additionally acts as immunomodulator focused on the innate immune system beyond its prominent effects on lymphocyte recirculation. J. Neuroinflamm. 2017, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Villar, M.; Raddassi, K.; Danielsen, A.C.; Guarnaccia, J.; Hafler, D.A. Fingolimod modulates T cell phenotype and regulatory T cell plasticity in vivo. J. Autoimmun. 2019, 96, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, S.; Staun-Ram, E.; Miller, A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL-10 and TGFβ in patients with Multiple Sclerosis. J. Autoimmun. 2016, 70, 40–51. [Google Scholar] [CrossRef]
- Di Dario, M.; Colombo, E.; Govi, C.; de Feo, D.; Messina, M.J.; Romeo, M.; Sangalli, F.; Moiola, L.; Rodegher, M.; Martino, G.; et al. Myeloid cells as target of fingolimod action in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e157. [Google Scholar] [CrossRef]
- Chun, J.; Giovannoni, G.; Hunter, S.F. Sphingosine 1-phosphate Receptor Modulator Therapy for Multiple Sclerosis: Differential Downstream Receptor Signalling and Clinical Profile Effects. Drugs 2021, 81, 207–231. [Google Scholar] [CrossRef]
- Comi, G.; Hartung, H.-P.; Bakshi, R.; Williams, I.M.; Wiendl, H. Benefit-Risk Profile of Sphingosine-1-Phosphate Receptor Modulators in Relapsing and Secondary Progressive Multiple Sclerosis. Drugs 2017, 77, 1755–1768. [Google Scholar] [CrossRef]
- Roy, R.; Alotaibi, A.A.; Freedman, M.S. Sphingosine 1-Phosphate Receptor Modulators for Multiple Sclerosis. CNS Drugs 2021, 35, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Schubart, A.; Antel, J.P. Central nervous system-directed effects of FTY720 (fingolimod). J. Neurol. Sci. 2008, 274, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.G.; Li, Y.; Ding, X.; Shang, X.; Lu, M.; Elias, S.B.; Chopp, M. Fingolimod treatment promotes proliferation and differentiation of oligodendrocyte progenitor cells in mice with experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2015, 76, 57–66. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Ciric, B.; Ma, C.-G.; Gran, B.; Rostami, A.; Zhang, G.-X. Effect of Fingolimod on Neural Stem Cells: A Novel Mechanism and Broadened Application for Neural Repair. Mol. Ther. 2017, 25, 401–415. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.-Y.; Ye, Z.-Q.; Ciric, B.; Ma, C.-G.; Rostami, A.; Li, X.; Zhang, G.-X. Combination Therapy with Fingolimod and Neural Stem Cells Promotes Functional Myelination in vivo Through a Non-immunomodulatory Mechanism. Front. Cell. Neurosci. 2019, 13, 14. [Google Scholar] [CrossRef]
- Choi, J.W.; Gardell, S.E.; Herr, D.; Rivera, R.; Lee, C.-W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2010, 108, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Bs, M.D.D.; Bs, E.C.; Chaabane, L.; Newcombe, J.; Martino, G.; Farina, C. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann. Neurol. 2014, 76, 325–337. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.-C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef]
- Gentile, A.; Musella, A.; Bullitta, S.; Fresegna, D.; de Vito, F.; Fantozzi, R.; Piras, E.; Gargano, F.; Borsellino, G.; Battistini, L.; et al. Siponimod (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J. Neuroinflamm. 2016, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.A.; Lee, D.S.; Sharma, A.; Wang, A.; Naouar, I.; Ma, X.I.; Pikor, N.; Nuesslein-Hildesheim, B.; Ramaglia, V.; Gommerman, J.L. Siponimod therapy implicates Th17 cells in a preclinical model of subpial cortical injury. JCI Insight 2020, 5, e132522. [Google Scholar] [CrossRef]
- Hundehege, P.; Cerina, M.; Eichler, S.; Thomas, C.; Herrmann, A.M.; Göbel, K.; Müntefering, T.; Fernandez-Orth, J.; Bock, S.; Narayanan, V.; et al. The next-generation sphingosine-1 receptor modulator BAF312 (siponimod) improves cortical network functionality in focal autoimmune encephalomyelitis. Neural Regen. Res. 2019, 14, 1950–1960. [Google Scholar] [CrossRef]
- Rossi, S.; Giudice, T.L.; de Chiara, V.; Musella, A.; Studer, V.; Motta, C.; Bernardi, G.; Martino, G.; Furlan, R.; Martorana, A.; et al. Oral fingolimod rescues the functional deficits of synapses in experimental autoimmune encephalomyelitis. Br. J. Pharmacol. 2012, 165, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Bassani, C.; de Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFκB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635. [Google Scholar] [CrossRef]
- Musella, A.; Gentile, A.; Guadalupi, L.; Rizzo, F.R.; de Vito, F.; Fresegna, D.; Bruno, A.; Dolcetti, E.; Vanni, V.; Vitiello, L.; et al. Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells 2020, 9, 1290. [Google Scholar] [CrossRef]
- Tiwari-Woodruff, S.; Yamate-Morgan, H.; Sekyi, M.; Lauderdale, K.; Hasselmann, J.; Schubart, A. The Sphingosine 1-phosphate (S1P) Receptor Modulator, Siponimod Decreases Oligodendrocyte Cell Death and Axon Demyelination in a Mouse Model of Multiple Sclerosis (P5.325). Neurology 2016, 86, P5.325. [Google Scholar]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1 ) and receptor-5 (S1P5 ) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Sun, Y.; Miao, J.; Gao, M.; Guo, L.; Song, X. Ponesimod modulates the Th1/Th17/Treg cell balance and ameliorates disease in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2021, 356, 577583. [Google Scholar] [CrossRef]
- Komiya, T.; Sato, K.; Shioya, H.; Inagaki, Y.; Hagiya, H.; Kozaki, R.; Imai, M.; Takada, Y.; Maeda, T.; Kurata, H.; et al. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin. Exp. Immunol. 2013, 171, 54–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rumah, K.R.; Linden, J.; Fischetti, V.A.; Vartanian, T. Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS ONE 2013, 8, e76359. [Google Scholar] [CrossRef]
- Rumah, K.R.; Ma, Y.; Linden, J.R.; Oo, M.L.; Anrather, J.; Schaeren-Wiemers, N.; Alonso, M.A.; Fischetti, V.A.; McClain, M.S.; Vartanian, T. The Myelin and Lymphocyte Protein MAL Is Required for Binding and Activity of Clostridium perfringens ε-Toxin. PLoS Pathog. 2015, 11, e1004896. [Google Scholar] [CrossRef]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. mBio 2015, 6, e02513. [Google Scholar] [CrossRef]
- Rumah, K.R.; Vartanian, T.K.; Fischetti, V.A. Oral Multiple Sclerosis Drugs Inhibit the In vitro Growth of Epsilon Toxin Producing Gut Bacterium, Clostridium perfringens. Front. Cell. Infect. Microbiol. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Kunisawa, J.; Kurashima, Y.; Gohda, M.; Higuchi, M.; Ishikawa, I.; Miura, F.; Ogahara, I.; Kiyono, H. Sphingosine 1-phosphate regulates peritoneal B-cell trafficking for subsequent intestinal IgA production. Blood 2007, 109, 3749–3756. [Google Scholar] [CrossRef]
- Jia, L.-l.; Zhang, M.; Liu, H.; Sun, J.; Pan, L.-l. Early-life fingolimod treatment improves intestinal homeostasis and pancreatic immune tolerance in non-obese diabetic mice. Acta Pharmacologica Sinica 2021, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, Y.; Andoh, A.; Yagi, Y.; Bamba, S.; Inatomi, O.; Tsujikawa, T.; Fujiyama, Y. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol. Rep. 2006, 16, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Jeldres, T.; Alvarez-Lobos, M.; Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 2021, 81, 985–1002. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.; Vermeire, S.; Ghosh, S.; Liu, W.J.; Petersen, A.; Charles, L.; Huang, V.; Usiskin, K.; et al. Long-Term Efficacy and Safety of Ozanimod in Moderately to Severely Active Ulcerative Colitis: Results from the Open-Label Extension of the Randomized, Phase 2 TOUCHSTONE Study. J. Crohns Colitis 2021, 15, 1120–1129. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Danese, S.; Wolf, D.C.; Liu, W.J.; Hua, S.Y.; Minton, N.; Olson, A.; D’Haens, G. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: A single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol. Hepatol. 2020, 5, 819–828. [Google Scholar] [CrossRef]
- Sharma, S.; Mathur, A.G.; Pradhan, S.; Singh, D.B.; Gupta, S. Fingolimod (FTY720): First approved oral therapy for multiple sclerosis. J. Pharmacol. Pharmacother. 2011, 2, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Al-Salama, Z.T. Siponimod: First Global Approval. Drugs 2019, 79, 1009–1015. [Google Scholar] [CrossRef]
- Markham, A. Ponesimod: First Approval. Drugs 2021, 81, 957–962. [Google Scholar] [CrossRef]
- Lamb, Y.N. Ozanimod: First Approval. Drugs 2020, 80, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Arnold, D.L.; Bar-Or, A.; Camm, J.; Derfuss, T.; Kieseier, B.C.; Sprenger, T.; Greenough, K.; Ni, P.; Harada, T. Safety and efficacy of amiselimod in relapsing multiple sclerosis (MOMENTUM): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2016, 15, 1148–1159. [Google Scholar] [CrossRef]
- Kappos, L.; Arnold, D.L.; Bar-Or, A.; Camm, A.J.; Derfuss, T.; Sprenger, T.; Davies, M.; Piotrowska, A.; Ni, P.; Harada, T. Two-year results from a phase 2 extension study of oral amiselimod in relapsing multiple sclerosis. Mult. Scler. J. 2018, 24, 1605–1616. [Google Scholar] [CrossRef]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Receptor | Associated Cell Types | Functions (Described up to Date) |
|---|---|---|
| S1PR1 | Immune system: T cells, B cells Macrophages Neutrophils DCs Monocytes Eosinophils Mast cells NK cells | Egress from lymph nodes, exit of mature T cells from thymus, migration of natural killer T cells from secondary lymphoid organs to circulation, transfer of immature B cells from bone marrow to circulation Macrophage recruitment Neutrophil migration, recruitment Trafficking of DCs Trafficking of monocytes Eosinophil recruitment Mast cell recruitment |
| GI: IECs | Upregulation of intestinal barrier proteins (claudin1, occludin) | |
| CNS: Astrocytes Oligodendrocytes Neurons Microglia | Activation, differentiation, proliferation of astrocytes, astrogliosis Differentiation of oligodendrocytes, process extension, survival of oligodendrocyte progenitors, myelination Growth cone formation, enhancement of neurite extension, synaptic transmission | |
| S1PR2 | Immune system: Macrophages Monocytes Mast cells Eosinophils | Enhance antibody-mediated phagocytosis, inhibit phagocytosis of bacteria and fungi Degranulation of mast cells |
| GI: IECs | Upregulation of c-Myc, cyclin D1, E-cadherin and Zona occludin 1, proliferation of IECs, absorption of NaCl, prevention of IECs apoptosis | |
| CNS: Neurons Microglia | Growth cone formation, inhibition of neurite extension, control of neural excitability | |
| S1PR3 | Immune system: Macrophages Monocytes Neutrophils DCs Eosinophils Mast cells | Leucocyte rolling on endothelial cells Macrophage’s chemotaxis and killing Neutrophil recruitment DC maturation Eosinophil recruitment |
| GI: IECs | ||
| CNS: Astrocytes Microglia | Activation, differentiation, proliferation of astrocytes, astrogliosis | |
| S1PR4 | Immune system: Macrophages Monocytes Neutrophils DCs Eosinophils Mast cells | Macrophage migration and cytokine release Neutrophil migration Plasmacytoid DC activation and differentiation |
| GI: IECs | ||
| CNS: | ||
| S1PR5 | Immune system: Patrolling monocytes NK cells | Egress of mature NK cells from lymph nodes and bone marrow |
| GI: IECs | ||
| CNS: Mature oligodendrocytes Neurons | Cell survival, process retraction, inhibition of OPC migration, myelination Growth cone formation, inhibition of neurite extension |
| Authors | Drug | MS Model | Main Findings |
|---|---|---|---|
| Choi et al., 2011 [126] | Fingolimod | EAE | The effectiveness of fingolimod is mediated by modulation of S1PR1 in astrocytes |
| Rossi et al., 2012 [132] | Fingolimod | EAE | Ameliorates pre- and postsynaptic glutamatergic transmission and restores clinical signs of disease |
| Colombo et al., 2014 [127] | Fingolimod | EAE | Fingolimod suppresses astrocytic activation (S1P, IL17, and IL1 induced NFκB translocation and NO production) and prevents astrocyte-induced neuronal death |
| Di Dario et al., 2015 [118] | Fingolimod | EAE | Suppression of release of proinflammatory cytokines (TNF-α, IL1β, IL6) leading to inhibition of myeloid cell activation (both in periphery and CNS) |
| Zhang et al., 2015 [123] | Fingolimod | EAE | Promotion of OPC proliferation and differentiation, decrease in disease severity |
| Zhang et al., 2017 [124] | Fingolimod | EAE | Combined administration of fingolimod and NSCs ameliorated clinical signs and CNS demyelination, promoted remyelination, prevented neurodegeneration and astrogliosis |
| Rothhammer et al., 2017 [128] | Fingolimod | EAE in NOD mice | Reduced CNS pathogenic innate immune activation |
| Gentile et al., 2016 [129] | Siponimod | EAE | Improved EAE clinical scores, attenuation of astrogliosis and microgliosis, prevented loss of striatal GABAergic neurons, reduced lymphocyte infiltration in striatum |
| Tiwari-Woodruff et al., 2016 [135] | Siponimod | Cuprizone mouse model | Prevents neurodegeneration and demyelination |
| Hundehege et al., 2019 [131] | Siponimod | Focal EAE | Partial restoration of neuronal network integrity |
| Ward et al., 2020 [130] | Siponimod | EAE | Reduced production of TH17 by T cells, diminished subpial demyelinating lesions |
| Scott et al., 2016 [136] | Ozanimod | EAE | Dose-dependent amelioration in clinical severity of EAE, transient peripheral lymphopenia |
| Musella et al., 2020 [134] | Ozanimod | EAE | Partial restore of striatal glutamatergic dysfunction caused by microglia/macrophage activation |
| Hou et al., 2021 [137] | Ponesimod | EAE | Restores the Th1/Th17/Treg balance and ameliorates disease severity |
| Komiya et al., 2013 [138] | ONO-4641 (Ceralifimod) | EAE in NOD mice | Prevents relapses of relapsing−remitting EAE, dose-dependent blockage of lymphocyte infiltration in CNS |
| Name | S1PR Subtype Modulation | Disease | Indication | Approval Phase |
|---|---|---|---|---|
| Fingolimod (FTY720) | S1PR1,3,4,5 | MS 1 | Relapsing forms of MS | Approved [149] |
| Siponimod (BAF312) | S1PR1,5 | MS | Relapsing forms of MS | Approved [150] |
| Ponesimod (ACT128800) | S1PR1 | MS | Relapsing forms of MS | Approved [151] |
| Ozanimod (RPC1063) | S1PR1,5 | MS IBD 2 | Relapsing forms of MS Moderate to severe UC 3 Moderate to severe CD 4 | Approved [152] Phase III (NCT02531126, NCT03915769) PhaseIII (NCT03467958, NCT03440385, NCT03440372, NCT03464097) |
| Amiselimod (MT-1303) | S1PR1,4,5 | MS IBD | RRMS Moderate to severe CD Mild to moderate UC | Phase II (completed) [153,154] Phase II (completed) (NCT02389790) Phase II (NCT04857112) |
| Etrasimod (APD334) | S1PR1,4,5 | IBD | Moderate to severe UC Moderate to severe CD | Phase III (NCT03950232) Phase IIb (NCT04173273) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatzikonstantinou, S.; Poulidou, V.; Arnaoutoglou, M.; Kazis, D.; Heliopoulos, I.; Grigoriadis, N.; Boziki, M. Signaling through the S1P−S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment. Cells 2021, 10, 3217. https://doi.org/10.3390/cells10113217
Chatzikonstantinou S, Poulidou V, Arnaoutoglou M, Kazis D, Heliopoulos I, Grigoriadis N, Boziki M. Signaling through the S1P−S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment. Cells. 2021; 10(11):3217. https://doi.org/10.3390/cells10113217
Chicago/Turabian StyleChatzikonstantinou, Simela, Vasiliki Poulidou, Marianthi Arnaoutoglou, Dimitrios Kazis, Ioannis Heliopoulos, Nikolaos Grigoriadis, and Marina Boziki. 2021. "Signaling through the S1P−S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment" Cells 10, no. 11: 3217. https://doi.org/10.3390/cells10113217
APA StyleChatzikonstantinou, S., Poulidou, V., Arnaoutoglou, M., Kazis, D., Heliopoulos, I., Grigoriadis, N., & Boziki, M. (2021). Signaling through the S1P−S1PR Axis in the Gut, the Immune and the Central Nervous System in Multiple Sclerosis: Implication for Pathogenesis and Treatment. Cells, 10(11), 3217. https://doi.org/10.3390/cells10113217