
Novel Mechanisms of Tumor Promotion by the Insulin Receptor Isoform A in Triple-Negative Breast Cancer Cells

 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  , ,
, ,  ,
, 


Abstract
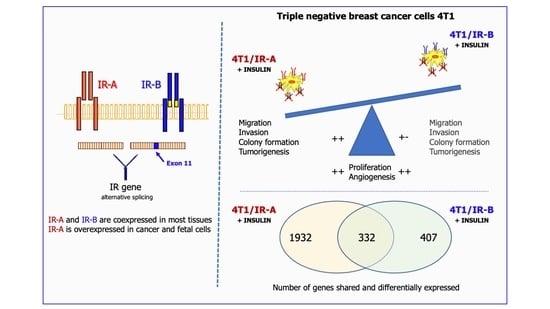
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Establishment of 4T1 Cells Overexpressing hIR-A or hIR-B Isoform
2.2. Cell Proliferation and Viability Assay
2.3. IGF2 Measurement in Cell Conditioned Medium
2.3.1. Biological Assay
2.3.2. IGF2 mRNA Measurement
2.4. Cell Migration and Invasion Assay
2.5. Mouse Allografts
2.6. Zebrafish Studies
2.7. RNA-Seq Data Analysis
2.8. Survival Analyses of TCGA BRCA Samples
2.9. Statistical Analysis
3. Results
3.1. Establishment and Characterization of Cells Overexpressing hIR-A or hIR-B Isoform
3.2. IR-A Expression Is Associated with Enhanced Migration, Invasion, and Anchorage-Independent Growth
3.3. IR-A Enhances BC Growth and Metastasis In Vivo
3.4. In Vivo Analysis of Tumor-Induced Angiogenesis
3.5. Gene Expression Regulation by IR-A and IR-B
3.6. Pathway Analysis
3.7. Validation of RNA Seq Analysis
3.8. Survival Analyses of Publicly Available Molecular Datasets
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, Y.-M.M.; White, A.J.; Nichols, H.B.; O’Brien, K.M.; Weinberg, C.R.; Sandler, D.P. The Association between Metabolic Health, Obesity Phenotype and the Risk of Breast Cancer. Int. J. Cancer 2017, 140, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Godsland, I.F. Insulin Resistance and Hyperinsulinaemia in the Development and Progression of Cancer. Clin. Sci. 2009, 118, 315–332. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Metabolic Inflammation and Insulin Resistance in Obesity. Circ. Res. 2020, 126, 1549–1564. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Morrione, A.; Belfiore, A. Insulin/IGF Signaling and Discoidin Domain Receptors: An Emerging Functional Connection. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118522. [Google Scholar] [CrossRef]
- Sciacca, L.; Costantino, A.; Pandini, G.; Mineo, R.; Frasca, F.; Scalia, P.; Sbraccia, P.; Goldfine, I.D.; Vigneri, R.; Belfiore, A. Insulin Receptor Activation by IGF-II in Breast Cancers: Evidence for a New Autocrine/paracrine Mechanism. Oncogene 1999, 18, 2471–2479. [Google Scholar] [CrossRef]
- Vella, V.; Nicolosi, M.L.; Giuliano, M.; Morrione, A.; Malaguarnera, R.; Belfiore, A. Insulin Receptor Isoform A Modulates Metabolic Reprogramming of Breast Cancer Cells in Response to IGF2 and Insulin Stimulation. Cells 2019, 8, 1017. [Google Scholar] [CrossRef]
- Sacco, A.; Morcavallo, A.; Pandini, G.; Vigneri, R.; Belfiore, A. Differential Signaling Activation by Insulin and Insulin-like Growth Factors I and II upon Binding to Insulin Receptor Isoform A. Endocrinology 2009, 150, 3594–3602. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morcavallo, A.; Genua, M.; Palummo, A.; Kletvikova, E.; Jiracek, J.; Brzozowski, A.M.; Iozzo, R.V.; Belfiore, A.; Morrione, A. Insulin and Insulin-like Growth Factor II Differentially Regulate Endocytic Sorting and Stability of Insulin Receptor Isoform A. J. Biol. Chem. 2012, 287, 11422–11436. [Google Scholar] [CrossRef]
- De Marco, P.; Cirillo, F.; Vivacqua, A.; Malaguarnera, R.; Belfiore, A.; Maggiolini, M. Novel Aspects Concerning the Functional Cross-Talk between the Insulin/IGF-I System and Estrogen Signaling in Cancer Cells. Front. Endocrinol. 2015, 6, 30. [Google Scholar] [CrossRef]
- Vella, V.; Malaguarnera, R.; Nicolosi, M.L.; Palladino, C.; Spoleti, C.; Massimino, M.; Vigneri, P.; Purrello, M.; Ragusa, M.; Morrione, A.; et al. Discoidin Domain Receptor 1 Modulates Insulin Receptor Signaling and Biological Responses in Breast Cancer Cells. Oncotarget 2017, 8, 43248–43270. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Pandini, G.; Sciacca, L.; Mineo, R.; Vigneri, R.; Pezzino, V.; Belfiore, A. A Novel Autocrine Loop Involving IGF-II and the Insulin Receptor Isoform-A Stimulates Growth of Thyroid Cancer. J. Clin. Endocrinol. Metab. 2002, 87, 245–254. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Nicolosi, M.L.; Lappano, R.; Ragusa, M.; Morrione, A.; Vella, V. A Novel Functional Crosstalk between DDR1 and the IGF Axis and Its Relevance for Breast Cancer. Cell Adh. Migr. 2018, 12, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.J.; LeRoith, D. Hyperinsulinaemia in Cancer. Nat. Rev. Cancer 2020, 20, 629–644. [Google Scholar] [CrossRef]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-Negative Breast Cancer: Treatment Challenges and Solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar]
- Maestri, E. The 3Rs Principle in Animal Experimentation: A Legal Review of the State of the Art in Europe and the Case in Italy. BioTech 2021, 10, 9. [Google Scholar] [CrossRef]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-Derived Xenograft in Zebrafish Embryos: A New Platform for Translational Research in Neuroendocrine Tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Alaimo, S.; Giugno, R.; Acunzo, M.; Veneziano, D.; Ferro, A.; Pulvirenti, A. Post-Transcriptional Knowledge in Pathway Analysis Increases the Accuracy of Phenotypes Classification. Oncotarget 2016, 7, 54572–54582. [Google Scholar] [CrossRef]
- Lawson, N.D.; Weinstein, B.M. In Vivo Imaging of Embryonic Vascular Development Using Transgenic Zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef]
- Carra, S.; Gaudenzi, G. New Perspectives in Neuroendocrine Neoplasms Research from Tumor Xenografts in Zebrafish Embryos. Minerva Endocrinol. 2020, 45, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for Integration and Interpretation of Large-Scale Molecular Data Sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, D.; Sheng, D.; Xu, J.; Chen, W.; Qin, Y.; Du, R.; Yang, X.; He, X.; Xie, N.; et al. NOTCH4 Maintains Quiescent Mesenchymal-like Breast Cancer Stem Cells via Transcriptionally Activating SLUG and GAS1 in Triple-Negative Breast Cancer. Theranostics 2020, 10, 2405–2421. [Google Scholar] [CrossRef]
- Tominaga, K.; Shimamura, T.; Kimura, N.; Murayama, T.; Matsubara, D.; Kanauchi, H.; Niida, A.; Shimizu, S.; Nishioka, K.; Tsuji, E.-I.; et al. Addiction to the IGF2-ID1-IGF2 Circuit for Maintenance of the Breast Cancer Stem-like Cells. Oncogene 2017, 36, 1276–1286. [Google Scholar] [CrossRef]
- Pidugu, V.K.; Pidugu, H.B.; Wu, M.-M.; Liu, C.-J.; Lee, T.-C. Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 2019, 6, 148. [Google Scholar] [CrossRef]
- Pan, H.; Wang, X.; Huang, W.; Dai, Y.; Yang, M.; Liang, H.; Wu, X.; Zhang, L.; Huang, W.; Yuan, L.; et al. Interferon-Induced Protein 44 Correlated with Immune Infiltration Serves as a Potential Prognostic Indicator in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 557157. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Li, L.; Xing, W.; Li, R.; Pei, C.; Dong, X.; Fu, Y.; Gu, C.; Guo, X.; Jia, Y.; et al. IRGM1 Enhances B16 Melanoma Cell Metastasis through PI3K-Rac1 Mediated Epithelial Mesenchymal Transition. Sci. Rep. 2015, 5, 12357. [Google Scholar] [CrossRef]
- Xiahou, Z.; Wang, X.; Shen, J.; Zhu, X.; Xu, F.; Hu, R.; Guo, D.; Li, H.; Tian, Y.; Liu, Y.; et al. NMI and IFP35 Serve as Proinflammatory DAMPs during Cellular Infection and Injury. Nat. Commun. 2017, 8, 950. [Google Scholar] [CrossRef] [PubMed]
- Kariri, Y.A.; Alsaleem, M.; Joseph, C.; Alsaeed, S.; Aljohani, A.; Shiino, S.; Mohammed, O.J.; Toss, M.S.; Green, A.R.; Rakha, E.A. The Prognostic Significance of Interferon-Stimulated Gene 15 (ISG15) in Invasive Breast Cancer. Breast Cancer Res. Treat. 2020. [Google Scholar] [CrossRef]
- Woodman, N.; Pinder, S.E.; Tajadura, V.; Le Bourhis, X.; Gillett, C.; Delannoy, P.; Burchell, J.M.; Julien, S. Two E-Selectin Ligands, BST-2 and LGALS3BP, Predict Metastasis and Poor Survival of ER-Negative Breast Cancer. Int. J. Oncol. 2016, 49, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Brockwell, N.K.; Rautela, J.; Owen, K.L.; Gearing, L.J.; Deb, S.; Harvey, K.; Spurling, A.; Zanker, D.; Chan, C.-L.; Cumming, H.E.; et al. Tumor Inherent Interferon Regulators as Biomarkers of Long-Term Chemotherapeutic Response in TNBC. NPJ Precis Oncol. 2019, 3, 21. [Google Scholar] [CrossRef]
- Greenwood, C.; Metodieva, G.; Al-Janabi, K.; Lausen, B.; Alldridge, L.; Leng, L.; Bucala, R.; Fernandez, N.; Metodiev, M.V. Stat1 and CD74 Overexpression Is Co-Dependent and Linked to Increased Invasion and Lymph Node Metastasis in Triple-Negative Breast Cancer. J. Proteom. 2012, 75, 3031–3040. [Google Scholar] [CrossRef]
- Khodarev, N.N. Intracellular RNA Sensing in Mammalian Cells: Role in Stress Response and Cancer Therapies. Int. Rev. Cell Mol. Biol. 2019, 344, 31–89. [Google Scholar]
- Chen, M.-C.; Baskaran, R.; Lee, N.-H.; Hsu, H.-H.; Ho, T.-J.; Tu, C.-C.; Lin, Y.-M.; Viswanadha, V.P.; Kuo, W.-W.; Huang, C.-Y. CXCL2/CXCR2 Axis Induces Cancer Stem Cell Characteristics in CPT-11-Resistant LoVo Colon Cancer Cells via Gαi-2 and Gαq/11. J. Cell. Physiol. 2019, 234, 11822–11834. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Fooladseresht, H.; Nemati, M.; Assadollahi, Z.; Sheikhi, A.; Ghaderi, A. Higher Circulating Levels of Chemokine CXCL10 in Patients with Breast Cancer: Evaluation of the Influences of Tumor Stage and Chemokine Gene Polymorphism. Cancer Biomark. 2016, 16, 545–554. [Google Scholar] [CrossRef]
- Rupertus, K.; Sinistra, J.; Scheuer, C.; Nickels, R.M.; Schilling, M.K.; Menger, M.D.; Kollmar, O. Interaction of the Chemokines I-TAC (CXCL11) and SDF-1 (CXCL12) in the Regulation of Tumor Angiogenesis of Colorectal Cancer. Clin. Exp. Metastasis 2014, 31, 447–459. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular Characterization of Basal-like and Non-Basal-like Triple-Negative Breast Cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Alikhani, N.; Tobin-Hess, A.; Blank, J.; Buffin, N.J.; Zelenko, Z.; Tennagels, N.; Werner, U.; LeRoith, D. Insulin Receptor Phosphorylation by Endogenous Insulin or the Insulin Analog AspB10 Promotes Mammary Tumor Growth Independent of the IGF-I Receptor. Diabetes 2013, 62, 3553–3560. [Google Scholar] [CrossRef][Green Version]
- Zelenko, Z.; Gallagher, E.J.; Antoniou, I.M.; Sachdev, D.; Nayak, A.; Yee, D.; LeRoith, D. EMT Reversal in Human Cancer Cells after IR Knockdown in Hyperinsulinemic Mice. Endocr. Relat. Cancer 2016, 23, 747–758. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutiérrez-Ángel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A Model of Breast Cancer Heterogeneity Reveals Vascular Mimicry as a Driver of Metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef]
- Smirnova, T.; Bonapace, L.; MacDonald, G.; Kondo, S.; Wyckoff, J.; Ebersbach, H.; Fayard, B.; Doelemeyer, A.; Coissieux, M.-M.; Heideman, M.R.; et al. Serpin E2 Promotes Breast Cancer Metastasis by Remodeling the Tumor Matrix and Polarizing Tumor Associated Macrophages. Oncotarget 2016, 7, 82289–82304. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix Metalloproteinases as Breast Cancer Drivers and Therapeutic Targets. Front. Biosci. 2015, 20, 1144–1163. [Google Scholar] [CrossRef]
- Ying, X.; Sun, Y.; He, P. Bone Morphogenetic Protein-7 Inhibits EMT-Associated Genes in Breast Cancer. Cell Physiol. Biochem. 2015, 37, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Huang, O.; Jiang, M.; Xie, Z.; Chen, D.; Zhang, X. Prognostic Value of Ephrin B Receptors in Breast Cancer: An Online Survival Analysis Using the Microarray Data of 3,554 patients. Oncol. Lett. 2019, 18, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Gaspari, M.; Pandini, G.; Palummo, A.; Cuda, G.; Larsen, M.R.; Vigneri, R.; Belfiore, A. Research Resource: New and Diverse Substrates for the Insulin Receptor Isoform A Revealed by Quantitative Proteomics After Stimulation With IGF-II or Insulin. Mol. Endocrinol. 2011, 25, 1456–1468. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.A.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An Interferon-Related Gene Signature for DNA Damage Resistance Is a Predictive Marker for Chemotherapy and Radiation for Breast Cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Forys, J.T.; Kuzmicki, C.E.; Saporita, A.J.; Winkeler, C.L.; Maggi, L.B., Jr.; Weber, J.D. ARF and p53 Coordinate Tumor Suppression of an Oncogenic IFN-β-STAT1-ISG15 Signaling Axis. Cell Rep. 2014, 7, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Medico, E.; Conte, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. Differential Gene Expression Induced by Insulin and Insulin-like Growth Factor-II through the Insulin Receptor Isoform A. J. Biol. Chem. 2003, 278, 42178–42189. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vella, V.; Giuliano, M.; La Ferlita, A.; Pellegrino, M.; Gaudenzi, G.; Alaimo, S.; Massimino, M.; Pulvirenti, A.; Dicitore, A.; Vigneri, P.; et al. Novel Mechanisms of Tumor Promotion by the Insulin Receptor Isoform A in Triple-Negative Breast Cancer Cells. Cells 2021, 10, 3145. https://doi.org/10.3390/cells10113145
Vella V, Giuliano M, La Ferlita A, Pellegrino M, Gaudenzi G, Alaimo S, Massimino M, Pulvirenti A, Dicitore A, Vigneri P, et al. Novel Mechanisms of Tumor Promotion by the Insulin Receptor Isoform A in Triple-Negative Breast Cancer Cells. Cells. 2021; 10(11):3145. https://doi.org/10.3390/cells10113145
Chicago/Turabian StyleVella, Veronica, Marika Giuliano, Alessandro La Ferlita, Michele Pellegrino, Germano Gaudenzi, Salvatore Alaimo, Michele Massimino, Alfredo Pulvirenti, Alessandra Dicitore, Paolo Vigneri, and et al. 2021. "Novel Mechanisms of Tumor Promotion by the Insulin Receptor Isoform A in Triple-Negative Breast Cancer Cells" Cells 10, no. 11: 3145. https://doi.org/10.3390/cells10113145
APA StyleVella, V., Giuliano, M., La Ferlita, A., Pellegrino, M., Gaudenzi, G., Alaimo, S., Massimino, M., Pulvirenti, A., Dicitore, A., Vigneri, P., Vitale, G., Malaguarnera, R., Morrione, A., Sims, A. H., Ferro, A., Maggiolini, M., Lappano, R., De Francesco, E. M., & Belfiore, A. (2021). Novel Mechanisms of Tumor Promotion by the Insulin Receptor Isoform A in Triple-Negative Breast Cancer Cells. Cells, 10(11), 3145. https://doi.org/10.3390/cells10113145