
Deletion of TSPO Causes Dysregulation of Cholesterol Metabolism in Mouse Retina

 , ,
, ,  ,
, 
Abstract
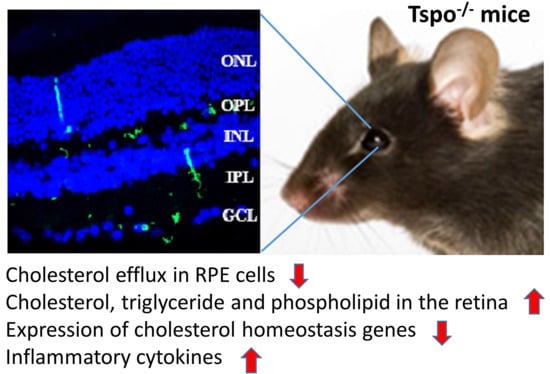
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Western Blotting
2.4. Haematoxylin and Eosin Staining (H&E)
2.5. Immunohistochemistry
2.6. Measurement of Cholesterol Efflux in Mouse Primary Retinal Pigment Epithelium (RPE) Cells
2.7. Gene Expression
2.8. Quantification of Total Cholesterol, Phospholipid and Triglyceride in Mouse Tissues
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Statistical Analysis
3. Results
3.1. Confirmation of TSPO Deletion
3.2. No Morphological Changes in Tspo KO Retinas
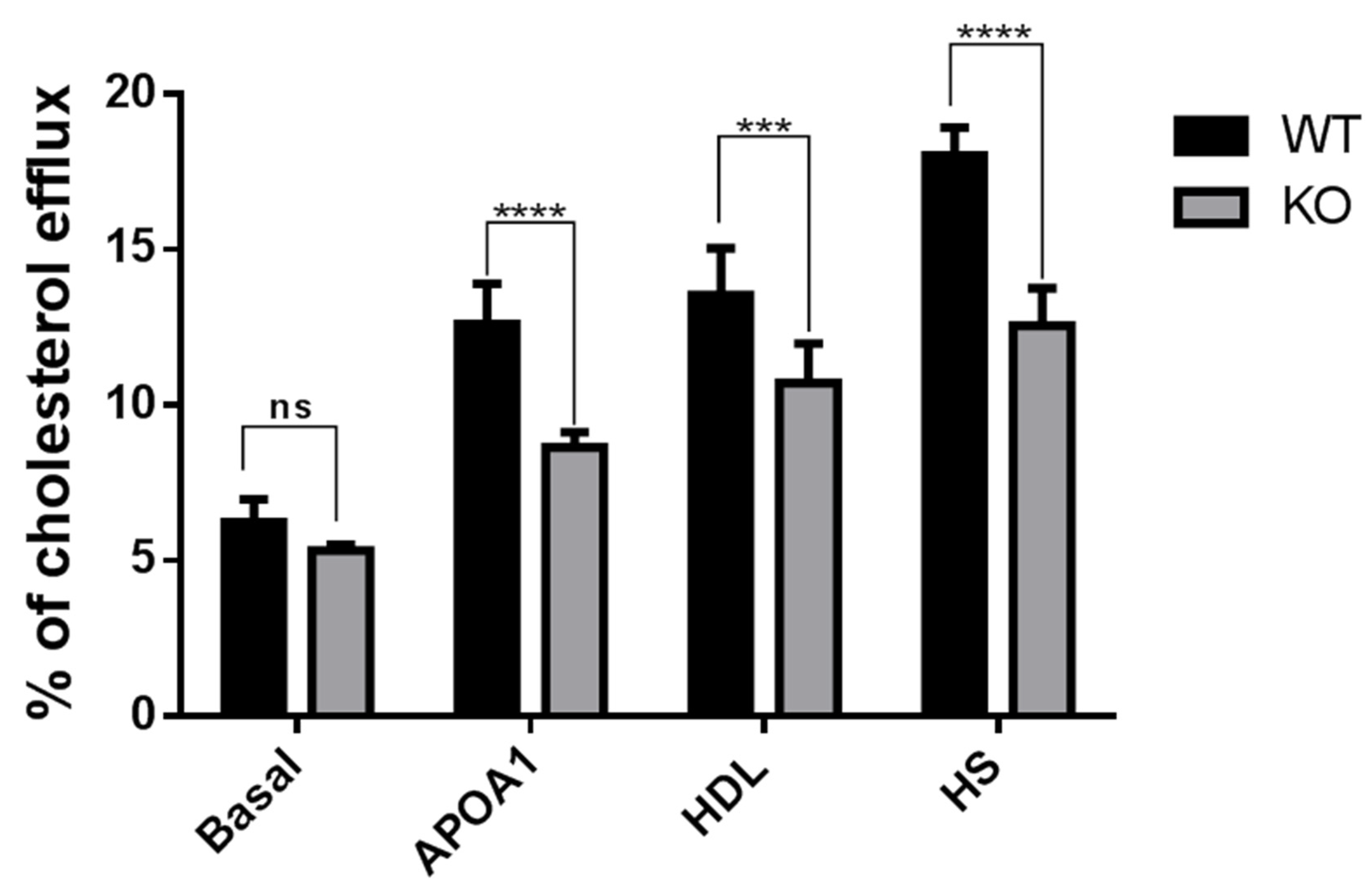
3.3. Cholesterol Efflux Reduced in Tspo KO Mouse RPE Cells
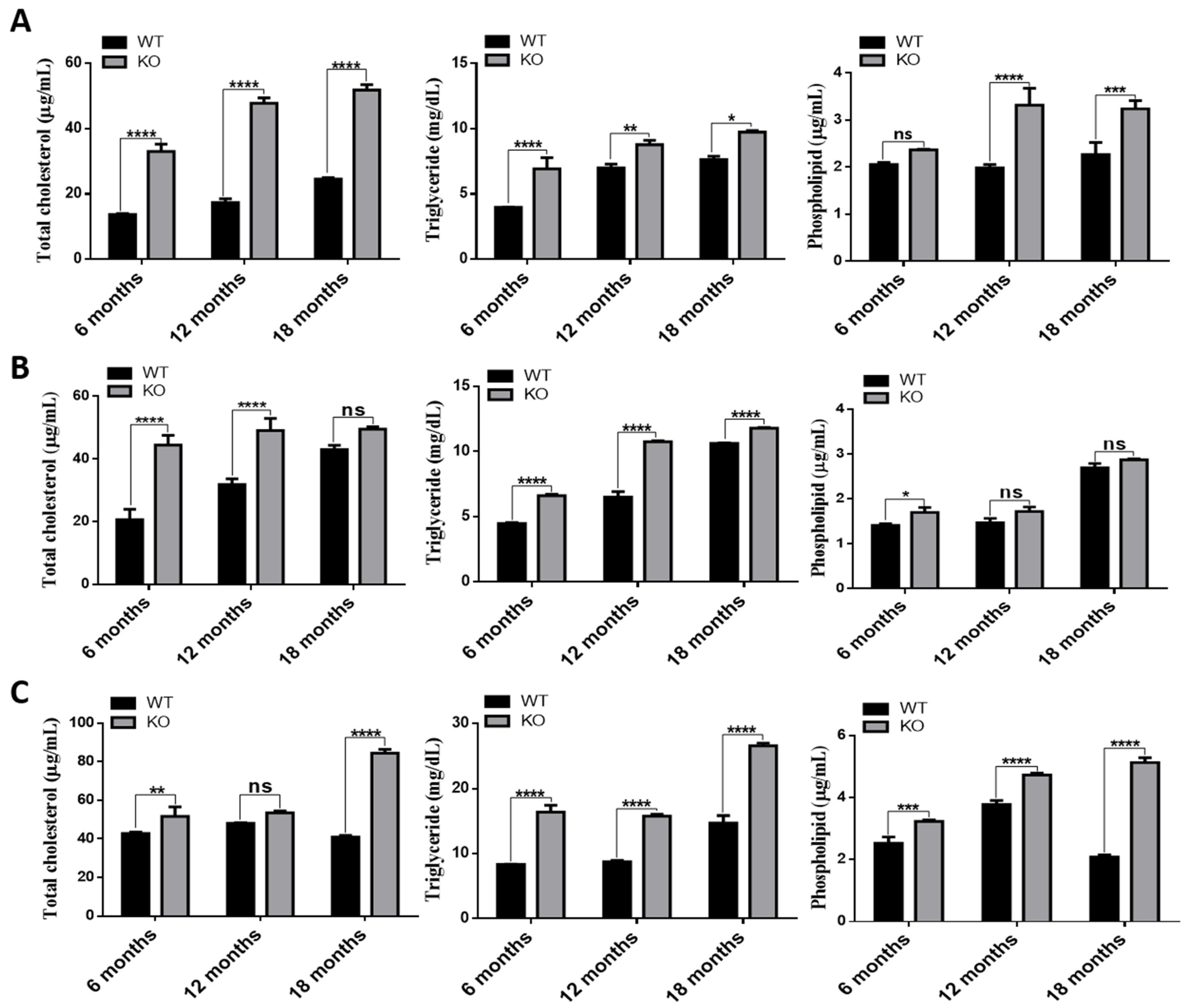
3.4. Increased Lipid Accumulation in Tspo KO Mouse Tissues
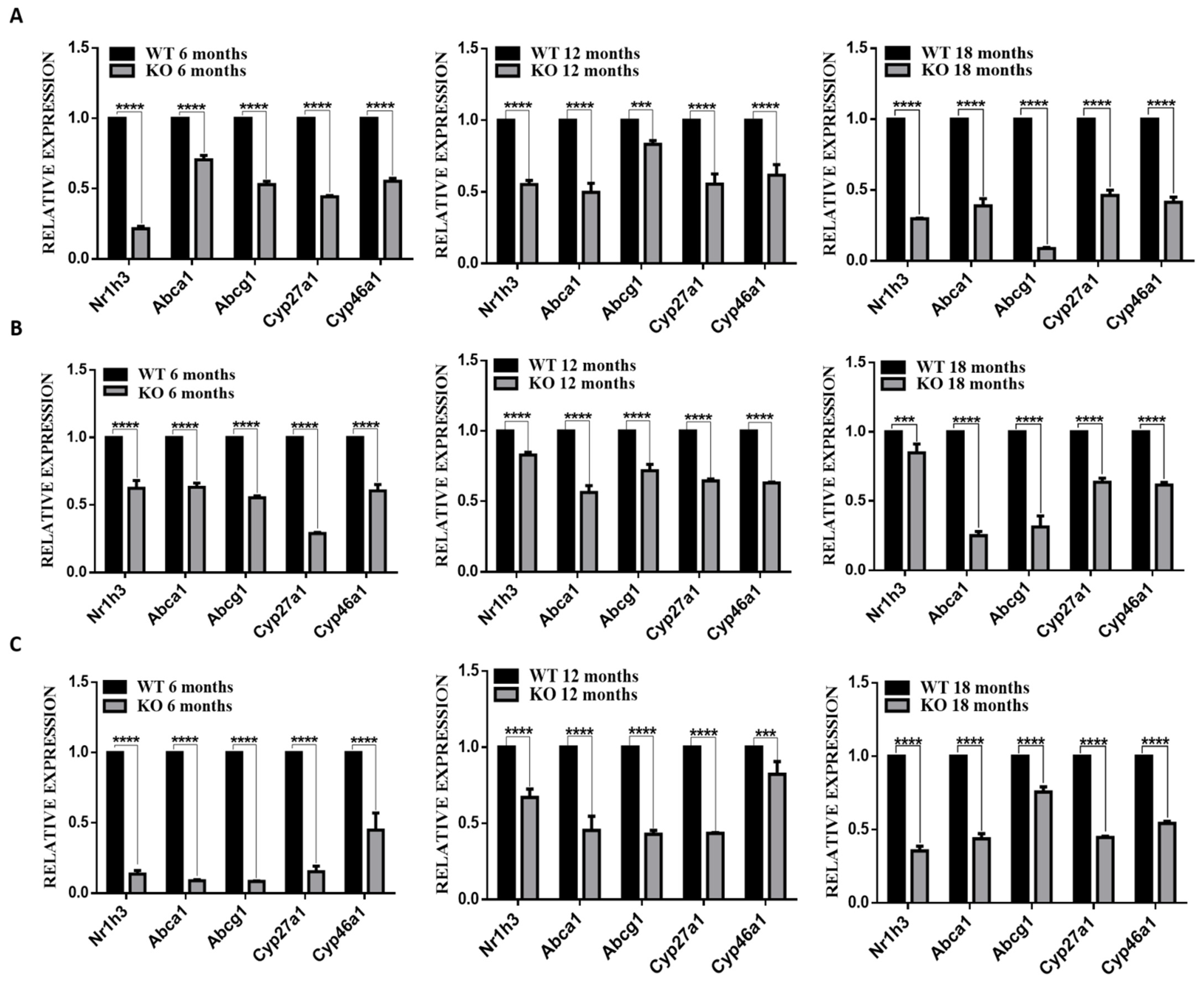
3.5. Deletion of TSPO Decreased Expression of Cholesterol Homeostasis Genes
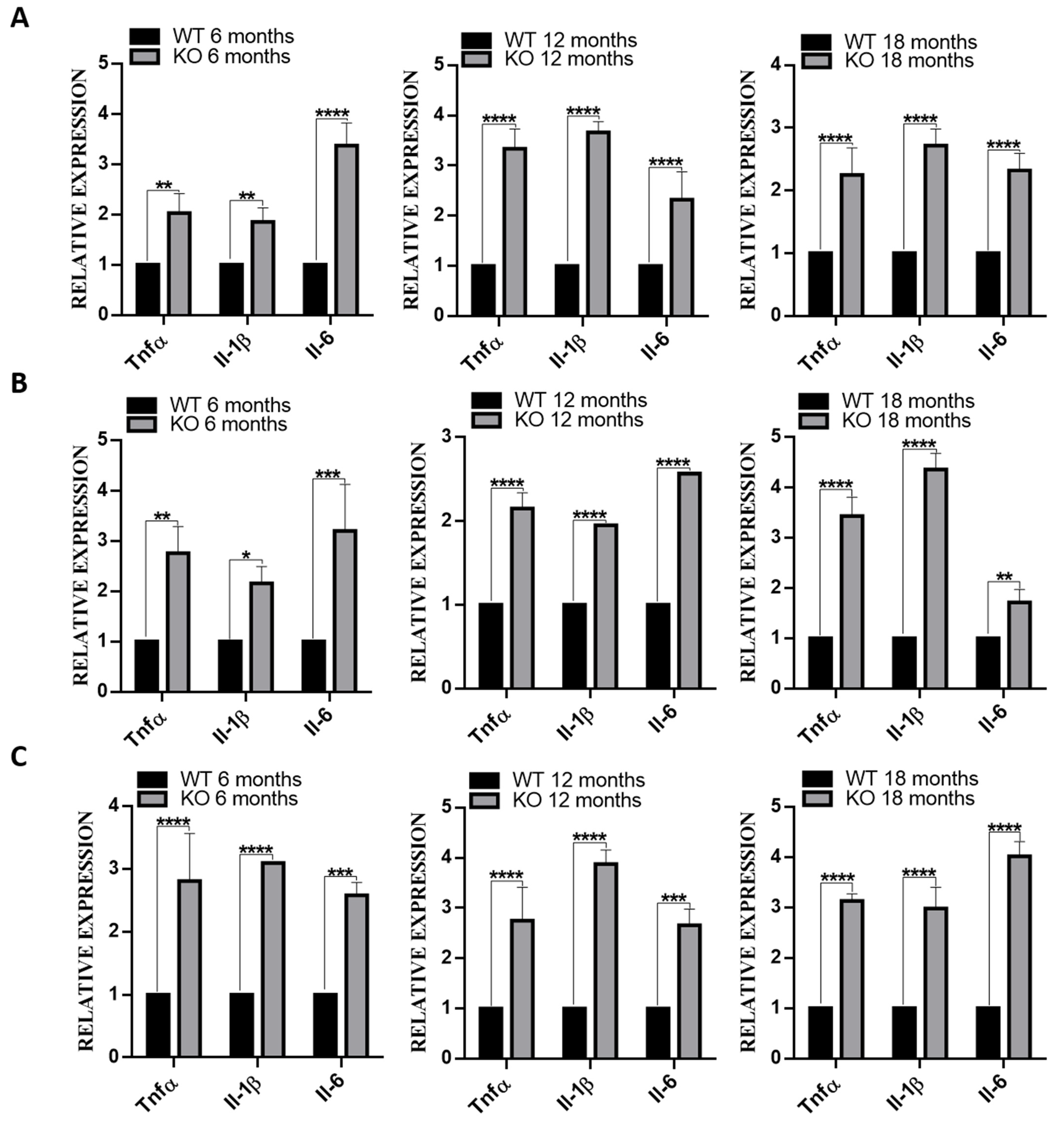
3.6. Deletion of TSPO Increased Inflammation in The Tissues of Tspo KO Mice
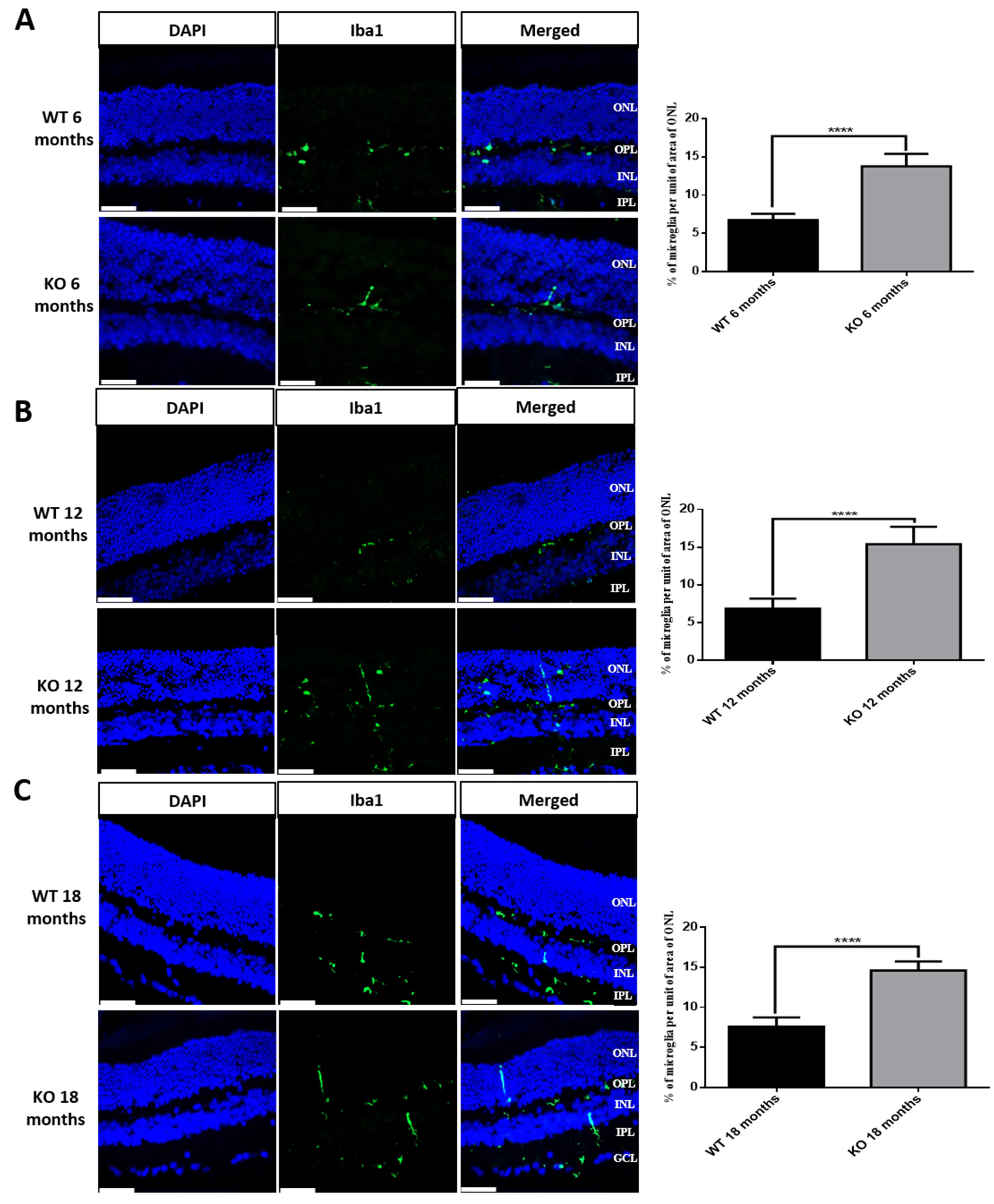
3.7. Microglial Activation in Tspo KO Mouse Retina
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grassmann, F.; Fauser, S.; Weber, B.H. The genetics of age-related macular degeneration (AMD)—Novel targets for designing treatment options? Eur. J. Pharm. Biopharm. 2015, 95, 194–202. [Google Scholar] [CrossRef]
- Jonasson, F.; Arnarsson, A.; Eiríksdottir, G.; Harris, T.B.; Launer, L.J.; Meuer, S.M.; Klein, B.E.; Klein, R.; Gudnason, V.; Cotch, M.F. Prevalence of age-related macular degeneration in old persons: Age, Gene/environment Susceptibility Reykjavik Study. Ophthalmology 2011, 118, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Hermann, M.; Caramoy, A.; Schröder, S.; Dröge, K.; Kirchhof, B.; Fauser, S. Prevalence of age-related macular degeneration in persons aged 90 years and older in Cologne. Acta ophthalmologica. 2012, 90, e500–e501. [Google Scholar] [CrossRef]
- Pikuleva, I.A.; Curcio, C.A. Cholesterol in the retina: The best is yet to come. Prog. Retin. Eye Res. 2014, 41, 64–89. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.L.; Li, W.; Shi, Y.H.; Le, G.W. Lipoic acid prevents high-fat diet– induced dyslipidemia and oxidative stress: A microarray analysis. Nutrition 2008, 24, 582–588. [Google Scholar] [CrossRef]
- Clemons, T.E.; Milton, R.C.; Klein, R.; Seddon, J.M.; Ferris, F.L., 3rd. Age-Related Eye Disease Study Research Group. Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology 2005, 112, 533–539. [Google Scholar]
- Rudolf, M.; Curcio, C.A. Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J. Histochem. Cytochem. 2009, 57, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant lipid and protein components of drusen. PLoS ONE 2010, 5, e10329. [Google Scholar] [CrossRef]
- Mares-Perlman, J.A.; Brady, W.E.; Klein, R.; VandenLangenberg, G.M.; Klein, B.E.; Palta, M. Dietary fat and age-related maculopathy. Arch. Ophthalmol. 1995, 113, 743–748. [Google Scholar] [CrossRef]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Malek, G.; Johnson, L.V.; Mace, B.E.; Saloupis, P.; Schmechel, D.E.; Rickman, D.W.; Toth, C.A.; Sullivan, P.M.; Rickman, C.B. Apolipoprotein E allele-dependent pathogenesis: A model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 11900–11905. [Google Scholar] [CrossRef] [Green Version]
- Toomey, C.B.; Kelly, U.; Saban, D.R.; Rickman, C.B. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc. Natl. Acad. Sci. USA 2015, 112, E3040–E3049. [Google Scholar] [CrossRef] [Green Version]
- Braestrup, C.; Squires, R.F. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H) diazepam binding. Proc. Natl. Acad. Sci. USA 1977, 74, 3805–3809. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a ther-apeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Jaremko, Ł.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Park, Y.; Nam, H.; Lee, J.W.; Yu, S.W. Translocator protein (TSPO): The new story of the old protein in neuroinflammation. BMB Rep. 2020, 53, 20–27. [Google Scholar] [CrossRef]
- Rupprecht, R.; Rammes, G.; Eser, D.; Baghai, T.C.; Schule, C.; Nothdurfter, C.; Troxler, T.; Gentsch, C.; Kalkman, H.O.; Chaperon, F.; et al. Translocator protein (18 kD) as targetfor anxiolytics without benzodiazepine-like side effects. Science 2009, 325, 490–493. [Google Scholar] [CrossRef] [Green Version]
- Biswas, L.; Zhou, X.; Dhillon, B.; Graham, A.; Shu, X. Retinal pigment epithelium cholesterol efflux mediated by the18kDa translocator protein, TSPO, a potential target for treating age-related macular degeneration. Hum. Mol. Genet. 2017, 26, 4327–4339. [Google Scholar] [CrossRef] [Green Version]
- Biswas, L.; Farhan, F.; Reilly, J.; Bartholomew, C.; Shu, X. TSPO ligands promote cholesterol efflux and suppress oxidative stress and inflammation in choroidal endothelial cells. Int. J. Mol. Sci. 2018, 19, 3740. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, K.S.; Craft, J.A.; Biswas, L.; Spencer, J.; Shu, X. Etifoxine reverses weight gain and alters the colonic bacterial community in a mouse model of obesity. Biochem. Pharmacol. 2020, 180, 114151. [Google Scholar] [CrossRef]
- Biswas, L.; Ibrahim, K.S.; Li, X.; Zhou, X.; Zeng, Z.; Craft, J.; Shu, X. Effect of a TSPO ligand on retinal pigment epithelial cholesterol homeostasis in high-fat fed mice, implication for age-related macular degeneration. Exp. Eye Res. 2021, 208, 108625. [Google Scholar] [CrossRef]
- Morohaku, K.; Pelton, S.H.; Daugherty, D.J.; Butler, W.R.; Deng, W.; Selvaraj, V. Translocator protein/peripheral benzodiazepine receptor is not required for steroidhormone biosynthesis. Endocrinology 2014, 155, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Tu, L.N.; Morohaku, K.; Manna, P.R.; Pelton, S.H.; Butler, W.R.; Stocco, D.M.; Selvaraj, V. Peripheral benzodiazepine receptor/translocator protein global knock-outmice are viable with no effects on steroid hormone biosynthesis. J. Biol. Chem. 2014, 289, 27444–27454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shahani, U.; Reilly, J.; Shu, X. Disease mechanisms and neuroprotection by tauroursodeoxycholic acid in Rpgr knockout mice. J. Cell Physiol. 2019, 234, 18801–18812. [Google Scholar] [CrossRef] [PubMed]
- Betlazar, C.; Harrison-Brown, M.; Middleton, R.J.; Banati, R.; Liu, G.J. Cellular sources and regional variations in the expression of the neuroinflammatory marker translocator protein (TSPO) in the normal brain. Int. J. Mol. Sci. 2018, 19, 2707. [Google Scholar] [CrossRef] [Green Version]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal microglia: Just bystander or target for therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef]
- Wang, S.K.; Xue, Y.; Cepko, C.L. Microglia modulation by TGF-β1 protects cones in mouse models of retinal degeneration. J. Clin. Investig. 2020, 130, 4360–4369. [Google Scholar] [CrossRef]
- Chung, J.Y.; Chen, H.; Midzak, A.; Burnett, A.L.; Papadopoulos, V.; Zirkin, B.R. Drug ligand-induced activation of translocator protein (TSPO) stimulates steroid production by aged brown Norway rat Leydig cells. Endocrinology 2013, 154, 2156–2165. [Google Scholar] [CrossRef] [Green Version]
- Hauet, T.; Yao, Z.X.; Bose, H.S.; Wall, C.T.; Han, Z.; Li, W.; Hales, D.B.; Miller, W.L.; Culty, M.; Papadopoulos, V. Peripheral-type benzodiazepine receptor-mediated action of steroidogenic acute regulatory protein on cholesterol entry into leydig cell mitochondria. Mol. Endocrinol. 2005, 19, 540–554. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Mukhin, A.G.; Costa, E.; Krueger, K.E. The peripheral-type benzodiazepine receptor is functionally linked to Leydig cell steroidogenesis. J. Biol. Chem. 1990, 265, 3772–3779. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Amri, H.; Li, H.; Boujrad, N.; Vidic, B.; Garnier, M. Targeted disruption of the peripheral-type benzodiazepine receptor gene inhibits steroidogenesis in the R2C Leydig tumor cell line. J. Biol. Chem. 1997, 272, 32129–32135. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Campioli, E.; Midzak, A.; Culty, M.; Papadopoulos, V. Conditional steroidogenic cell-targeted deletion of TSPO unveils a crucial role in viability and hormone-dependent steroid formation. Proc. Natl. Acad. Sci. USA 2015, 112, 7261–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Campioli, E.; Sottas, C.; Zirkin, B.; Papadopoulos, V. Amhr2-cre–mediated global Tspo knockout. J. Endocr. Soc. 2020, 4, bvaa001. [Google Scholar] [CrossRef]
- Barron, A.M.; Ji, B.; Kito, S.; Suhara, T.; Higuchi, M. Steroidogenic abnormalities intranslocator protein knockout mice and significance in the aging male. Biochem. J. 2018, 475, 75–85. [Google Scholar] [CrossRef]
- Owen, D.R.; Fan, J.; Campioli, E.; Venugopal, S.; Midzak, A.; Daly, E.; Harlay, A.; Issop, L.; Libri, V.; Kalogiannopoulou, D.; et al. TSPO mutations in rats and a human polymorphism impair the rate of steroid synthesis. Biochem. J. 2017, 474, 3985–3999. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Alhasani, R.H.; Zhou, X.; Reilly, J.; Zeng, Z.; Strang, N.; Shu, X. Oxysterols and retinal degeneration. Br. J. Pharmacol. 2021, 178, 3205–3219. [Google Scholar] [CrossRef]
- Dimitrova-Shumkovska, J.; Krstanoski, L.; Veenman, L. Diagnostic and therapeutic potential of TSPO studies regarding neurodegenerative diseases, psychiatric disorders, alcohol use disorders, traumatic brain injury, and stroke: An update. Cells 2020, 9, 870. [Google Scholar] [CrossRef] [Green Version]
- Veenman, L. Raloxifene as treatment for various types of brain injuries and neurodegenerative diseases: A good start. Int. J. Mol. Sci. 2020, 21, 7586. [Google Scholar] [CrossRef]
- Karlstetter, M.; Nothdurfter, C.; Aslanidis, A.; Moeller, K.; Horn, F.; Scholz, R.; Neumann, H.; Weber, B.H.; Rupprecht, R.; Langmann, T. Translocator protein (18 kDa)(TSPO) is expressed in reactive retinal microglia and modulates microglial in-flammation and phagocytosis. J. Neuroinflammation 2014, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Scholz, R.; Caramoy, A.; Bhuckory, M.B.; Rashid, K.; Chen, M.; Xu, H.; Grimm, C.; Langmann, T. Targeting translocator protein (18 kDa) (TSPO) dampens pro-inflammatory microglia reactivity in the retina and protects from degeneration. J. Neuroinflammation 2015, 12, 201. [Google Scholar] [CrossRef] [Green Version]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Coffey, P.J.; Gias, C.; McDermott, C.J.; Lundh, P.; Pickering, M.C.; Sethi, C.; Bird, A.; Fitzke, F.W.; Maass, A.; Chen, L.L.; et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. USA 2007, 104, 16651–16656. [Google Scholar] [CrossRef] [Green Version]
- Landowski, M.; Kelly, U.; Klingeborn, M.; Groelle, M.; Ding, J.D.; Grigsby, D.; Rickman, C.B. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 3703–3711. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhan, F.; Almarhoun, M.; Wong, A.; Findlay, A.S.; Bartholomew, C.; Williams, M.T.S.; Hurd, T.W.; Shu, X. Deletion of TSPO Causes Dysregulation of Cholesterol Metabolism in Mouse Retina. Cells 2021, 10, 3066. https://doi.org/10.3390/cells10113066
Farhan F, Almarhoun M, Wong A, Findlay AS, Bartholomew C, Williams MTS, Hurd TW, Shu X. Deletion of TSPO Causes Dysregulation of Cholesterol Metabolism in Mouse Retina. Cells. 2021; 10(11):3066. https://doi.org/10.3390/cells10113066
Chicago/Turabian StyleFarhan, Fahad, Mohammad Almarhoun, Aileen Wong, Amy S. Findlay, Chris Bartholomew, Mark T. S. Williams, Toby W. Hurd, and Xinhua Shu. 2021. "Deletion of TSPO Causes Dysregulation of Cholesterol Metabolism in Mouse Retina" Cells 10, no. 11: 3066. https://doi.org/10.3390/cells10113066
APA StyleFarhan, F., Almarhoun, M., Wong, A., Findlay, A. S., Bartholomew, C., Williams, M. T. S., Hurd, T. W., & Shu, X. (2021). Deletion of TSPO Causes Dysregulation of Cholesterol Metabolism in Mouse Retina. Cells, 10(11), 3066. https://doi.org/10.3390/cells10113066