DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies



Abstract
:1. Introduction
1.1. Alzheimer’s Disease and Other Tauopathies
Clinics of AD
1.2. Pathomechanisms of AD—A Role of Aβ and TAU in Disease Progression (Neuroanatomics, Clinics and Imaging Level)
1.3. The Role of TAU in Neuronal Dysfunction on a Cellular & Molecular Level
1.4. Established Genetic Mechanisms of AD and Tauopathies
2. Implication of DNA Methylation in AD and Tauopathies
2.1. Age-Dependent Changes of DNA Methylation Marks and the Relevance for AD and Tauopathies
2.2. Evidence for the Implication of Altered DNA Methylation Signatures in AD and Tauopathies
2.2.1. DNA Methylation Changes Lead to Pathological Phosphorylation of TAU
2.2.2. Altered DNA Methylation Signatures as a Consequence of Disease Pathophysiology, Such as Aβ Burden and TAU-Phosphorylation
2.2.3. Aβ Peptide and TAU-Phosphorylation-Driven Changes in the Expression and Localization of DNA Repair Related Proteins
2.2.4. Aβ-Associated Changes in DNA Methylation of Cell Cycle-Related Genes
3. Epigenetic Treatment?—The Potential and Limitations of DNA Methylation-Based Therapy Approaches
4. Altered DNA Methylation Signatures as Potential Biomarkers for AD/Tauopathies Disease and Disease Progression?
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Mandelkow, E. Mechanisms of Axonal Sorting of Tau and Influence of the Axon Initial Segment on Tau Cell Polarity. In Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2019; Volume 1184, pp. 69–77. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Van Swieten, J.C.; Goedert, M. Tau gene mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17). Neurogenetics 2000, 2, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S.; et al. Relative Frequencies of Alzheimer Disease, Lewy Body, Vascular and Frontotemporal Dementia, and Hippocampal Sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2016, 16, 203–212. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, W.; Yang, Y.; Murzin, A.; Falcon, B.; Kotecha, A.; Van Beers, M.; Tarutani, A.; Kametani, F.; Garringer, H.J.; et al. Structure-based Classification of Tauopathies. bioRxiv 2021, 1–27. [Google Scholar]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-Tau (FTLD-Tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, D.; Irwin, D.J. Emerging Diagnostic and Therapeutic Strategies for Tauopathies. Curr. Neurol. Neurosci. Rep. 2017, 17. [Google Scholar] [CrossRef]
- Irwin, D.J. Tauopathies as clinicopathological entities. Park. Relat. Disord. 2016, 22, S29–S33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arena, J.D.; Smith, D.H.; Lee, E.B.; Gibbons, G.S.; Irwin, D.J.; Robinson, J.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Stewart, W.; Johnson, V.E. Tau immunophenotypes in chronic traumatic encephalopathy recapitulate those of ageing and Alzheimer’s disease. Brain 2020, 143, 1572–1587. [Google Scholar] [CrossRef]
- Götz, J.; Halliday, G.; Nisbet, R.M. Molecular Pathogenesis of the Tauopathies. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 239–261. [Google Scholar] [CrossRef]
- Lebouvier, T.; Pasquier, F.; Buée, L. Update on tauopathies. Curr. Opin. Neurol. 2017, 30, 589–598. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolev, I.O.; Symonds, L.L.; Bozoki, A.C. Predicting progression from mild cognitive impairment to Alzheimer’s dementia using clinical, MRI, and plasma biomarkers via probabilistic pattern classification. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, S.; Tideman, P.; Cullen, N.; Zetterberg, H.; Blennow, K.; Dage, J.L.; Stomrud, E.; Janelidze, S.; Mattsson-Carlgren, N.; Hansson, O.; et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat. Med. 2021, 27, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease: Mechanism of disease. N. Engl. J. Med. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, T.D. Alzheimer Disease Overview. 1993. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1161/ (accessed on 7 November 2021).
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2010, 12, 67–72. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Hammond, T.C.; Xing, X.; Wang, C.; Ma, D.; Nho, K.; Crane, P.K.; Elahi, F.; Ziegler, D.A.; Liang, G.; Cheng, Q.; et al. β-amyloid and tau drive early Alzheimer’s disease decline while glucose hypometabolism drives late decline. Commun. Biol. 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Choi, J.Y.; Hwang, M.S.; Lee, J.H.; Kim, Y.J.; Lee, H.M.; Lyoo, C.H.; Ryu, Y.H.; Lee, M.S. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016, 87, 375–383. [Google Scholar] [CrossRef]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Biel, D.; Brendel, M.; Rubinski, A.; Buerger, K.; Janowitz, D.; Dichgans, M.; Franzmeier, N. Tau-PET and in vivo Braak-staging as a prognostic marker in Alzheimer’s disease. medRxiv 2021. [Google Scholar]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, J.W.; Young, A.L.; Oxtoby, N.P.; Smith, R.; Ossenkoppele, R.; Strandberg, O.T.; La Joie, R.; Aksman, L.M.; Grothe, M.J.; Iturria-Medina, Y.; et al. Characterizing the spatiotemporal variability of Alzheimer’s disease pathology. medRxiv 2017. [Google Scholar] [CrossRef]
- Drubin, D.G. Tau protein function in living cells. J. Cell Biol. 2004, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, R.L.; Donlon, T.A.; Kurnit, D.M.; Harris, P.; Kosik, K.S. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Mol. Brain Res. 2003, 1, 271–280. [Google Scholar] [CrossRef]
- Andreadis, A. Tau splicing and the intricacies of dementia. J. Cell. Physiol. 2012, 227, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Trabzuni, D.; Wray, S.; Vandrovcova, J.; Ramasamy, A.; Walker, R.; Smith, C.; Luk, C.; Gibbs, J.R.; Dillman, A.; Hernandez, D.G.; et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum. Mol. Genet. 2012, 21, 4094–4103. [Google Scholar] [CrossRef] [Green Version]
- Doran, M.; Du Plessis, D.G.; Ghadiali, E.J.; Mann, D.M.A.; Pickering-Brown, S.; Larner, A.J. Familial early-onset dementia with tau intron 10 + 16 mutation with clinical features similar to those of Alzheimer disease. Arch. Neurol. 2007, 64, 1535–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, H.R.; Osaki, Y.; Holton, J.; Lees, A.J.; Wood, N.W.; Revesz, T.; Quinn, N. Tau exon 10 + 16 mutation FTDP-17 presenting clinically as sporadic young onset PSP. Neurology 2003, 61, 102–104. [Google Scholar] [CrossRef]
- Zempel, H.; Dennissen, F.J.A.; Kumar, Y.; Luedtke, J.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J. Biol. Chem. 2017, 292, 12192–12207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, S.; Bell, M.; Klimek, J.; Zempel, H. Differential Effects of the Six Human TAU Isoforms: Somatic Retention of 2N-TAU and Increased Microtubule Number Induced by 4R-TAU. Front. Neurosci. 2021, 15, 547. [Google Scholar] [CrossRef]
- Ali, F.; Josephs, K. Rare Tauopathies. Semin. Neurol. 2019, 39, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Crowther, R.A.; Spillantini, M.G. Tau mutations cause frontotemporal dementias. Neuron 1998, 21, 955–958. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Murley, A.G.; Coyle-Gilchrist, I.; Rouse, M.A.; Simon Jones, P.; Li, W.; Wiggins, J.; Lansdall, C.; Rodríguez, P.V.; Wilcox, A.; Tsvetanov, K.A.; et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain 2020, 143, 1555–1571. [Google Scholar] [CrossRef] [PubMed]
- Bellenguez, C.; Küçükali, F.; Jansen, I.; Andrade, V.; Morenau-Grau, S.; Amin, N.; Grenier-Boley, B.; Boland, A.; Kleineidam, L.; Holmans, P.; et al. Large meta-analysis of genome-wide association studies expands knowledge of the genetic etiology of Alzheimer’s disease and highlights potential translational opportunities. medRxiv 2020, 17, 10. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat. Genet. 2021, 53, 392–402. [Google Scholar] [CrossRef]
- Tacik, P.; Sanchez-Contreras, M.; Rademakers, R.; Dickson, D.W.; Wszolek, Z.K. Genetic disorders with tau pathology: A review of the literature and report of two patients with tauopathy and positive family histories. Proc. Neurodegener. Dis. 2016, 16, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Tacik, P.; Wszolek, Z.; SanchezContreras, M.; Rademakers, R.; Dickson, D. Literature review of genetic disorders with tau pathology. Neurology 2016. [Google Scholar]
- Mulroy, E.; Jaunmuktane, Z.; Balint, B.; Erro, R.; Latorre, A.; Bhatia, K.P. Some New and Unexpected Tauopathies in Movement Disorders. Mov. Disord. Clin. Pract. 2020, 7, 616–626. [Google Scholar] [CrossRef]
- Darwich, N.F.; Phan, J.M.; Kim, B.; Suh, E.; Papatriantafyllou, J.D.; Changolkar, L.; Nguyen, A.T.; O’Rourke, C.M.; He, Z.; Porta, S.; et al. Autosomal dominant VCP hypomorph mutation impairs disaggregation of PHF-tau. Science 2020, 370. [Google Scholar] [CrossRef]
- Huin, V.; Deramecourt, V.; Caparros-Lefebvre, D.; Maurage, C.A.; Duyckaerts, C.; Kovari, E.; Pasquier, F.; Buée-Scherrer, V.; Labreuche, J.; Behal, H.; et al. The MAPT gene is differentially methylated in the progressive supranuclear palsy brain. Mov. Disord. 2016, 31, 1883–1890. [Google Scholar] [CrossRef]
- Wen, Y.; Zhou, Y.; Jiao, B.; Shen, L. Genetics of progressive supranuclear palsy: A review. J. Parkinsons. Dis. 2021, 11, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this is a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Sherr, G.L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.V.C. Get Out and Stay Out: New Insights Into DNA Methylation Reprogramming in Mammals. Front. Cell Dev. Biol. 2021, 8. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. Neuronal DNA Methyltransferases: Epigenetic Mediators between Synaptic Activity and Gene Expression? Neuroscientist 2018, 24, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; Dsouza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Hudson, N.O.; Whitby, F.G.; Buck-Koehntop, B.A. Structural insights into methylated DNA recognition by the C-terminal zinc fingers of the DNA reader protein ZBTB38. J. Biol. Chem. 2018, 293, 19835–19843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Kaas, G.A.; Zhong, C.; Eason, D.E.; Ross, D.L.; Vachhani, R.V.; Ming, G.l.; King, J.R.; Song, H.; Sweatt, J.D. TET1 controls CNS 5-Methylcytosine Hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013, 79, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Symmank, J.; Zimmer, G. Regulation of neuronal survival by DNA methyltransferases. Neural. Regen. Res. 2017, 12, 1768–1775. [Google Scholar] [CrossRef]
- Symmank, J.; Bayer, C.; Schmidt, C.; Hahn, A.; Pensold, D.; Zimmer-Bensch, G. DNMT1 modulates interneuron morphology by regulating Pak6 expression through crosstalk with histone modifications. Epigenetics 2018, 13, 536–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer-Bensch, G. Emerging Roles of Long Non-Coding RNAs as Drivers of Brain Evolution. Cells 2019, 8, 1399. [Google Scholar] [CrossRef] [Green Version]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, A.M.; Sewal, A.S.; Rapp, P.R. Epigenetic contributions to cognitive aging: Disentangling mindspan and lifespan. Learn. Mem. 2014, 21, 569–574. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowbotham, D.A. Epigenetic Changes in Aging and Age-related Disease. J. Aging Sci. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Grant, W.B.; Campbell, A.; Itzhaki, R.F.; Savory, J. The significance of environmental factors in the etiology of Alzheimer’s disease. J. Alzheimer’s Dis. 2002, 4, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.M.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar] [CrossRef]
- Laplant, Q.; Vialou, V.; Covington, H.E.; Dumitriu, D.; Feng, J.; Warren, B.L.; Maze, I.; Dietz, D.M.; Watts, E.L.; Iñiguez, S.D.; et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Wilf, R.; Menon, T.; Panikker, P.; Sarthi, J.; Elefant, F. Epigenetic control of learning and memory in drosophila by Tip60 HAT action. Genetics 2014, 198, 1571–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A.; et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Cui, D.; Xu, X. Dna methyltransferases, dna methylation, and age-associated cognitive function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, A.M.M.; Hemstedt, T.J.; Bading, H. Rescue of aging-associated decline in Dnmt3a2 expression restores cognitive abilities. Nat. Neurosci. 2012, 15, 1111–1113. [Google Scholar] [CrossRef]
- Johnson, A.A.; Akman, K.; Calimport, S.R.G.; Wuttke, D.; Stolzing, A.; De Magalhães, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.; Pensold, D.; Bayer, C.; Tittelmeier, J.; González-Bermúdez, L.; Marx-Blümel, L.; Linde, J.; Groß, J.; Salinas-Riester, G.; Lingner, T.; et al. DNA Methyltransferase 1 (DNMT1) Function Is Implicated in the Age-Related Loss of Cortical Interneurons. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- McKinney, B.C.; Lin, C.W.; Rahman, T.; Oh, H.; Lewis, D.A.; Tseng, G.; Sibille, E. DNA methylation in the human frontal cortex reveals a putative mechanism for age-by-disease interactions. Transl. Psychiatry 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Ukitsu, M.; Genda, Y. The methylation status of cytosines in a τ gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neurosci. Lett. 1999, 275, 89–92. [Google Scholar] [CrossRef]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y.; Yoshimura, M.; Genda, Y.; Ukitsu, M. Reduction with age in methylcytosine in the promoter region -224~-101 of the amyloid precursor protein gene in autopsy human cortex. Mol. Brain Res. 1999, 70, 288–292. [Google Scholar] [CrossRef]
- Nagata, K.; Mano, T.; Murayama, S.; Saido, T.C.; Iwata, A. DNA methylation level of the neprilysin promoter in Alzheimer’s disease brains. Neurosci. Lett. 2018, 670, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hellström-Lindahl, E.; Ravid, R.; Nordberg, A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: Inverse correlation with Aβ levels. Neurobiol. Aging 2008, 29, 210–221. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Harris, S.E.; Shah, S.; McRae, A.F.; von Zglinicki, T.; Martin-Ruiz, C.; Wray, N.R.; Visscher, P.M.; Deary, I.J. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int. J. Epidemiol. 2016, 45, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef] [Green Version]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany. NY). 2015, 7, 1198–1211. [Google Scholar] [CrossRef]
- Degerman, S.; Josefsson, M.; Nordin Adolfsson, A.; Wennstedt, S.; Landfors, M.; Haider, Z.; Pudas, S.; Hultdin, M.; Nyberg, L.; Adolfsson, R. Maintained memory in aging is associated with young epigenetic age. Neurobiol. Aging 2017, 55, 167–171. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Metpally, R.P.; Carey, D.J.; Crist, R.C.; Berrettini, W.H.; Wilson, G.D.; Imam, K.; et al. Artificial intelligence and leukocyte epigenomics: Evaluation and prediction of late-onset Alzheimer’s disease. PLoS ONE 2021, 16. [Google Scholar] [CrossRef]
- Pellegrini, C.; Pirazzini, C.; Sala, C.; Sambati, L.; Yusipov, I.; Kalyakulina, A.; Ravaioli, F.; Kwiatkowska, K.M.; Durso, D.F.; Ivanchenko, M.; et al. A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer’s Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Y.; Readhead, B.; Chen, K.; Su, Y.; Reiman, E.M.; Dudley, J.T. Longitudinal data in peripheral blood confirm that PM20D1 is a quantitative trait locus (QTL) for Alzheimer’s disease and implicate its dynamic role in disease progression. Clin. Epigenetics 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation. Neurobiol. Aging 2010, 31, 2025–2037. [Google Scholar] [CrossRef] [Green Version]
- Chouliaras, L.; Mastroeni, D.; Delvaux, E.; Grover, A.; Kenis, G.; Hof, P.R.; Steinbusch, H.W.M.; Coleman, P.D.; Rutten, B.P.F.; van den Hove, D.L.A. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 2091–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Gasparoni, G.; Bultmann, S.; Lutsik, P.; Kraus, T.F.J.; Sordon, S.; Vlcek, J.; Dietinger, V.; Steinmaurer, M.; Haider, M.; Mulholland, C.B.; et al. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin 2018, 11. [Google Scholar] [CrossRef]
- Foraker, J.; Millard, S.P.; Leong, L.; Thomson, Z.; Chen, S.; Keene, C.D.; Bekris, L.M.; Yu, C.E.; Fischer, A. The APOE Gene is Differentially Methylated in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 745–755. [Google Scholar] [CrossRef]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.C.; Jiang, T.; Yang, A.F.; Du, Y.J.; Wu, M.; Kong, L.H. Epigenetic modulation on tau phosphorylation in Alzheimer’s disease. Neural Plast. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayas, C.L.; Ávila, J. GSK-3 and tau: A key duet in alzheimer’s disease. Cells 2021, 10, 721. [Google Scholar] [CrossRef]
- Nicolia, V.; Ciraci, V.; Cavallaro, R.A.; Ferrer, I.; Scarpa, S.; Fuso, A. GSK3β 5’-flanking DNA Methylation and Expression in Alzheimer’s Disease Patients. Curr. Alzheimer Res. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, C.; Zi, X.; Tu, Q.; Guo, K. Epigenetic modulation of Cdk5 contributes to memory deficiency induced by amyloid fibrils. Exp. Brain Res. 2015, 233, 165–173. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Aso, E.; Heyn, H.; Matsuda, T.; Bock, C.; Ferrer, I.; Esteller, M. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus 2014. [Google Scholar] [CrossRef] [Green Version]
- Lonze, B.E.; Riccio, A.; Cohen, S.; Ginty, D.D. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 2002, 34, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Yu, J.T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef]
- Tanila, H. The role of BDNF in Alzheimer’s disease. Neurobiol. Dis. 2017, 97, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.H.; Yang, Y.; Sun, Y.X.; Zhang, C.D. Exogenous brain-derived neurotrophic factor attenuates cognitive impairment induced by okadaic acid in a rat model of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 2173–2181. [Google Scholar] [CrossRef]
- Ambigapathyy, G.; Zhengy, Z.; Keifer, J. Regulation of BDNF chromatin status and promoter accessibility in a neural correlate of associative learning. Epigenetics 2015, 10, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Do Carmo, S.; Hanzel, C.E.; Jacobs, M.L.; MacHnes, Z.; Iulita, M.F.; Yang, J.; Yu, L.; Ducatenzeiler, A.; Danik, M.; Breuillaud, L.S.; et al. Rescue of Early bace-1 and Global DNA Demethylation by S-Adenosylmethionine Reduces Amyloid Pathology and Improves Cognition in an Alzheimer’s Model. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-homologous end joining induced alterations in DNA methylation: A source of permanent epigenetic change. Oncotarget 2017, 8, 40359–40372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobile, V.; Pucci, C.; Chiurazzi, P.; Neri, G.; Tabolacci, E. Dna methylation, mechanisms of FMR1 inactivation and therapeutic perspectives for fragile X syndrome. Biomolecules 2021, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Song, H.; Croteau, D.L.; Akbari, M.; Bohr, V.A. Genome instability in Alzheimer disease. Mech. Ageing Dev. 2017, 161, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R.; et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E9645–E9654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N.; et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant accumulation of BRCA1 in Alzheimer disease and other tauopathies. J. Neuropathol. Exp. Neurol. 2020, 79, 22–33. [Google Scholar] [CrossRef]
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau aggregates in human tauopathies. Brain Sci. 2020, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.G. Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl. Cell Differ. 2011, 53, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Taher, N.; McKenzie, C.; Garrett, R.; Baker, M.; Fox, N.; Isaacs, G.D. Amyloid-β alters the DNA methylation status of cell-fate genes in an Alzheimer’s disease model. J. Alzheimer’s Dis. 2014, 38, 831–844. [Google Scholar] [CrossRef]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Khan, U.A.; Liu, L.; Provenzano, F.A.; Berman, D.E.; Profaci, C.P.; Sloan, R.; Mayeux, R.; Duff, K.E.; Small, S.A. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 2014, 17, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide12 DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Héberlé, É.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Fuso, A.; Seminara, L.; Cavallaro, R.A.; D’Anselmi, F.; Scarpa, S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol. Cell. Neurosci. 2005, 28, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, S.; Cavallaro, R.A.; D’Anselmi, F.; Fuso, A. Gene silencing through methylation: An epigenetic intervention on Alzheimer disease. J. Alzheimer’s Dis. 2006, 9, 407–414. [Google Scholar] [CrossRef]
- Chan, A.; Shea, T.B. Effects of dietary supplementation with N-acetyl cysteine, acetyl-L-carnitine and S-adenosyl methionine on cognitive performance and aggression in normal mice and mice expressing human ApoE4. NeuroMolecular Med. 2008, 10, 46. [Google Scholar] [CrossRef]
- Fuso, A. The “golden age” of DNA methylation in neurodegenerative diseases. Proc. Clin. Chem. Lab. Med. 2013, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Scarpa, S. One-carbon metabolism and Alzheimer’s disease: Is it all a methylation matter? Neurobiol. Aging 2011, 32, 1192–1195. [Google Scholar] [CrossRef]
- Momparler, R.L.; Momparler, L.F.; Samson, J. Comparison of the antileukemic activity of 5-aza-2′-deoxycytidine, 1-β-d-arabinofuranosylcytosine and 5-azacytidine against L1210 leukemia. Leuk. Res. 1984, 8, 1043–1049. [Google Scholar] [CrossRef]
- Issa, J.P.J.; Garcia-Manero, G.; Giles, F.J.; Mannari, R.; Thomas, D.; Faderl, S.; Bayar, E.; Lyons, J.; Rosenfeld, C.S.; Cortes, J.; et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood 2004, 103, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Lübbert, M.; Daskalakis, M.; Sander, P.N.; Kündgen, A. Azanucleoside DNA Methyltransferase inhibitor drugs: Update on clinical applications in Myelodysplastic syndromes and acute myeloid leukemia. In Proceedings of the Epigenetics and Human Health; Springer Nature: Cham, Switzerland, 2016; pp. 197–221. [Google Scholar] [CrossRef]
- Sandi, C.; Sandi, M.; Virmouni, S.A.; Al-Mahdawi, S.; Pook, M.A. Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; De Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3177–3183. [Google Scholar] [CrossRef] [Green Version]
- Winquist, E.; Knox, J.; Ayoub, J.P.; Wood, L.; Wainman, N.; Reid, G.K.; Pearce, L.; Shah, A.; Eisenhauer, E. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig. New Drugs 2006, 24, 159–167. [Google Scholar] [CrossRef]
- Stewart, D.J.; Donehower, R.C.; Eisenhauer, E.A.; Wainman, N.; Shah, A.K.; Bonfils, C.; MacLeod, A.R.; Besterman, J.M.; Reid, G.K. A phase I pharmacokinetic and pharmacodynamic study of the DNA methyltransferase 1 inhibitor MG98 administered twice weekly. Ann. Oncol. 2003, 14, 766–774. [Google Scholar] [CrossRef]
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-specific targeting of DNA methylation in the mammalian genome. Cancers 2019, 11, 1515. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Xu, Y.; Liu, Z.; Liu, W.; Jiang, L.; Zhang, R.; Cui, D.; Zhang, Q.; Xu, S. Elevation of peripheral BDNF promoter methylation predicts conversion from amnestic mild cognitive impairment to Alzheimer’s disease: A 5-year longitudinal study. J. Alzheimer’s Dis. 2017, 56, 391–401. [Google Scholar] [CrossRef]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Usefulness of DNA methylation levels in COASY and SPINT1 gene promoter regions as biomarkers in diagnosis of Alzheimer’s disease and Amnestic mild cognitive impairment. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercorio, R.; Pergoli, L.; Galimberti, D.; Favero, C.; Carugno, M.; Dalla Valle, E.; Barretta, F.; Cortini, F.; Scarpini, E.; Valentina, V.B.; et al. PICALM gene methylation in blood of Alzheimer’s disease patients is associated with cognitive decline. J. Alzheimer’s Dis. 2018, 65, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, A.; Arosio, B.; Falconi, A.; Micioni Di Bonaventura, M.V.; Karimi, M.; Mari, D.; Casati, M.; Maccarrone, M.; D’Addario, C. Global changes in DNA methylation in Alzheimer’s disease peripheral blood mononuclear cells. Brain. Behav. Immun. 2015, 45, 139–144. [Google Scholar] [CrossRef] [PubMed]
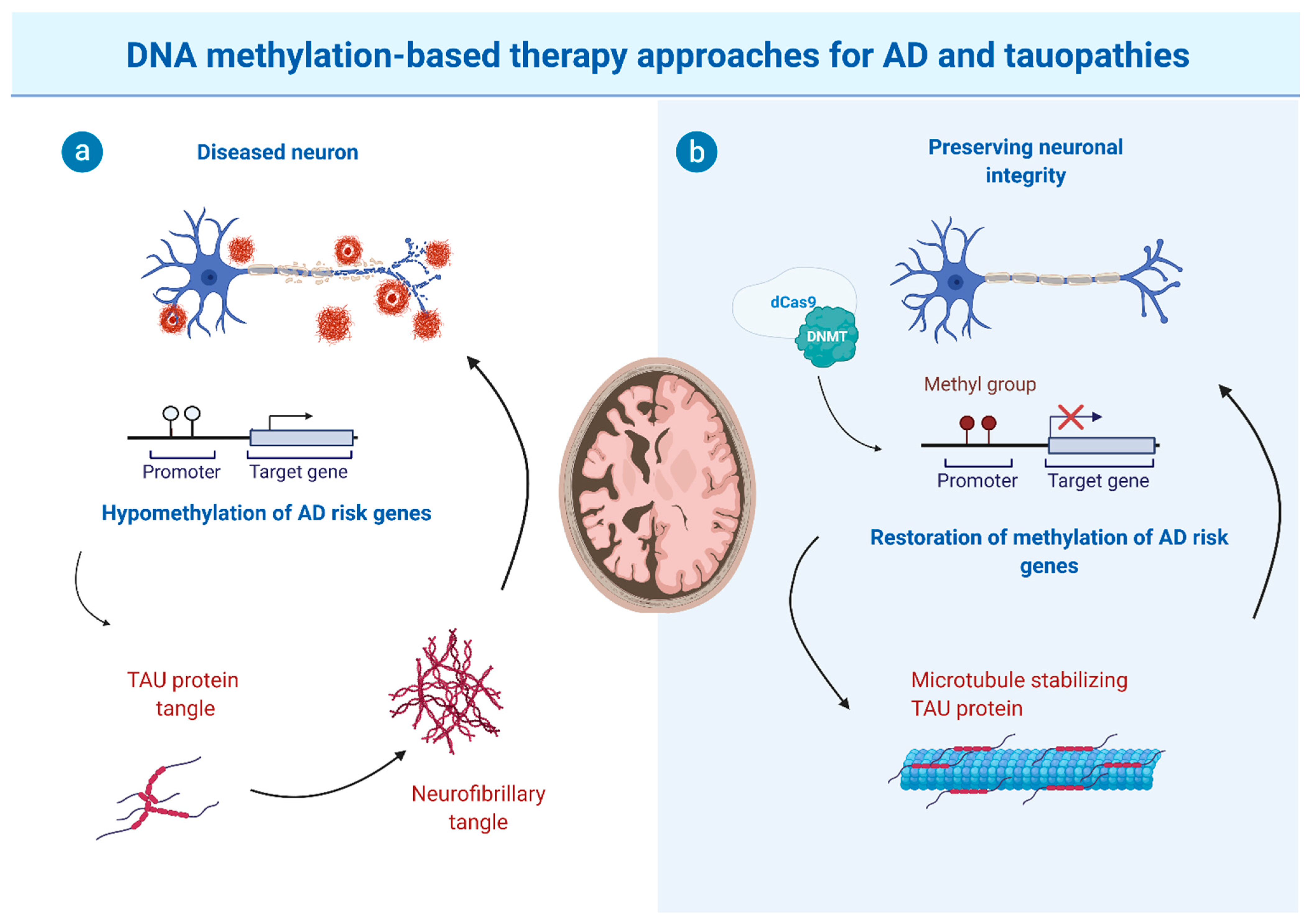

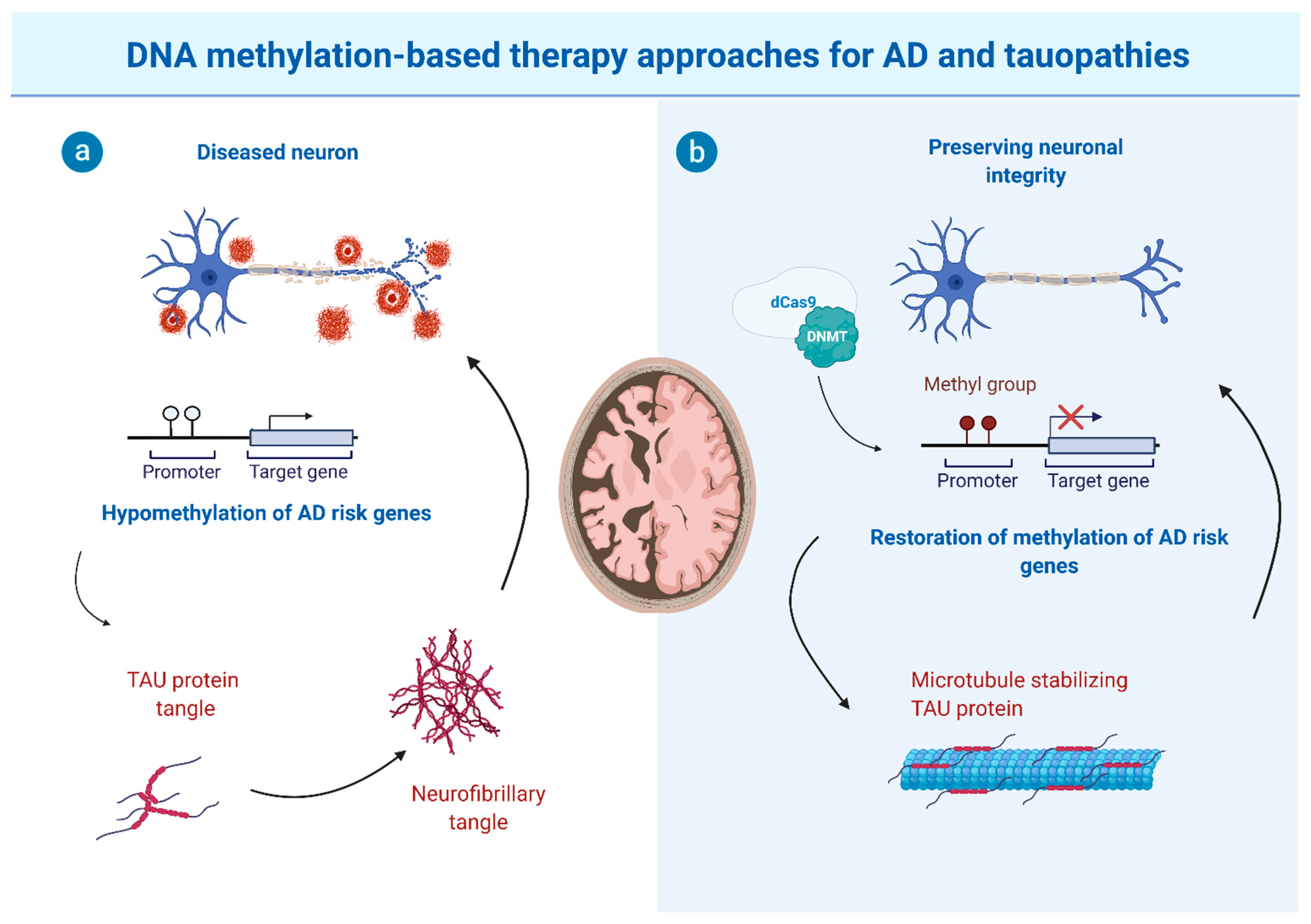{kind=link}
| Disease Entity | Clinic Description/Overview | Etiology | Secondary to or Coexisting with |
|---|---|---|---|
| Familial FTLD-TAU due to coding mutations in MAPT | Very heterogenous group of aging associated tauopathies, which comprise i.a. formerly FTDP17(t) and patients diagnosed with PSP | Genetic: MAPT | - |
| Vacuolar tauopathy | FTLD-like syndrome due to defective TAU disaggregation | Genetic: VCP [55] | - |
| Other forms of FTLD-TAU (like) tauopathies | Heterogenous group of aging-associated tauopathies, like CBD, PiD, GGT, AGD, PART, ARTAG, most of which are further subclassified | Mostly sporadic, (epi)genetic causes unclear | - |
| Progressive supranuclear palsy (PSP) | Rare neurodegenerative disorder, but a common atypical Parkinson syndrome with cognitive, motor, behavior and language abnormalities, often diagnosed as AD | Epigenetic: Hypomethylation of MAPT [56] Genetic: MAPT Sporadic: GWAS with loci close to MAPT, STX6, EIF2AK3, MOBP, DUSP, SLCO1A2, RUNX2, i.a. [57] | - |
| PSP look-alike syndromes | Clinically similar to PSP, rare | Genetic: LRRK2, DCTN1, BSN | mostly unclear |
| Familial Alzheimer Disease | Age of Onset usually between 40 and 70 years, fast progression | Genetic: APP, PSEN1, PSEN2, up to ~75 risk modifying genes | Amyloid-pathology |
| Familial Parkinson Disease | Various group of familial Parkinson Syndromes | Genetic: SNCA, PRKN, LRRK2, other | alpha-Synuclein deposits |
| Familial FTLD-ALS Syndromes | Syndromes with manifestations ranging from pure ALS to pure FTLD or overlapping phenotypes | Genetic: GRN, C9ORF72, TARDBP, other | Deposits of dipeptide repeats, RNA inclusions, TDP-43 |
| Hereditary cerebral amyloid angiopathy | Familial forms of dementia (fam. British and fam. Danish dementia) | Genetic: ITM2B | Amyloid-pathology |
| Niemann Pick Disease Type C | Lysosomal storage disease with hepatosplenomegaly, progressive dementia, and premature death ranging from infancy to late adulthood | Genetic: NPC1, NPC2 | Cholesterol accumulations |
| Kufs Disease | A neurodegenerative lysosomal storage disease/neuronal ceroid lipofuscinosis | Genetic: CLN6 (PPT1, DNAJC5, CTSF) | Lipofuscin accumulations |
| Christianson Syndrome | X-linked mental retardation syndrome with microcephaly, muscle hypotonia, movement disorder, and epilepsy | Genetic: SLC9A6 | -/unclear |
| Mental Retardation, X-linked, syndromic, Hedera type | X-linked mental retardation syndrome with global developmental delay, parkinsonism, spasticity, and progressive neurodegeneration | Genetic: ATP6AP2 | SQSTM1 depositions |
| Myotonic Dystrophy Type 1 & 2 | Most common forms of muscular dystrophy characterized by muscle weakness, progressive muscle loss, and may include cataracts, diabetes, and dementia at late stages | Genetic: DMPK, CNBP | RNA nuclear inclusions |
| (Infantile) Sialic Acid Storage Disease | NDD with lysosomal dysfunction presenting in infancy in its severe form or in adulthood with progressive brain atrophy | Genetic: SLC17A5 | -/unclear |
| PKAN | NDD with brain iron accumulation | Genetic: PANK2 | Iron depositions |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmer-Bensch, G.; Zempel, H. DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies. Cells 2021, 10, 3064. https://doi.org/10.3390/cells10113064
Zimmer-Bensch G, Zempel H. DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies. Cells. 2021; 10(11):3064. https://doi.org/10.3390/cells10113064
Chicago/Turabian StyleZimmer-Bensch, Geraldine, and Hans Zempel. 2021. "DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies" Cells 10, no. 11: 3064. https://doi.org/10.3390/cells10113064
APA StyleZimmer-Bensch, G., & Zempel, H. (2021). DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies. Cells, 10(11), 3064. https://doi.org/10.3390/cells10113064