
PI3K Pathway Inhibition with NVP-BEZ235 Hinders Glycolytic Metabolism in Glioblastoma Multiforme Cells



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Cultures, and Drug Treatment
2.2. RNA Extraction and RNA Sequencing
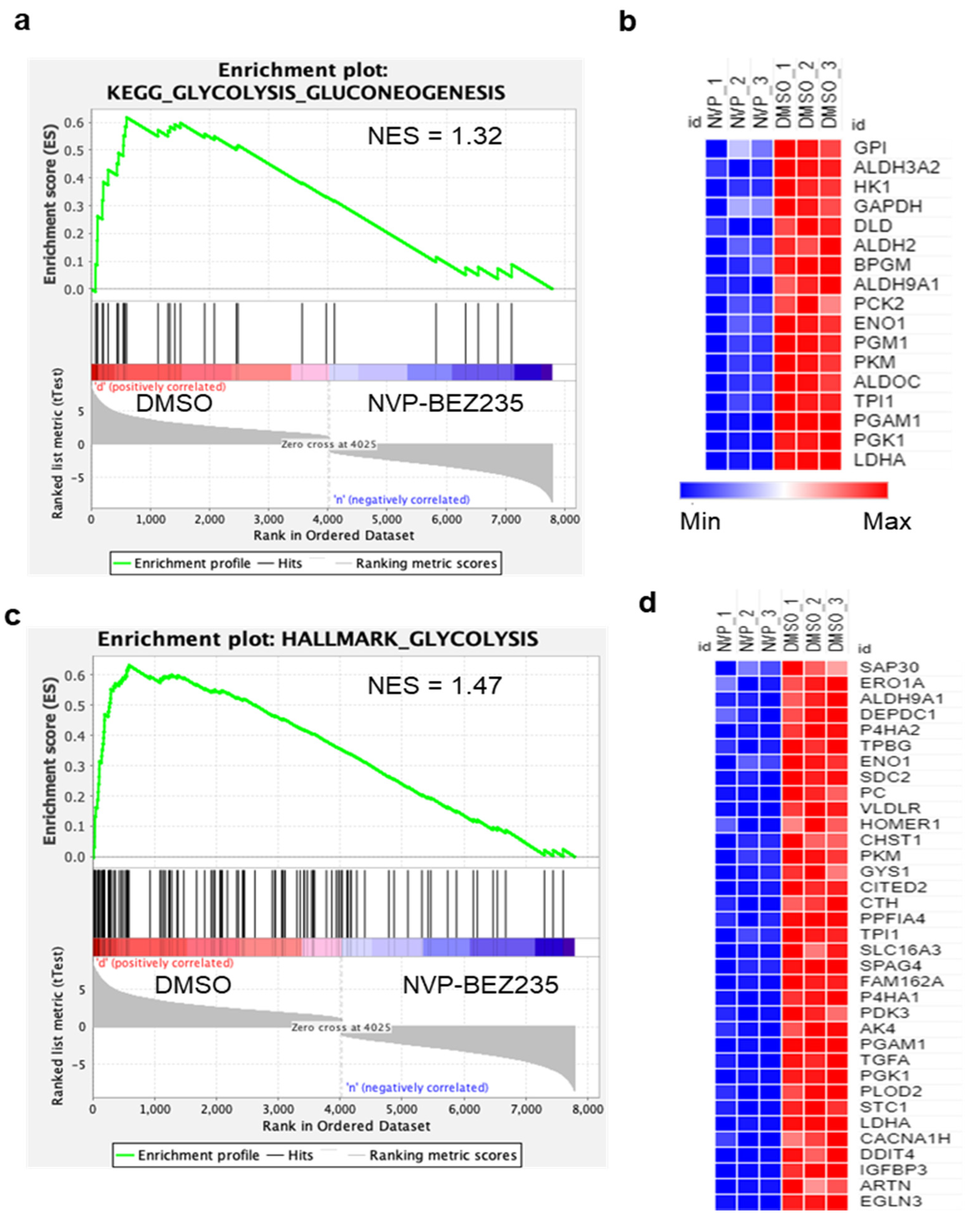
2.3. Bioinformatics Analysis
2.4. Quantitative Real-Time PCR (qRT-PCR)
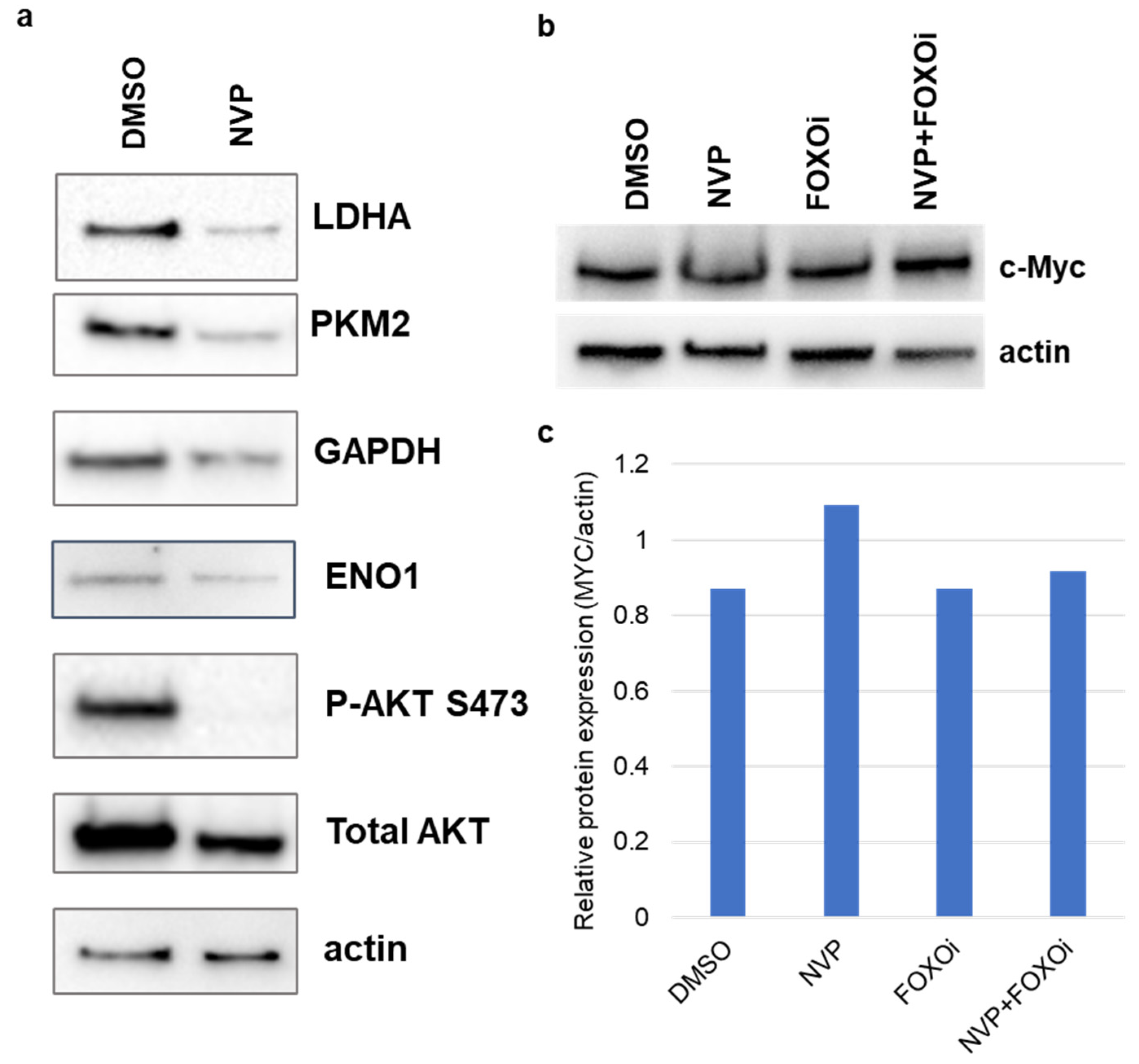
2.5. Western Blot
2.6. Quantitative Glutamate Assay
2.7. NAD+/NADH Cell-Based Assay
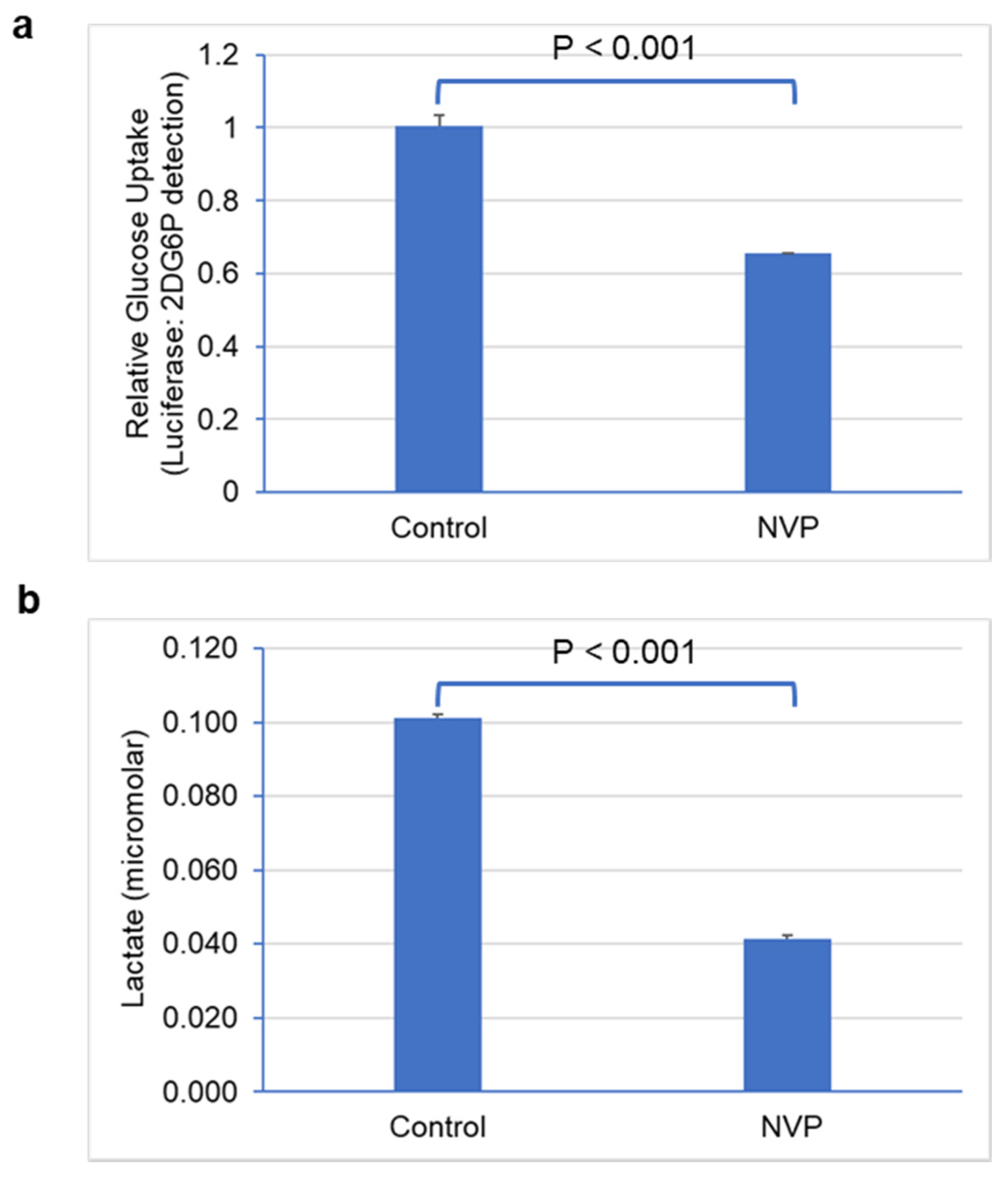
2.8. Glucose Uptake and Lactate Secretion Assays
3. Results
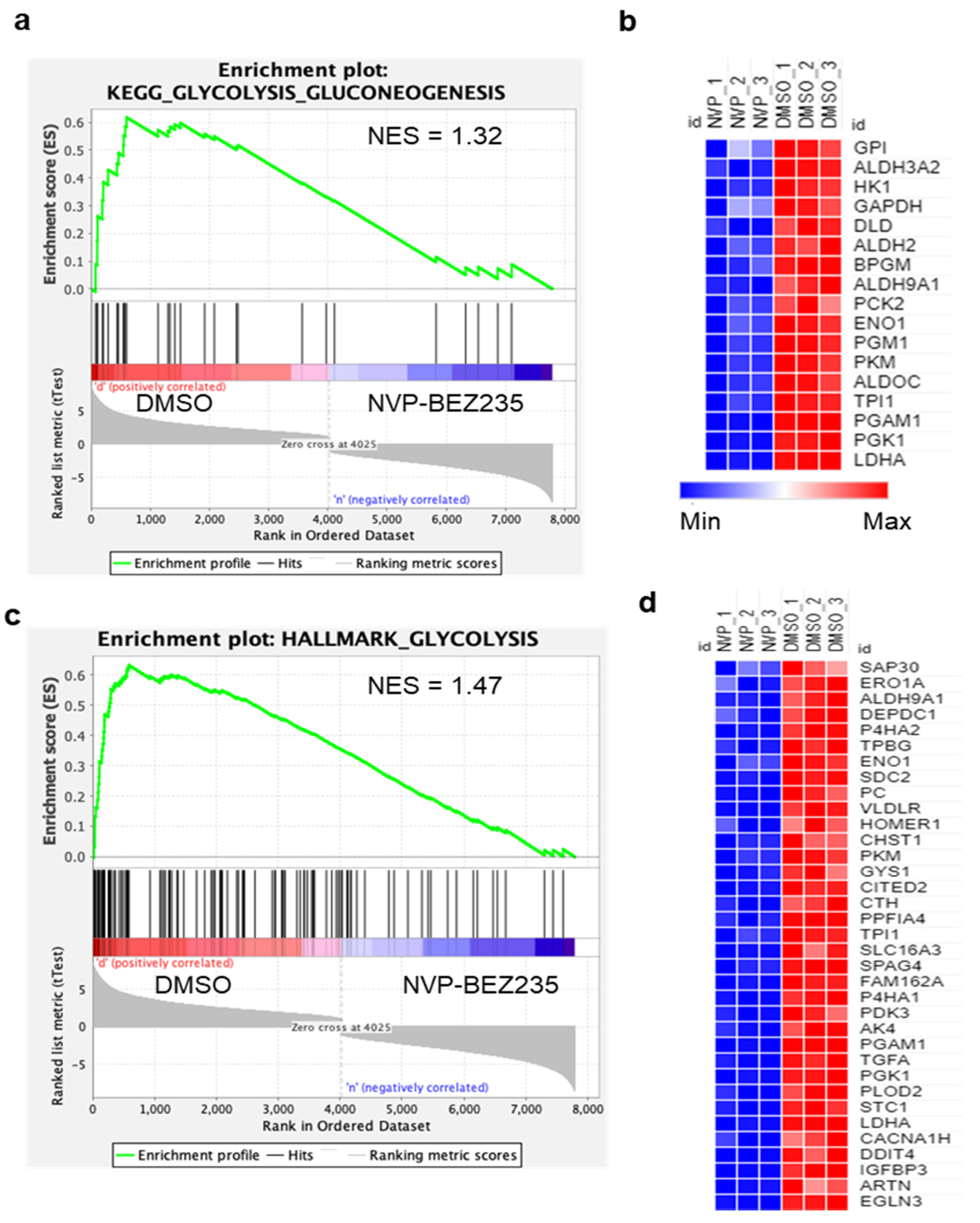
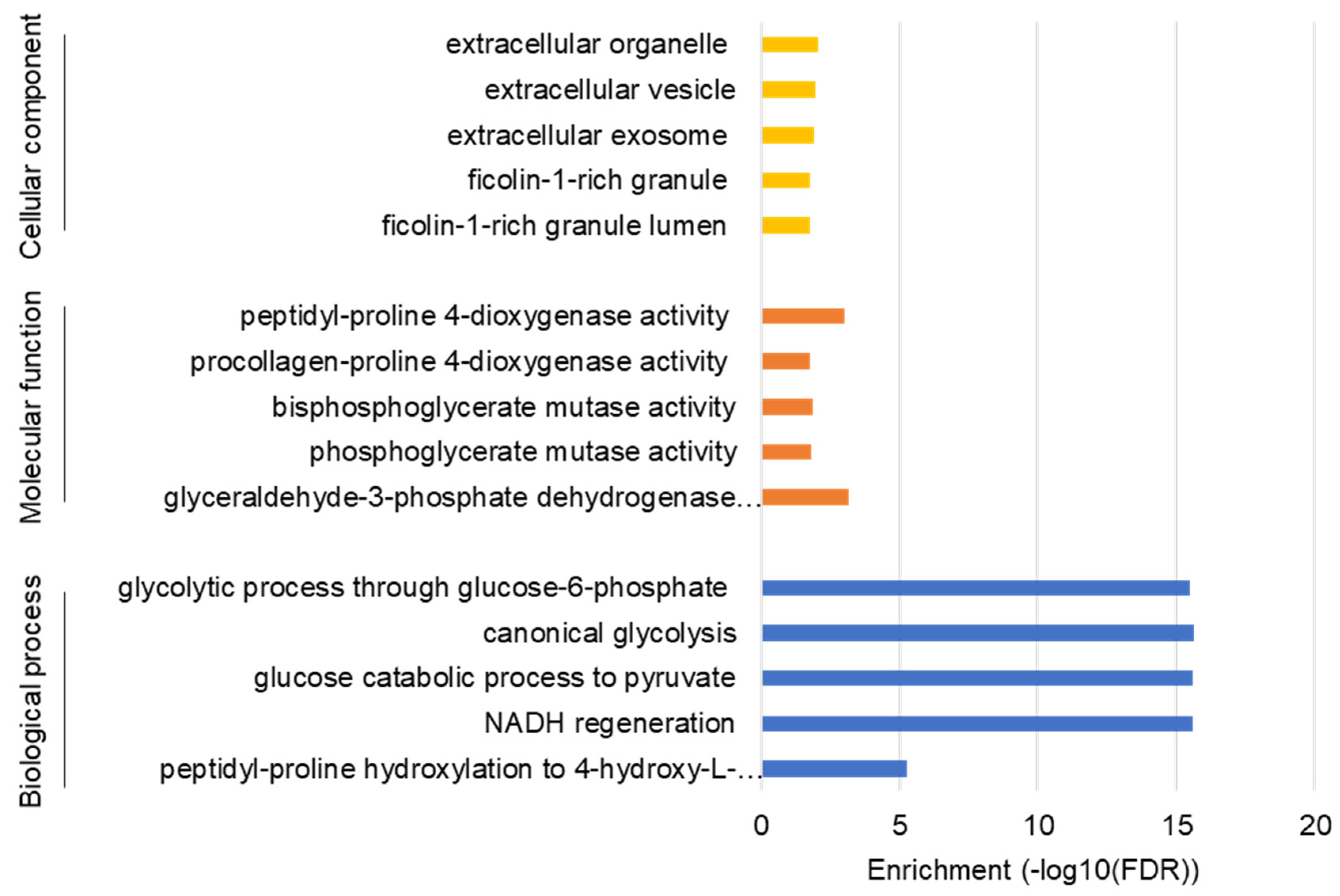
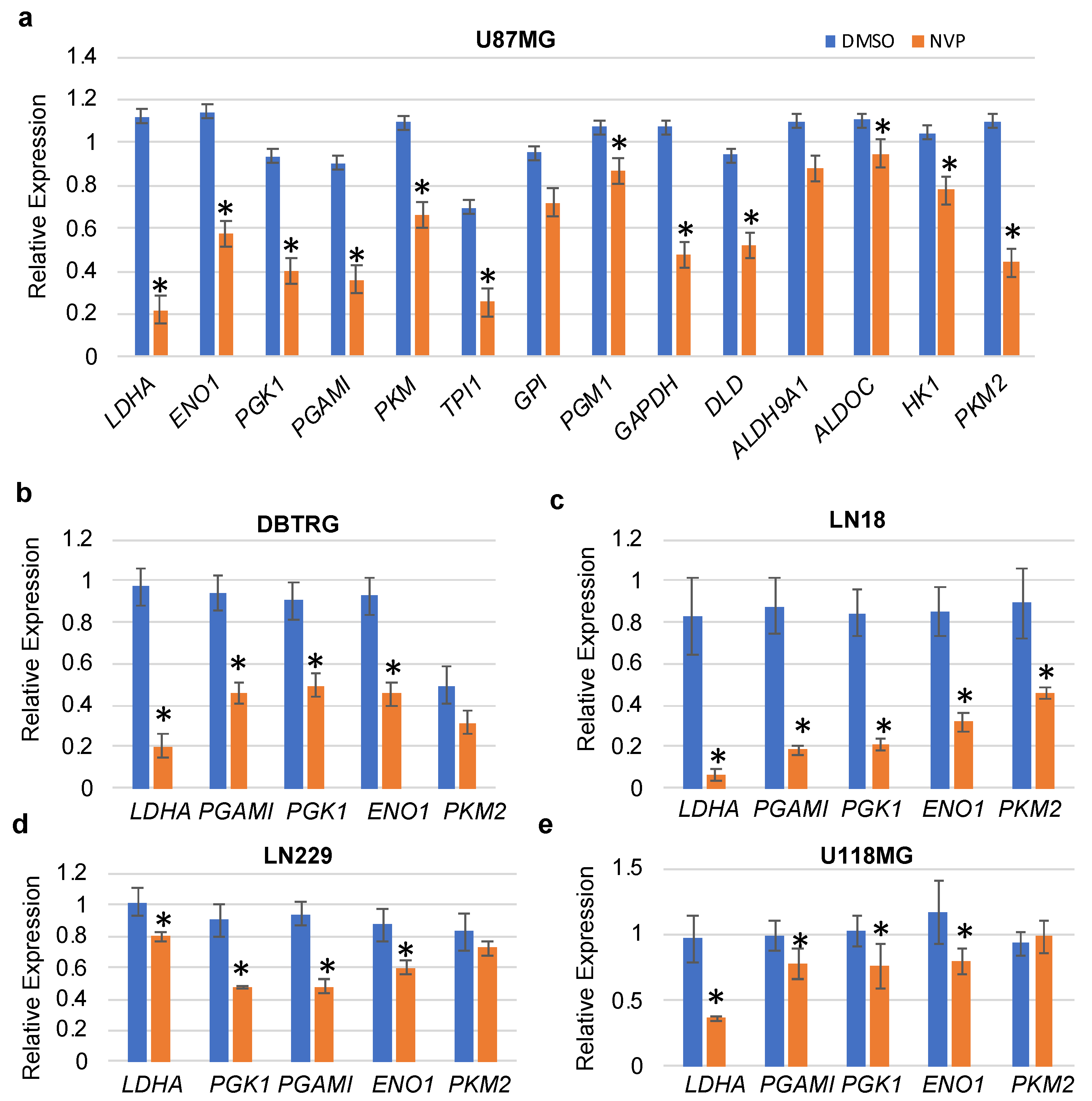
3.1. NVP-BEZ235 Treatment Reduced Glycolytic Gene Expression in GBM Cells
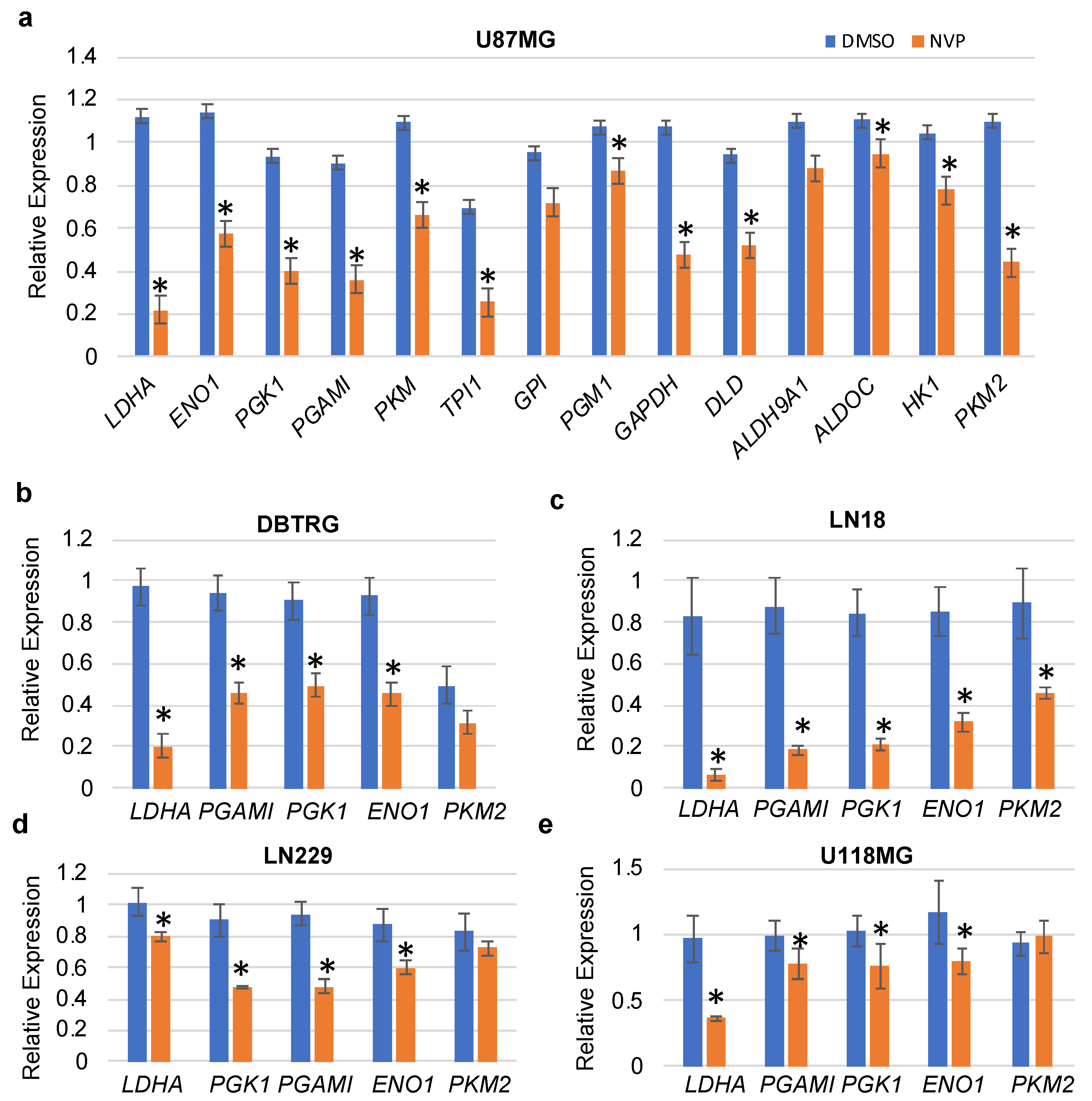
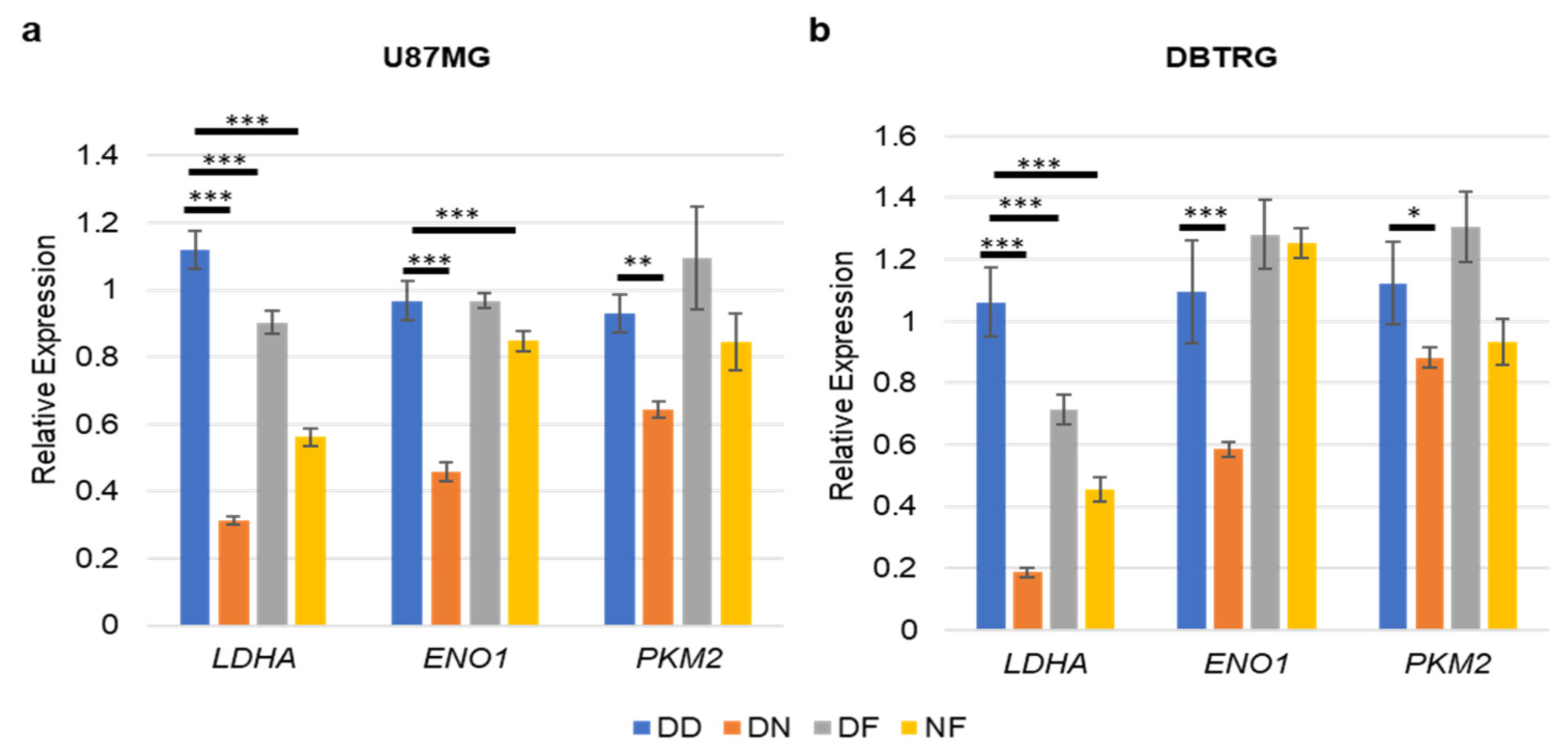
3.2. NVP-BEZ235 Treatment Reduced Glycolytic Genes in Numerous GBM Cell Lines
3.3. NVP-BEZ235 Treatment Led to Reduced NAD+ and Glutamate in U87MG Cells
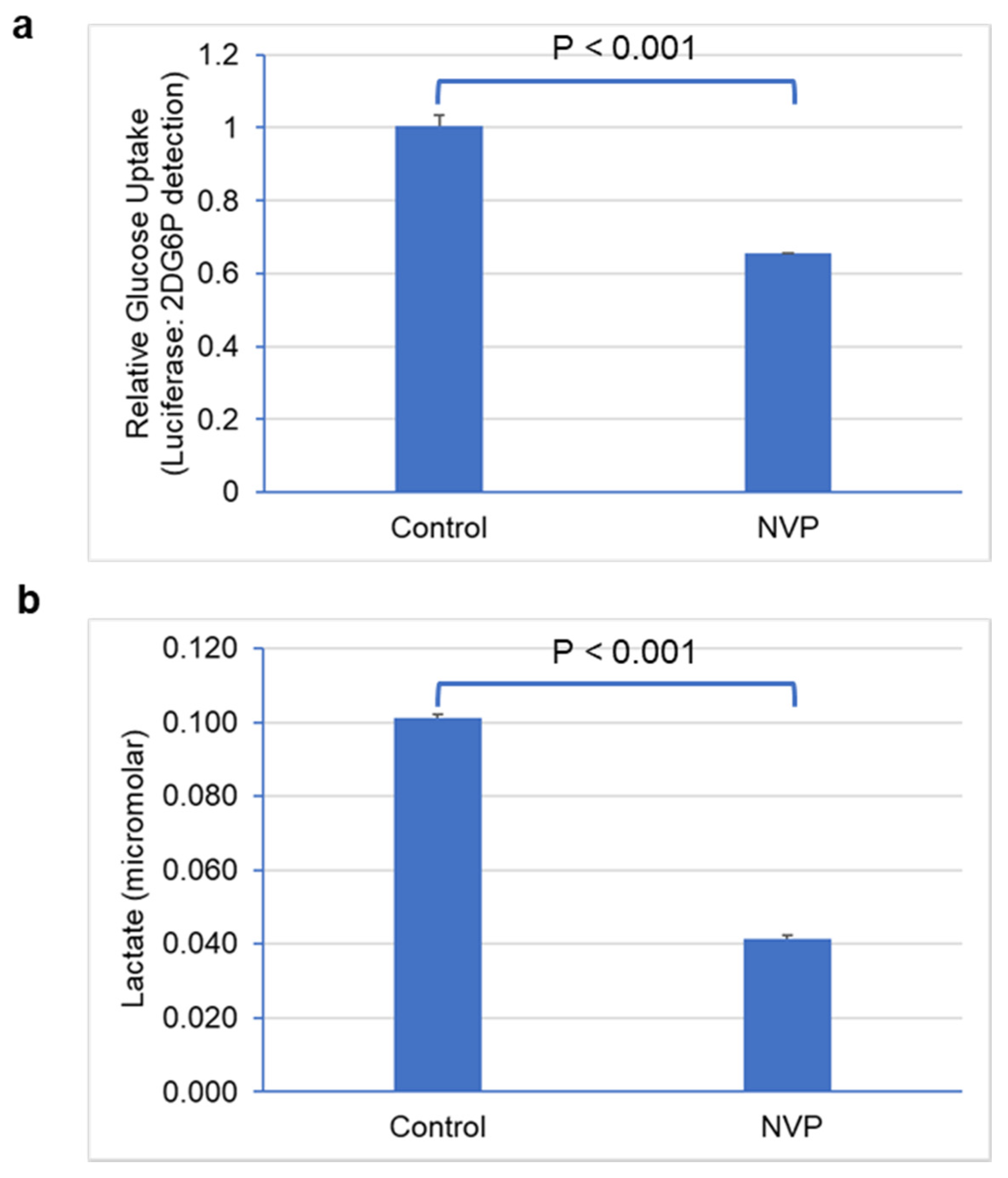
3.4. Glucose Uptake and Lactate Secretion Were Reduced under NVP-BEZ235 Treatment in U87MG Cells
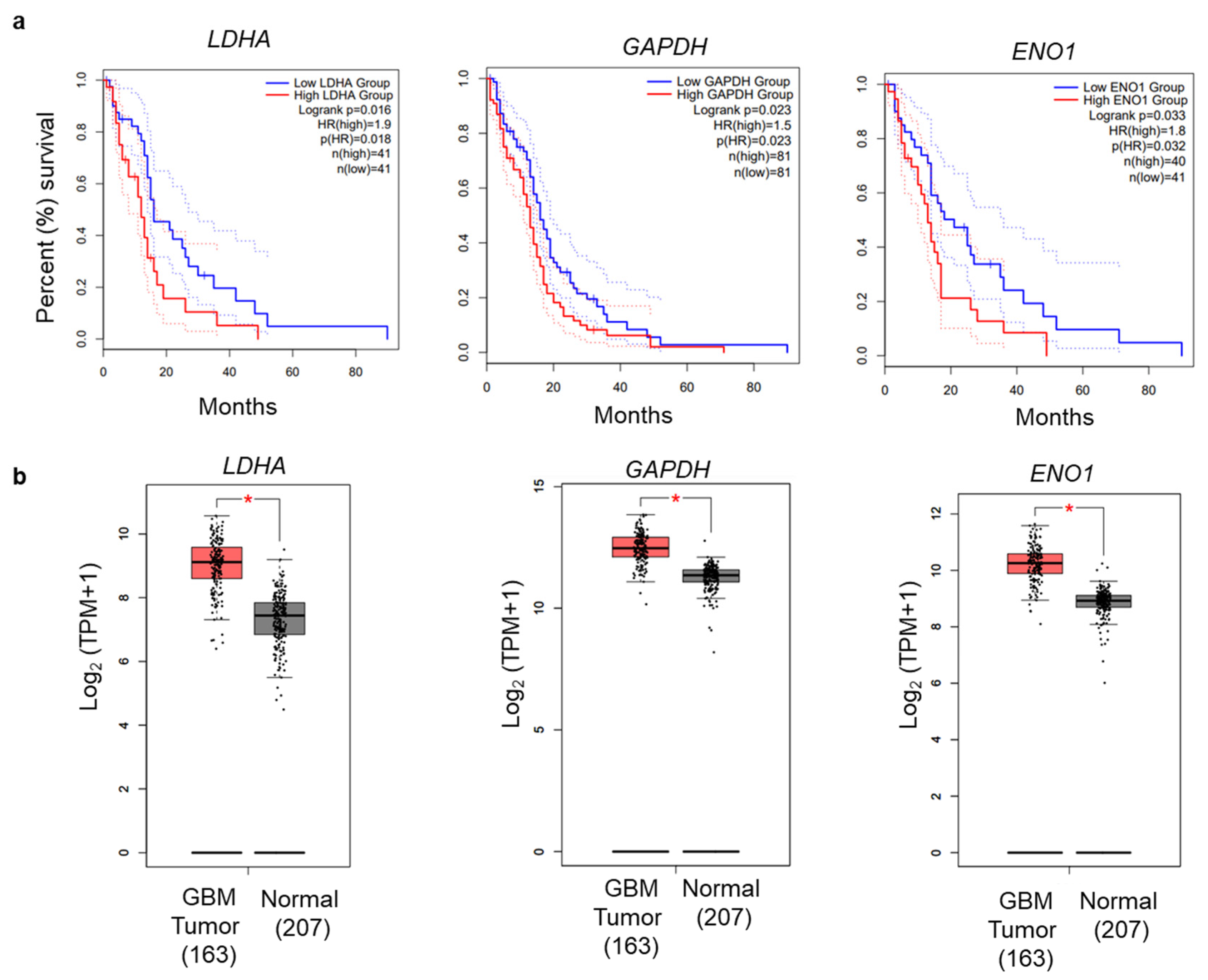
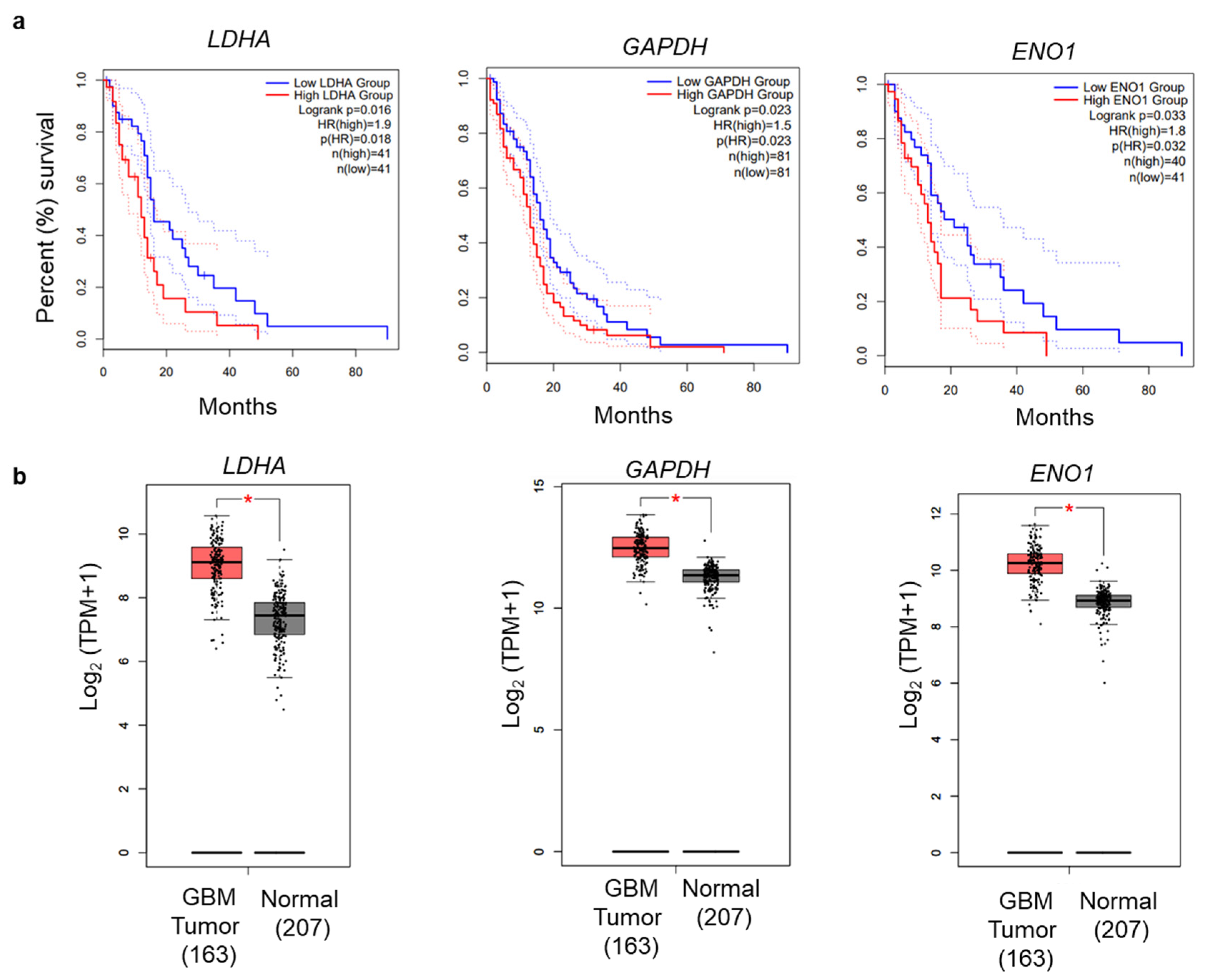
3.5. Glycolytic Genes Are Associated with Poor Prognosis in GBM
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Batsios, G.; Viswanath, P.; Subramani, E.; Najac, C.; Gillespie, A.M.; Santos, R.D.; Molloy, A.R.; Pieper, R.O.; Ronen, S.M. PI3K/mTOR inhibition of IDH1 mutant glioma leads to reduced 2HG production that is associated with increased survival. Sci. Rep. 2019, 9, 10521. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Wan, X.; Wei, X. PROX1 promotes human glioblastoma cell proliferation and invasion via activation of the nuclear factor-κB signaling pathway. Mol. Med. Rep. 2016, 15, 963–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsir, T.; Eriksson, A.; Orrego, A.; Lindström, M.S.; Nistér, M. Expression of PROX1 Is a Common Feature of High-Grade Malignant Astrocytic Gliomas. J. Neuropathol. Exp. Neurol. 2010, 69, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Fu, X.; Wang, J.; Guan, R.; Sun, Y.; To, S.T. Inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt–MAPK pathway. J. Cell. Physiol. 2019, 234, 5888–5903. [Google Scholar] [CrossRef]
- Atif, F.; Yousuf, S.; Espinosa-Garcia, C.; Sergeeva, E.; Stein, D.G. Progesterone Treatment Attenuates Glycolytic Metabolism and Induces Senescence in Glioblastoma. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Balça-Silva, J.; Matias, D.; Carmo, A.; Dubois, L.G.F.; Gonçalves, A.C.; Girao, H.; Canedo, N.H.S.; Correia, A.H.; De Souza, J.M.; Ribeiro, A.B.S.; et al. Glioblastoma entities express subtle differences in molecular composition and response to treatment. Oncol. Rep. 2017, 38, 1341–1352. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Touat, M.; Alexander, B.M.; Mellinghoff, I.K.; Ramkissoon, S.; McCluskey, C.S.; Pelton, K.; Haidar, S.; Basu, S.S.; Gaffey, S.C.; et al. Buparlisib in Patients With Recurrent Glioblastoma Harboring Phosphatidylinositol 3-Kinase Pathway Activation: An Open-Label, Multicenter, Multi-Arm, Phase II Trial. J. Clin. Oncol. 2019, 37, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Cacace, A.; Sboarina, M.; Vazeille, T.; Sonveaux, P. Glutamine activates STAT3 to control cancer cell proliferation independently of glutamine metabolism. Oncogene 2017, 36, 2074–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Nilsson, A.; Haanstra, J.R.; Engqvist, M.; Gerding, A.; Bakker, B.M.; Klingmüller, U.; Teusink, B.; Nielsen, J. Quantitative analysis of amino acid metabolism in liver cancer links glutamate excretion to nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10294–10304. [Google Scholar] [CrossRef]
- Venneti, S.; Thompson, C.B. Metabolic Modulation of Epigenetics in Gliomas. Brain Pathol. 2013, 23, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liang, Q.; Wang, L. Icaritin inhibits glioblastoma cell viability and glycolysis by blocking the IL-6/Stat3 pathway. J. Cell. Biochem. 2019, 120, 7257–7264. [Google Scholar] [CrossRef]
- Oudard, S.; Arvelo, F.; Miccoli, L.; Apiou, F.; Dutrillaux, A.M.; Poisson, M.; Dutrillaux, B.; Poupon, M.F. High glycolysis in gliomas despite low hexokinase transcription and activity correlated to chromosome 10 loss. Br. J. Cancer 1996, 74, 839–845. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Pinedo, C.; El Mjiyad, N.; Ricci, J.E. Cancer metabolism: Current perspectives and future directions. Cell Death Dis. 2012, 3, e248. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Cui, H. Epigenetic modulation of metabolism in glioblastoma. Semin. Cancer Biol. 2019, 57, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Martinez, E.; Vazquez, N.; Lopez, A.; Fanniel, V.; Sanchez, L.; Marks, R.; Hinojosa, L.; Cuello, V.; Cuevas, M.; Rodriguez, A.; et al. The PI3K pathway impacts stem gene expression in a set of glioblastoma cell lines. J. Cancer Res. Clin. Oncol. 2020, 146, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-F.; Wang, J.; Shao, W.; Wu, C.-P.; Chen, Z.-P.; To, S.-S.T.; Li, W.-P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 1476. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol. 2013, 3, 96. [Google Scholar] [CrossRef] [Green Version]
- Eckerdt, F.D.; Bell, J.B.; Gonzalez, C.; Oh, M.S.; Perez, R.E.; Mazewski, C.; Fischietti, M.; Goldman, S.; Nakano, I.; Platanias, L.C. Combined PI3Kα-mTOR Targeting of Glioma Stem Cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2019, 20, 74–88. [Google Scholar] [CrossRef]
- Dang, C.V.; Le, A.; Gao, P. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzalin, A.; Brambilla, F.; Arbustini, E.; Basello, K.; Speciani, A.; Mauri, P.; Bezzi, P.; Magrassi, L. A New Pathway Promotes Adaptation of Human Glioblastoma Cells to Glucose Starvation. Cells 2020, 9, 1249. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2019, 20, 57–70. [Google Scholar] [CrossRef]
- Santo, E.E.; Stroeken, P.; Sluis, P.V.; Koster, J.; Versteeg, R.; Westerhout, E.M. FOXO3a Is a Major Target of Inactivation by PI3K/AKT Signaling in Aggressive Neuroblastoma. Cancer Res. 2013, 73, 2189–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhan, M.; Silva, M.; Xingan, X.; Huang, Y.; Zheng, W. Role of FOXO Transcription Factors in Cancer Metabolism and Angiogenesis. Cells 2020, 9, 1586. [Google Scholar] [CrossRef]
- Jacobs, F.; van der Heide, L.P.; Wijchers, P.J.; Burbach, J.H.; Hoekman, M.F.; Smidt, M.P. FoxO6, a Novel Member of the FoxO Class of Transcription Factors with Distinct Shuttling Dynamics. J. Biol. Chem. 2003, 278, 35959–35967. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Brosens, J.J.; Lam, E.W.-F. Resist or die: FOXO transcription factors determine the cellular response to chemotherapy. Cell Cycle 2008, 7, 3133–3136. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-Y.; Wu, H.-C.; Wu, J.-E.; Huang, K.-Y.; Yang, S.-C.; Chen, S.-X.; Tsao, C.-J.; Hsu, K.-F.; Chen, Y.-L.; Hong, T.-M. The dual PI3K/mTOR inhibitor BEZ235 restricts the growth of lung cancer tumors regardless of EGFR status, as a potent accompanist in combined therapeutic regimens. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef]
- Lin, A.; Piao, H.-L.; Zhuang, L.; Sarbassov, S.D.; Ma, L.; Gan, B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014, 74, 1682–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzen, D.; Divé, I.; Lorenz, N.I.; Luger, A.-L.; Steinbach, J.P.; Ronellenfitsch, M.W. Second Generation mTOR Inhibitors as a Double-Edged Sword in Malignant Glioma Treatment. Int. J. Mol. Sci. 2019, 20, 4474. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, N.; Lopez, A.; Cuello, V.; Persans, M.; Schuenzel, E.; Innis-Whitehouse, W.; Keniry, M. NVP-BEZ235 or JAKi Treatment leads to decreased survival of examined GBM and BBC cells. Cancer Treat. Res. Commun. 2021, 27, 100340. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachmann, S.M.; Hofmann, I.; Schnell, C.; Fritsch, C.; Wee, S.; Lane, H.; Wang, S.; Garcia-Echeverria, C.; Maira, S.-M. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 22299–22304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Wang, N.; Wang, X.; Zhang, Y.; Lin, S.; Mao, G.; Liu, D.; Dang, C.; Zhou, Z. A glycolysis-related gene signature predicts prognosis of patients with esophageal adenocarcinoma. Aging 2020, 12, 25828–25844. [Google Scholar] [CrossRef]
- Masui, K.; Tanaka, K.; Akhavan, D.; Babic, I.; Gini, B.; Matsutani, T.; Iwanami, A.; Liu, F.; Villa, G.R.; Gu, Y.; et al. mTOR Complex 2 Controls Glycolytic Metabolism in Glioblastoma through FoxO Acetylation and Upregulation of c-Myc. Cell Metab. 2013, 18, 726–739. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.D.V.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Pérès, S.; Paulevé, L.; Nogueira, M.L.; Steyaert, J.-M.; Schwartz, L. The Redox Status of Cancer Cells Supports Mechanisms behind the Warburg Effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology 2016, 18, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Shi, Y.; Li, Y.; He, Z.-C.; Zhou, K.; Zhang, X.-N.; Yang, K.-D.; Wu, J.-R.; Kung, H.-F.; Ping, Y.-F.; et al. A glycolysis-based ten-gene signature correlates with the clinical outcome, molecular subtype and IDH1 mutation in glioblastoma. J. Genet. Genom. 2017, 44, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Colardo, M.; Segatto, M.; Di Bartolomeo, S. Targeting RTK-PI3K-mTOR Axis in Gliomas: An Update. Int. J. Mol. Sci. 2021, 22, 4899. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, Z.; Pei, H.; Wang, H.; Li, L.; Xia, Q. FoxO3a induces temozolomide resistance in glioblastoma cells via the regulation of β-catenin nuclear accumulation. Oncol. Rep. 2017, 37, 2391–2397. [Google Scholar] [CrossRef] [Green Version]
- Paik, J.-H.; Kollipara, R.; Chu, G.; Ji, H.; Xiao, Y.; Ding, Z.; Miao, L.; Tothova, Z.; Horner, J.W.; Carrasco, D.R.; et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Regulate Endothelial Cell Homeostasis. Cell 2007, 128, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Link, W.; Fernandez-Marcos, P.J. FOXO transcription factors at the interface of metabolism and cancer. Int. J. Cancer 2017, 141, 2379–2391. [Google Scholar] [CrossRef] [Green Version]
- Di, H.; Zhang, X.; Guo, Y.; Shi, Y.; Fang, C.; Yuan, Y.; Wang, J.; Shang, C.; Guo, W.; Li, C. Silencing LDHA inhibits proliferation, induces apoptosis and increases chemosensitivity to temozolomide in glioma cells. Oncol. Lett. 2018, 15, 5131–5136. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, R.; Furuichi, K.; Yamaguchi, M.; Miyazaki, N.; Aoki, H.; Chibana, H.; Ito, K.; Aoki, S. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Sci. Rep. 2019, 9, 18699. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Wang, Y.; Chen, X.; Shi, Y.; Wang, J.; Zhang, J.; Qian, J.; Li, R.; Tao, T.; Wei, W.; et al. PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop. Oncotarget 2015, 6, 13006–13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsi, L.; Mescola, A.; Alessandrini, A. Glutamate Receptors and Glioblastoma Multiforme: An Old "Route" for New Perspectives. Int. J. Mol. Sci. 2019, 20, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottahedeh, J.; Haffner, M.C.; Grogan, T.R.; Hashimoto, T.; Crowell, P.D.; Beltran, H.; Sboner, A.; Bareja, R.; Esopi, D.; Isaacs, W.B.; et al. CD38 is methylated in prostate cancer and regulates extracellular NAD+. Cancer Metab. 2018, 6, 13. [Google Scholar] [CrossRef]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar] [PubMed]
- Ansari, K.I.; Ogawa, D.; Rooj, A.K.; Lawler, S.E.; Krichevsky, A.M.; Johnson, M.D.; Chiocca, E.A.; Bronisz, A.; Godlewski, J. Glucose-Based Regulation of miR-451/AMPK Signaling Depends on the OCT1 Transcription Factor. Cell Rep. 2015, 11, 902–909. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene | Correlation Coefficient (R) | p-Value |
|---|---|---|---|
| LDHA | ENO1 | 0.56 | <0.05 |
| LDHA | GAPDH | 0.64 | <0.05 |
| ENO1 | GAPDH | 0.55 | <0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udawant, S.; Litif, C.; Lopez, A.; Gunn, B.; Schuenzel, E.; Keniry, M. PI3K Pathway Inhibition with NVP-BEZ235 Hinders Glycolytic Metabolism in Glioblastoma Multiforme Cells. Cells 2021, 10, 3065. https://doi.org/10.3390/cells10113065
Udawant S, Litif C, Lopez A, Gunn B, Schuenzel E, Keniry M. PI3K Pathway Inhibition with NVP-BEZ235 Hinders Glycolytic Metabolism in Glioblastoma Multiforme Cells. Cells. 2021; 10(11):3065. https://doi.org/10.3390/cells10113065
Chicago/Turabian StyleUdawant, Shreya, Carl Litif, Alma Lopez, Bonnie Gunn, Erin Schuenzel, and Megan Keniry. 2021. "PI3K Pathway Inhibition with NVP-BEZ235 Hinders Glycolytic Metabolism in Glioblastoma Multiforme Cells" Cells 10, no. 11: 3065. https://doi.org/10.3390/cells10113065
APA StyleUdawant, S., Litif, C., Lopez, A., Gunn, B., Schuenzel, E., & Keniry, M. (2021). PI3K Pathway Inhibition with NVP-BEZ235 Hinders Glycolytic Metabolism in Glioblastoma Multiforme Cells. Cells, 10(11), 3065. https://doi.org/10.3390/cells10113065