STAT6 Deficiency Attenuates Myeloid Fibroblast Activation and Macrophage Polarization in Experimental Folic Acid Nephropathy


 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Antibodies and Reagents
2.3. Immunohistochemistry
2.4. Immunofluorescence
2.5. Quantitative Real-Time RT-PCR
2.6. Western Blotting
2.7. Assessment of Renal Fibrosis
2.8. Measurement of Kidney Function
2.9. Statistical Analysis
3. Results
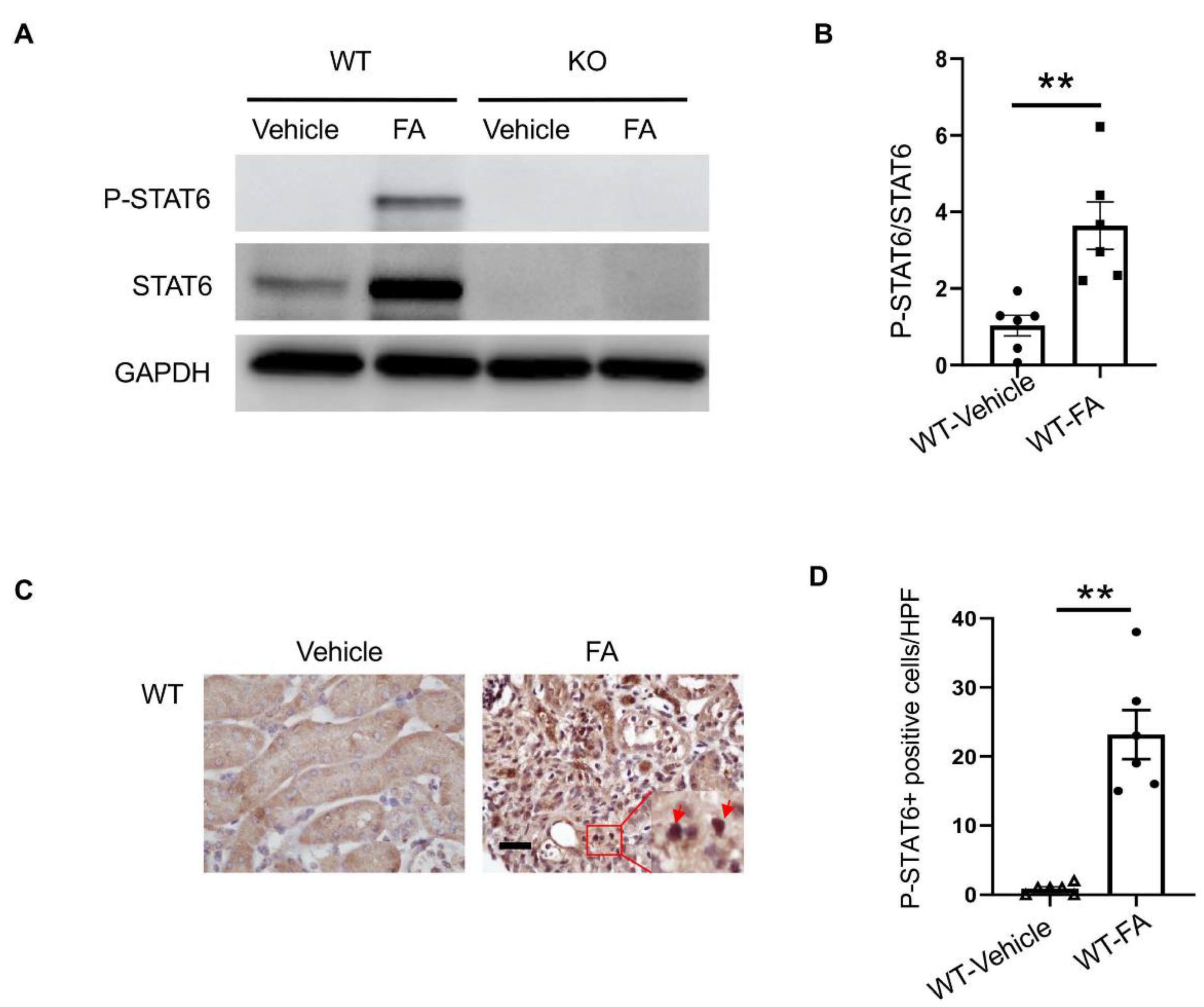
3.1. STAT6 Is Activated in the Kidney with Folic Acid Nephropathy
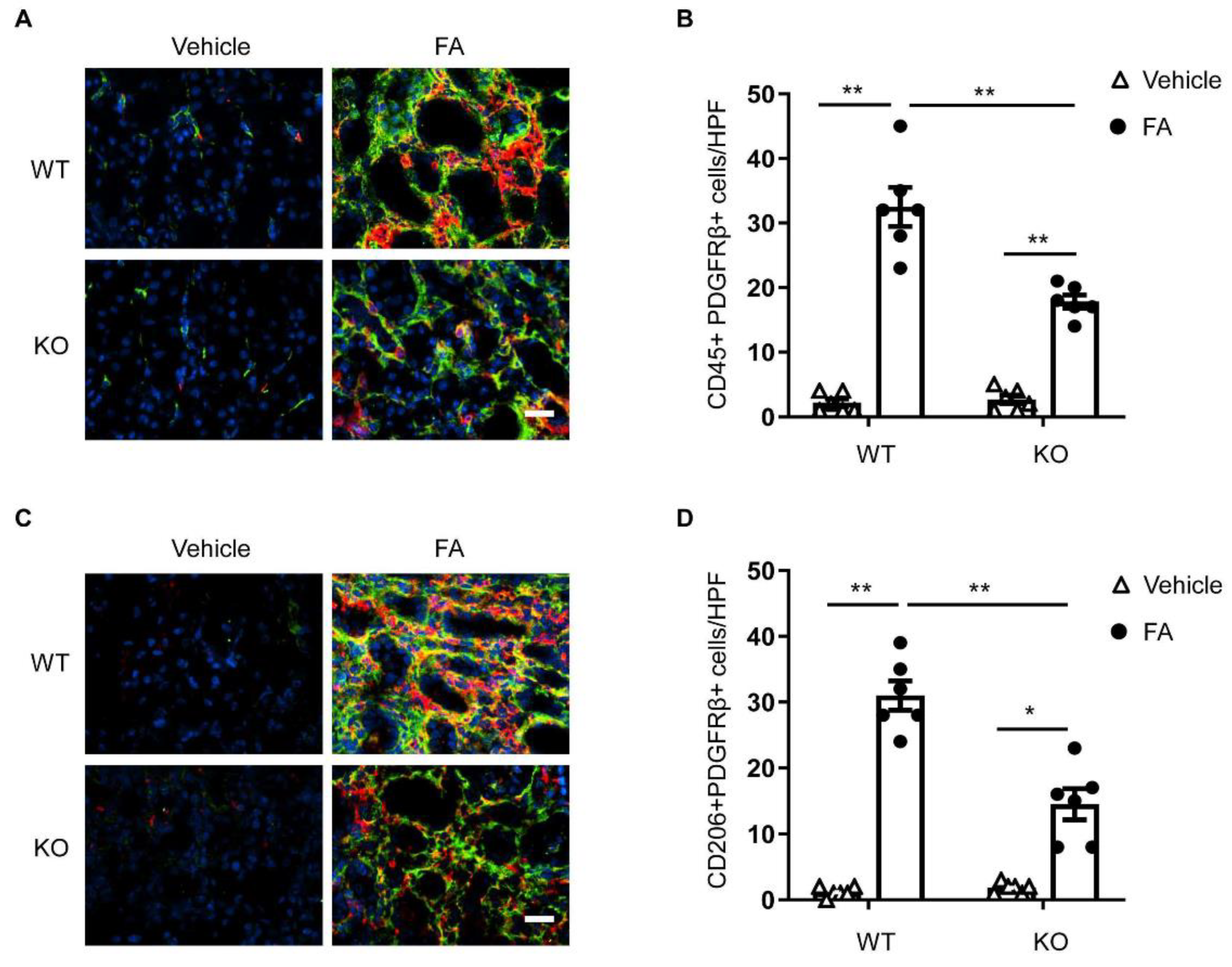
3.2. STAT6 Deficiency Impairs Myeloid Fibroblast Accumulation
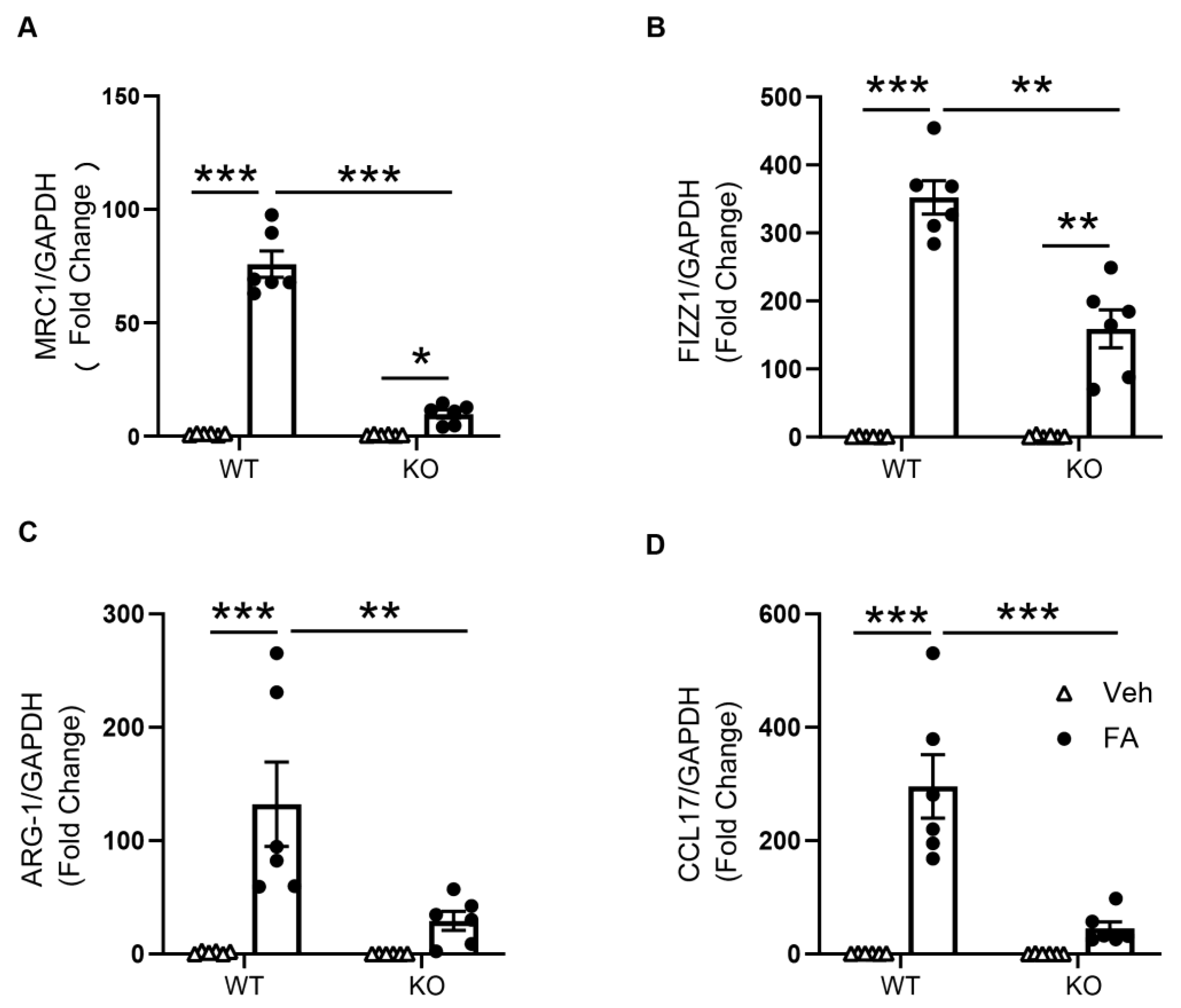
3.3. STAT6 Deficiency Attenuates M2 Macrophage Polarization
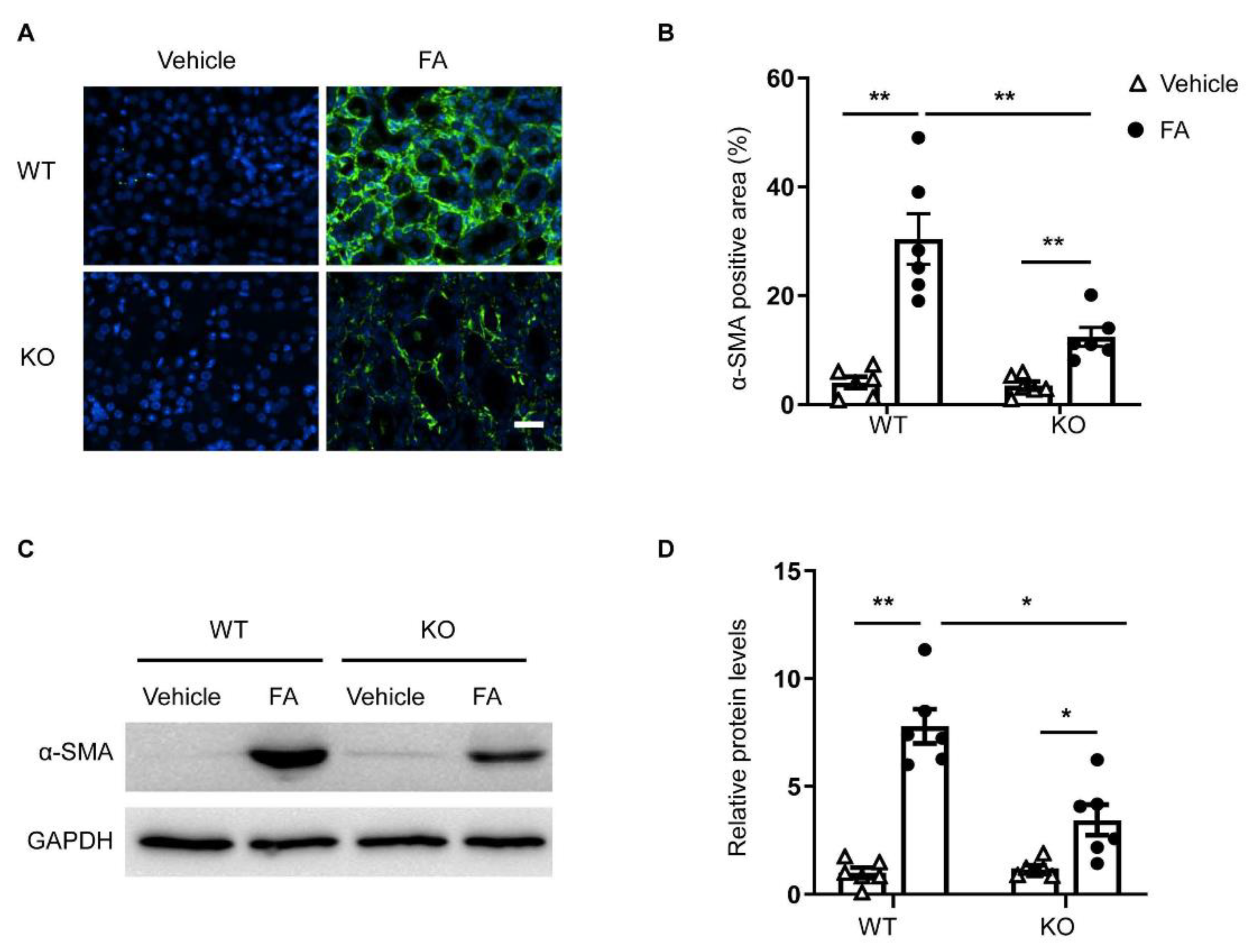
3.4. STAT6 Deficiency Inhibits Myofibroblast Transformation
3.5. STAT6 Deficiency Attenuates ECM Protein Production
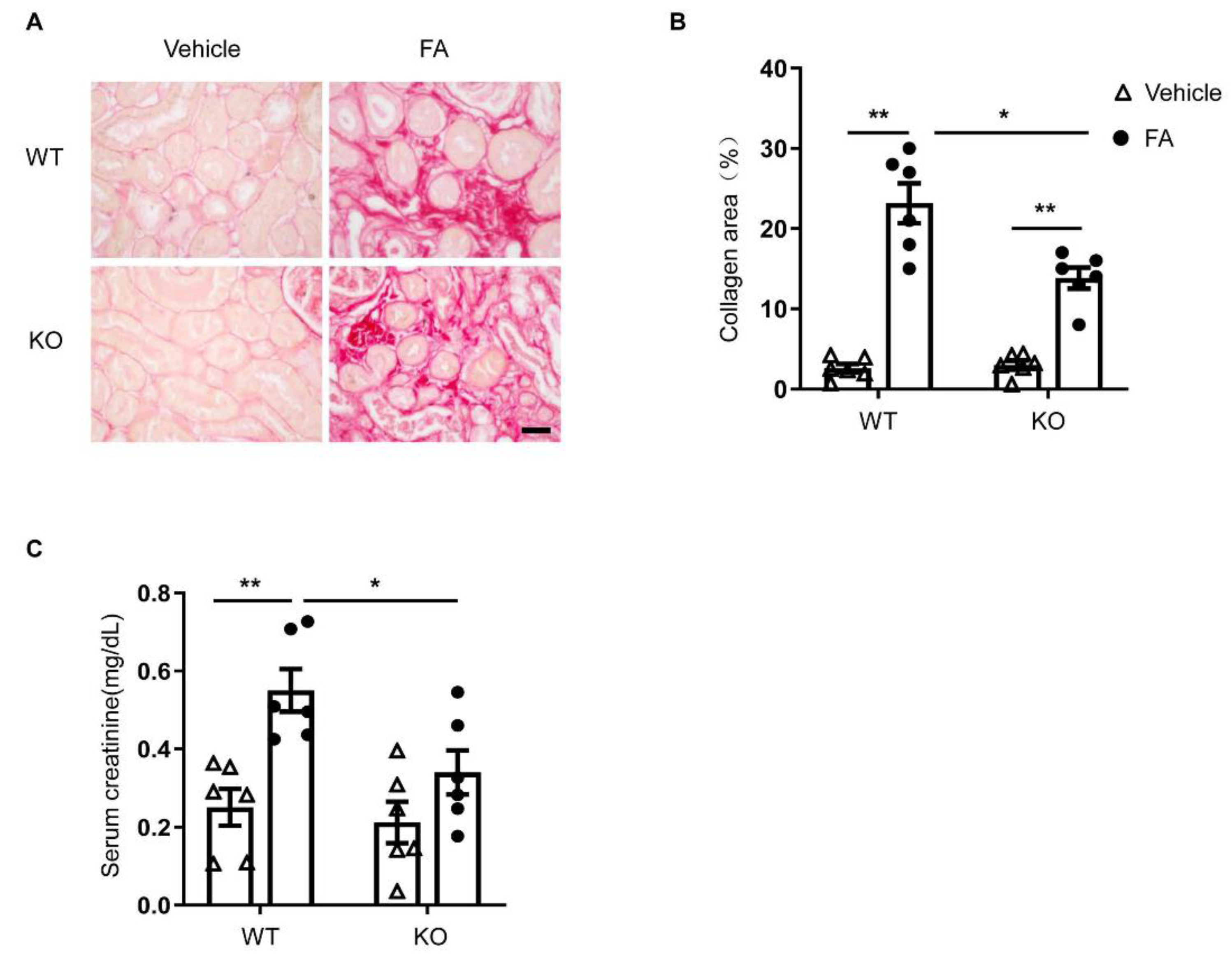
3.6. STAT6 Deficiency Ameliorates Renal Fibrosis and Protects Renal Function
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weiner, D.E. Public health consequences of chronic kidney disease. Clin. Pharmacol. Ther. 2009, 86, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Kottgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Entman, M.L.; Wang, Y. CCR2 Regulates the Uptake of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. PLoS ONE 2013, 8, e77493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab. Invest. 2014, 94, 545–556. [Google Scholar] [CrossRef] [Green Version]
- An, C.; Jia, L.; Wen, J.; Wang, Y. Targeting Bone Marrow-Derived Fibroblasts for Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 305–322. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T.; Yokoyama, H.; Lipp, M.; Ueha, S.; Matsushima, K.; Kaneko, S. Secondary lymphoid tissue chemokine (SLC/CCL21)/CCR7 signaling regulates fibrocytes in renal fibrosis. Proc. Natl. Acad. Sci. USA 2006, 103, 14098–14103. [Google Scholar] [CrossRef] [Green Version]
- Niedermeier, M.; Reich, B.; Rodriguez Gomez, M.; Denzel, A.; Schmidbauer, K.; Gobel, N.; Talke, Y.; Schweda, F.; Mack, M. CD4+ T cells control the differentiation of Gr1+ monocytes into fibrocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 17892–17897. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lin, S.C.; Chen, G.; He, L.; Hu, Z.; Chan, L.; Trial, J.; Entman, M.L.; Wang, Y. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1644–1659. [Google Scholar] [CrossRef] [Green Version]
- Blakaj, A.; Bucala, R. Fibrocytes in health and disease. Fibrogenesis Tissue Repair 2012, 5, S6. [Google Scholar] [CrossRef] [Green Version]
- An, C.; Jiao, B.; Du, H.; Tran, M.; Zhou, D.; Wang, Y. Myeloid PTEN deficiency aggravates renal inflammation and fibrosis in angiotensin II-induced hypertension. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Z.; Yang, J.; Mitch, W.E.; Wang, Y. JAK3/STAT6 Stimulates Bone Marrow-Derived Fibroblast Activation in Renal Fibrosis. J. Am. Soc. Nephrol. 2015, 26, 3060–3071. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Lin, S.C.; Chen, J.; He, L.; Dong, F.; Xu, J.; Han, S.; Du, J.; Entman, M.L.; Wang, Y. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1876–1886. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; An, C.; Jin, X.; Hu, Z.; Wang, Y. Disruption of CXCR6 Ameliorates Kidney Inflammation and Fibrosis in Deoxycorticosterone Acetate/Salt Hypertension. Sci. Rep. 2020, 10, 133. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jin, H.; Ullenbruch, M.; Hu, B.; Hashimoto, N.; Moore, B.; McKenzie, A.; Lukacs, N.W.; Phan, S.H. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: Role of IL-4/IL-13 and mediation via STAT-6. J. Immunol. 2004, 173, 3425–3431. [Google Scholar] [CrossRef] [Green Version]
- Walford, H.H.; Doherty, T.A. STAT6 and lung inflammation. Jak-Stat. 2013, 2, e25301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, H.; Zhang, Z.; Yan, J.; Wang, Y.; Hu, Z.; Mitch, W.E.; Wang, Y. The IL-4 receptor alpha has a critical role in bone marrow-derived fibroblast activation and renal fibrosis. Kidney Int. 2017, 92, 1433–1443. [Google Scholar] [CrossRef]
- Jiao, B.; An, C.; Tran, M.; Du, H.; Wang, P.; Zhou, D.; Wang, Y. Pharmacological Inhibition of STAT6 Ameliorates Myeloid Fibroblast Activation and Alternative Macrophage Polarization in Renal Fibrosis. Front. Immunol. 2021, 12, 735014. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.Y.; Wang, X.; Vijayan, S.; Tang, Y.; Kim, Y.O.; Padberg, K.; Regen, T.; Molokanova, O.; Chen, T.; Bopp, T.; et al. IL-4 Receptor Alpha Signaling through Macrophages Differentially Regulates Liver Fibrosis Progression and Reversal. EBioMedicine 2018, 29, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Ma, Q.; Zhu, Z.D.; Li, G.; Wang, Y.; Li, Q.Q.; Chen, Y.F.; Shang, Z.Z.; Zhang, J.; Zhao, L. Mechanism of Corilagin interference with IL-13/STAT6 signaling pathways in hepatic alternative activation macrophages in schistosomiasis-induced liver fibrosis in mouse model. Eur. J. Pharm. 2016, 793, 119–126. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Entman, M.L.; Wang, Y. Critical Role of CXCL16 in Hypertensive Kidney Injury and Fibrosis. Hypertension 2013, 62, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Ma, Z.; Peng, H.; He, L.; Hu, Z.; Wang, Y. CXCL16 Deficiency Attenuates Renal Injury and Fibrosis in Salt-Sensitive Hypertension. Sci. Rep. 2016, 6, 28715. [Google Scholar] [CrossRef]
- Ma, Z.; Jin, X.; He, L.; Wang, Y. CXCL16 regulates renal injury and fibrosis in experimental renal artery stenosis. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H815–H821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jia, L.; Hu, Z.; Entman, M.L.; Mitch, W.E.; Wang, Y. AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int. 2018, 93, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; An, C.; Jiao, B.; Safirstein, R.L.; Wang, Y. AMP-activated protein kinase contributes to cisplatin-induced renal epithelial cell apoptosis and acute kidney injury. Am. J. Physiol. Ren. Physiol. 2020, 319, F1073–F1080. [Google Scholar] [CrossRef]
- Jin, X.; Chen, J.; Hu, Z.; Chan, L.; Wang, Y. Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int. 2013, 83, 604–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Jia, L.; Hu, Z.; Wang, Y. Pharmacological Inhibition of PTEN Aggravates Acute Kidney Injury. Sci. Rep. 2017, 7, 9503. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef]
- Tampe, D.; Zeisberg, M. Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef]
- Farris, A.B.; Colvin, R.B. Renal interstitial fibrosis: Mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens 2012, 21, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Reich, B.; Schmidbauer, K.; Rodriguez Gomez, M.; Johannes Hermann, F.; Gobel, N.; Bruhl, H.; Ketelsen, I.; Talke, Y.; Mack, M. Fibrocytes develop outside the kidney but contribute to renal fibrosis in a mouse model. Kidney Int. 2013, 84, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhang, Z.; Jia, L.; Wang, Y. Role of Bone Marrow-Derived Fibroblasts in Renal Fibrosis. Front. Physiol. 2016, 7, 61. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yan, J.; Jin, X.; Entman, M.L.; Wang, Y. The chemokine receptor CXCR6 contributes to recruitment of bone marrow-derived fibroblast precursors in renal fibrosis. Kidney Int. 2014, 86, 327–337. [Google Scholar] [CrossRef] [Green Version]
- An, C.; Wen, J.; Hu, Z.; Mitch, W.E.; Wang, Y. Phosphoinositide 3-kinase gamma deficiency attenuates kidney injury and fibrosis in angiotensin II-induced hypertension. Nephrol. Dial. Transpl. 2020, 35, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Huang, B. The multi-differentiation potential of peripheral blood mononuclear cells. Stem Cell Res. Ther. 2012, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Wurster, A.L.; Tanaka, T.; Grusby, M.J. The biology of Stat4 and Stat6. Oncogene 2000, 19, 2577–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goenka, S.; Kaplan, M.H. Transcriptional regulation by STAT6. Immunol. Res. 2011, 50, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.J.; Ip, S.; Teh, S.J.; Wong, C.; Jirik, F.R.; Grusby, M.J.; Teh, H.S. A role for T helper 2 cells in mediating skin fibrosis in tight-skin mice. Cell. Immunol. 1999, 196, 60–68. [Google Scholar] [CrossRef]
- Gordon, S.; Pluddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yang, T.; Li, L.; Sun, L.; Hou, Y.; Hu, X.; Zhang, L.; Tian, H.; Zhao, Q.; Peng, J.; et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 2014, 5, 4696. [Google Scholar] [CrossRef]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Muller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef]
- Vernon, M.A.; Mylonas, K.J.; Hughes, J. Macrophages and renal fibrosis. Semin. Nephrol. 2010, 30, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. 2014, 4, 34–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, B.; An, C.; Du, H.; Tran, M.; Wang, P.; Zhou, D.; Wang, Y. STAT6 Deficiency Attenuates Myeloid Fibroblast Activation and Macrophage Polarization in Experimental Folic Acid Nephropathy. Cells 2021, 10, 3057. https://doi.org/10.3390/cells10113057
Jiao B, An C, Du H, Tran M, Wang P, Zhou D, Wang Y. STAT6 Deficiency Attenuates Myeloid Fibroblast Activation and Macrophage Polarization in Experimental Folic Acid Nephropathy. Cells. 2021; 10(11):3057. https://doi.org/10.3390/cells10113057
Chicago/Turabian StyleJiao, Baihai, Changlong An, Hao Du, Melanie Tran, Penghua Wang, Dong Zhou, and Yanlin Wang. 2021. "STAT6 Deficiency Attenuates Myeloid Fibroblast Activation and Macrophage Polarization in Experimental Folic Acid Nephropathy" Cells 10, no. 11: 3057. https://doi.org/10.3390/cells10113057
APA StyleJiao, B., An, C., Du, H., Tran, M., Wang, P., Zhou, D., & Wang, Y. (2021). STAT6 Deficiency Attenuates Myeloid Fibroblast Activation and Macrophage Polarization in Experimental Folic Acid Nephropathy. Cells, 10(11), 3057. https://doi.org/10.3390/cells10113057