
Cell-to-Cell Interactions Mediating Functional Recovery after Stroke

 ,
, 
 , and
, and 
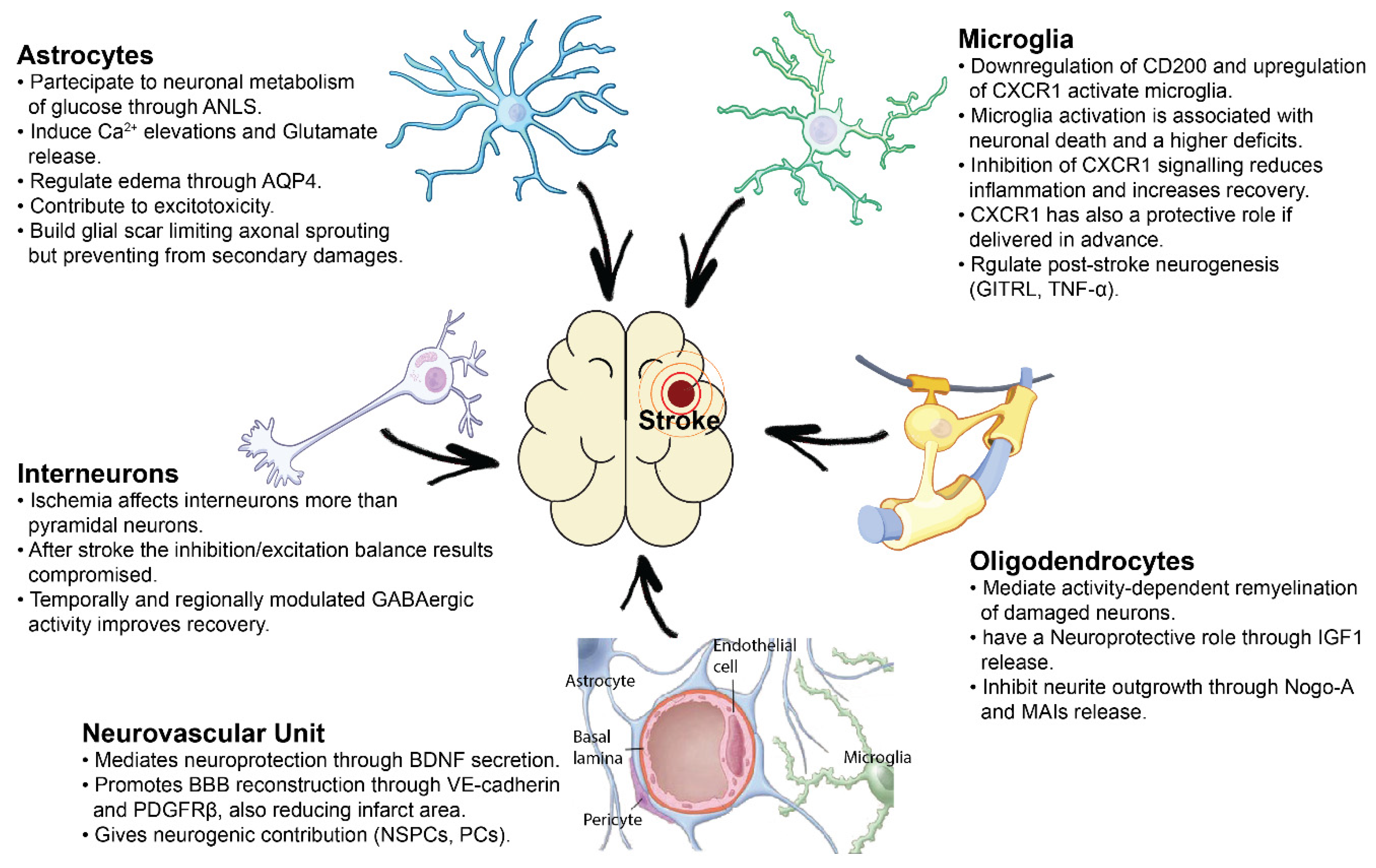{kind=link}
Abstract
1. Introduction
2. Cellular and Molecular Support from the Neurovascular Unit after Ischemia
3. Pathophysiological Role of Astrocytes and Reactive Gliosis after Brain Ischemia
4. Role of Oligodendrocytes in Neural Support and Sprouting Inhibition
5. Microglial-Mediated Inflammation in Stroke: A Double-Edged Sword
6. Pyramidal and GABA-Ergic Neural Interactions in Post-Stroke Plasticity
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wafa, H.A.; Wolfe, C.D.A.; Emmett, E.; Roth, G.A.; Johnson, C.O.; Wang, Y. Burden of Stroke in Europe. Stroke 2020, 51, 2418–2427. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Timmis, A.; Townsend, N.; Gale, C.P.; Torbica, A.; Lettino, M.; Petersen, S.E.; Mossialos, E.A.; Maggioni, A.P.; Kazakiewicz, D.; May, H.T.; et al. European society of cardiology: Cardiovascular disease statistics 2019. Eur. Heart J. 2020, 41, 12–85. [Google Scholar] [CrossRef]
- Bernhardt, J.; English, C.; Johnson, L.; Cumming, T.B. Early Mobilization after Stroke: Early Adoption but Limited Evidence. Stroke 2015, 07, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Furuichi, Y.; Katsuta, K.; Maeda, M.; Ueyama, N.; Moriguchi, A.; Matsuoka, N.; Goto, T.; Yanagihara, T. Neuroprotective action of tacrolimus (FK506) in focal and global cerebral ischemia in rodents: Dose dependency, therapeutic time window and long-term efficacy. Brain Res. 2003, 965, 137–145. [Google Scholar] [CrossRef]
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.J.; Teves, L.; Tymianski, M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nat. Cell Biol. 2012, 483, 213–217. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, X.; Liu, T.; Shao, J.; Fu, N.; Yan, A.; Geng, K.; Xia, W. Adjudin-preconditioned neural stem cells enhance neuroprotection after ischemia reperfusion in mice. Stem Cell Res. Ther. 2017, 8, 1–22. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, D.; Huber, M.; Shi, J.; An, H.; Wei, W.; He, X.; Ding, Y.; Ji, X. New Endovascular Approach for Hypothermia With Intrajugular Cooling and Neuroprotective Effect in Ischemic Stroke. Stroke 2020, 51, 628–636. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Halsey, A.; Wang, M.X.; Tornroth-Horsefield, S.; Conner, A.C.; Badaut, J.; Iliff, J.J.; Bill, R.M. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain 2021. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef] [PubMed]
- Sylvain, N.J.; Salman, M.M.; Pushie, M.J.; Hou, H.; Meher, V.; Herlo, R.; Peeling, L.; Kelly, M.E. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim. Biophys. Acta-Biomembr. 2021, 1863, 183573. [Google Scholar] [CrossRef]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke a guideline for healthcare professionals from the American Heart Association/American Stroke A. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Armstead, W.M.; Raghupathi, R. Endothelin and the neurovascular unit in pediatric traumatic brain injury. Neurol. Res. 2011, 33, 127–132. [Google Scholar] [CrossRef][Green Version]
- Abbott, N.J.; Friedman, A. Overview and introduction: The blood-brain barrier in health and disease. Epilepsia 2012, 53, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakamura, H.; Kishima, H. Therapeutic strategy against ischemic stroke with the concept of neurovascular unit. Neurochem. Int. 2019, 126, 246–251. [Google Scholar] [CrossRef]
- Guo, S.; Kim, W.J.; Lok, J.; Lee, S.R.; Besancon, E.; Luo, B.H.; Stins, M.F.; Wang, X.; Dedhar, S.; Lo, E.H. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 7582–7587. [Google Scholar] [CrossRef]
- Ozaki, T.; Muramatsu, R.; Sasai, M.; Yamamoto, M.; Kubota, Y.; Fujinaka, T.; Yoshimine, T.; Yamashita, T. The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci. Rep. 2016, 6, 25893. [Google Scholar] [CrossRef]
- Nakano-Doi, A.; Sakuma, R.; Matsuyama, T.; Nakagomi, T. Ischemic stroke activates the VE-cadherin promoter and increases VE-cadherin expression in adult mice. Histol. Histopathol. 2018, 33, 507–521. [Google Scholar] [CrossRef]
- Arimura, K.; Ago, T.; Kamouchi, M.; Nakamura, K.; Ishitsuka, K.; Kuroda, J.; Sugimori, H.; Ooboshi, H.; Sasaki, T.; Kitazono, T. PDGF Receptor {\&}{\#}946; Signaling in Pericytes Following Ischemic Brain Injury. Curr. Neurovasc. Res. 2012, 9, 1–9. [Google Scholar] [CrossRef]
- Shen, J.; Ishii, Y.; Xu, G.; Dang, T.C.; Hamashima, T.; Matsushima, T.; Yamamoto, S.; Hattori, Y.; Takatsuru, Y.; Nabekura, J.; et al. PDGFR-Β as a positive regulator of tissue repair in a mouse model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2012, 32, 353–367. [Google Scholar] [CrossRef]
- Makihara, N.; Arimura, K.; Ago, T.; Tachibana, M.; Nishimura, A.; Nakamura, K.; Matsuo, R.; Wakisaka, Y.; Kuroda, J.; Sugimori, H.; et al. Involvement of platelet-derived growth factor receptor $β$ in fibrosis through extracellular matrix protein production after ischemic stroke. Exp. Neurol. 2015, 264, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A pericyte origin of spinal cord scar tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia Enhances the Generation of Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef]
- Dar, A.; Domev, H.; Ben-Yosef, O.; Tzukerman, M.; Zeevi-Levin, N.; Novak, A.; Germanguz, I.; Amit, M.; Itskovitz-Eldor, J. Multipotent vasculogenic pericytes from human pluripotent stem cells promote recovery of murine ischemic limb. Circulation 2012, 125, 87–99. [Google Scholar] [CrossRef]
- Klein, D.; Weißhardt, P.; Kleff, V.; Jastrow, H.; Jakob, H.G.; Ergün, S. Vascular wall-resident CD44+ multipotent stem cells give rise to pericytes and smooth muscle cells and contribute to new vessel maturation. PLoS ONE 2011, 6, e20540. [Google Scholar] [CrossRef] [PubMed]
- Dore-Duffy, P.; Katychev, A.; Wang, X.; Van Buren, E. CNS microvascular pericytes exhibit multipotential stem cell activity. J. Cereb. Blood Flow Metab. 2006, 26, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Olson, J.D.; Mintz, A.; Delbono, O. Type-2 pericytes participate in normal and tumoral angiogenesis. Am. J. Physiol. Cell Physiol. 2014, 307, C25–C38. [Google Scholar] [CrossRef]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Enikolopov, G.N.; Mintz, A.; Delbono, O. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res. 2013, 10, 67–84. [Google Scholar] [CrossRef]
- Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Sakuma, R.; Lu, S.; Narita, A.; Kawahara, M.; Taguchi, A.; Matsuyama, T. Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells 2015, 33, 1962–1974. [Google Scholar] [CrossRef]
- Palmer, T.D.; Willhoite, A.R.; Gage, F.H. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 2000, 425, 479–494. [Google Scholar] [CrossRef]
- Louissaint, A.; Rao, S.; Leventhal, C.; Goldman, S.A. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 2002, 34, 945–960. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Dickinson-Anson, H.; Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996, 16, 2027–2033. [Google Scholar] [CrossRef]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef]
- Teng, H.; Zhang, Z.G.; Wang, L.; Zhang, R.L.; Zhang, L.; Morris, D.; Gregg, S.R.; Wu, Z.; Jiang, A.; Lu, M.; et al. Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J. Cereb. Blood Flow Metab. 2008, 28, 764–771. [Google Scholar] [CrossRef]
- Nakagomi, T.; Taguchi, A.; Fujimori, Y.; Saino, O.; Nakano-Doi, A.; Kubo, S.; Gotoh, A.; Soma, T.; Yoshikawa, H.; Nishizaki, T.; et al. Isolation and characterization of neural stem/progenitor cells from post-stroke cerebral cortex in mice. Eur. J. Neurosci. 2009, 29, 1842–1852. [Google Scholar] [CrossRef]
- Nakagomi, N.; Nakagomi, T.; Kubo, S.; Nakano-Doi, A.; Saino, O.; Takata, M.; Yoshikawa, H.; Stern, D.M.; Matsuyama, T.; Taguchi, A. Endothelial cells support survival, proliferation, and neuronal differentiation of transplanted adult ischemia-induced neural stem/progenitor cells after cerebral infarction. Stem Cells 2009, 27, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Doi, A.; Nakagomi, T.; Fujikawa, M.; Nakagomi, N.; Kubo, S.; Lu, S.; Yoshikawa, H.; Soma, T.; Taguchi, A.; Matsuyama, T. Bone marrow mononuclear cells promote proliferation of endogenous neural stem cells through vascular niches after cerebral infarction. Stem Cells 2010, 28, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Ago, T.; Wakisaka, Y.; Kuroda, J.; Shijo, M.; Yoshikawa, Y.; Komori, M.; Nishimura, A.; Makihara, N.; Nakamura, K.; et al. Early Reperfusion after Brain Ischemia Has Beneficial Effects beyond Rescuing Neurons. Stroke 2017, 48, 2222–2230. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakagomi, N.; Doe, N.; Nakano-Doi, A.; Sawano, T.; Takagi, T.; Matsuyama, T.; Yoshimura, S.; Nakagomi, T. Early Reperfusion Following Ischemic Stroke Provides Beneficial Effects, Even After Lethal Ischemia with Mature Neural Cell Death. Cells 2020, 9, 1374. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Tanaka, Y.; Nakagomi, N.; Matsuyama, T.; Yoshimura, S. How long are reperfusion therapies beneficial for patients after stroke onset? Lessons from lethal ischemia following early reperfusion in a mouse model of stroke. Int. J. Mol. Sci. 2020, 21, 6360. [Google Scholar] [CrossRef]
- Sokoloff, L.; Reivich, M.; Kennedy, C.; Des Rosiers, M.; Patlak, C.; Pettigrew, K.; Sakurada, O.; Shinohara, M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: Theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 1977, 28, 897–916. [Google Scholar] [CrossRef]
- Raichle, M.E.; Mintun, M.A. Brain Work and Brain Imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Ring, A.; Sonnewald, U.; Waagepetersen, H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009, 109, 87–93. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of astroglia. Adv. Exp. Med. Biol. 2019, 1175, 45–91. [Google Scholar]
- Zonta, M.; Angulo, M.; Gobbo, S.; Rosengarten, B.; Hossmann, K.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef]
- Huang, Y.H.; Bergles, D.E. Glutamate transporters bring competition to the synapse. Curr. Opin. Neurobiol. 2004, 14, 346–352. [Google Scholar] [CrossRef]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef]
- Wang, X.; Lou, N.; Xu, Q.; Tian, G.F.; Peng, W.G.; Han, X.; Kang, J.; Takano, T.; Nedergaard, M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006, 9, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.D.; Kafitz, K.W.; Rose, C.R. Developmental profile and mechanisms of GABA-induced calcium signaling in hippocampal astrocytes. Glia 2008, 56, 1127–1137. [Google Scholar] [CrossRef]
- Haydon, P.G. Glia: Listening and talking to the synapse. Nat. Rev. Neurosci. 2001, 2, 185–193. [Google Scholar] [CrossRef]
- Lia, A.; Henriques, V.; Zonta, M.; Chiavegato, A.; Carmignoto, G.; Gómez-Gonzalo, M.; Losi, G. Calcium Signals in Astrocyte Microdomains, a Decade of Great Advances. Front. Cell. Neurosci. 2021, 15, 177. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Zhang, Z.; Tao, Z.; Braams, S.; Rauen, T. Glutamate forward and reverse transport: From molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 2008, 60, 609–619. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Ding, S. Ca2+ Signaling in Astrocytes and its Role in Ischemic Stroke. Adv. Neurobiol. 2014, 11, 189–211. [Google Scholar] [PubMed]
- Dong, Q.P.; He, J.Q.; Chai, Z. Astrocytic Ca2+ waves mediate activation of extrasynaptic NMDA receptors in hippocampal neurons to aggravate brain damage during ischemia. Neurobiol. Dis. 2013, 58, 68–75. [Google Scholar] [CrossRef]
- Ding, S.; Wang, T.; Cui, W.; Haydon, P.G. Photothrombosis ischemia stimulates as sustained astrocytic Ca2+ signaling in vivo. Glia 2009, 57, 767–776. [Google Scholar] [CrossRef]
- Kitchen, P.; Day, R.E.; Salman, M.M.; Conner, M.T.; Bill, R.M.; Conner, A.C. Beyond water homeostasis: Diverse functional roles of mammalian aquaporins. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 2410–2421. [Google Scholar] [CrossRef]
- Zelenina, M. Regulation of brain aquaporins. Neurochem. Int. 2010, 57, 468–488. [Google Scholar] [CrossRef] [PubMed]
- Oshio, K.; Binder, D.K.; Yang, B.; Schecter, S.; Verkman, A.S.; Manley, G.T. Expression of aquaporin water channels in mouse spinal cord. Neuroscience 2004, 127, 685–693. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef]
- Barreto, G.E.; Sun, X.; Xu, L.; Giffard, R.G. Astrocyte proliferation following stroke in the mouse depends on distance from the infarct. PLoS ONE 2011, 6, e27881. [Google Scholar] [CrossRef]
- Mestriner, R.G.; Pagnussat, A.S.; Boisserand, L.S.B.; Valentim, L.; Netto, C.A. Skilled reaching training promotes astroglial changes and facilitated sensorimotor recovery after collagenase-induced intracerebral hemorrhage. Exp. Neurol. 2011, 227, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.C.; Scheibe, J.; Glocke, I.; Weise, G.; Exp, A.D.-A.N. undefined 2013 Object-based analysis of astroglial reaction and astrocyte subtype morphology after ischemic brain injury. Acta Neurobiol. Exp. 2013, 73, 79–87. [Google Scholar]
- Li, H.; Zhang, N.; Lin, H.Y.; Yu, Y.; Cai, Q.Y.; Ma, L.; Ding, S. Histological, cellular and behavioral assessments of stroke outcomes after photothrombosis-induced ischemia in adult mice. BMC Neurosci. 2014, 15, 58. [Google Scholar] [CrossRef]
- McKeon, R.J.; Schreiber, R.C.; Rudge, J.S.; Silver, J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J. Neurosci. 1991, 11, 3398–3411. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, Y.; Cui, Y.; Roberts, C.; Lu, M.; Wilhelmsson, U.; Pekny, M.; Chopp, M. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia 2014, 62, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, Y.; Li, S. Molecular mechanisms of scar-sourced axon growth inhibitors. Brain Res. 2015, 1619, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Overman, J.J.; Clarkson, A.; Wanner, I.B.; Overman, W.T.; Eckstein, I.; Maguire, J.L.; Dinov, I.; Toga, A.W.; Carmichael, S.T. PNAS Plus: A role for ephrin-A5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc. Natl. Acad. Sci. USA 2012, 109, E2230–E2239. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Nakano, T.; Irie, K.; Higuchi, S.; Fujioka, M.; Orito, K.; Iwasaki, K.; Jin, G.; Lo, E.H.; Mishima, K.; et al. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2010, 30, 871–882. [Google Scholar] [CrossRef]
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M. V Reactive Astrocytes Protect Tissue and Preserve Function after Spinal Cord Injury. J. Neurosci. 2004, 24, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Höftberger, R.; Lassmann, H. Inflammatory demyelinating diseases of the central nervous system. Handb. Clin. Neurol. 2018, 145, 285–299. [Google Scholar]
- Pantoni, L.; Garcia, J.H. Pathogenesis of leukoaraiosis: A review. Stroke 1997, 28, 652–659. [Google Scholar] [CrossRef]
- Medana, I.M.; Esiri, M.M. Axonal damage: A key predictor of outcome in human CNS diseases. Brain 2003, 126, 515–530. [Google Scholar] [CrossRef]
- Zhang, R.; Chopp, M.; Zhang, Z.G. Oligodendrogenesis after cerebral ischemia. Front. Cell. Neurosci. 2013, 7, 1–7. [Google Scholar] [CrossRef]
- Kang, S.H.; Fukaya, M.; Yang, J.K.; Rothstein, J.D.; Bergles, D.E. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 2010, 68, 668–681. [Google Scholar] [CrossRef]
- Mandai, K.; Matsumoto, M.; Kitagawa, K.; Matsushita, K.; Ohtsuki, T.; Mabuchi, T.; Colman, D.R.; Kamada, T.; Yanagihara, T. Ischemic damage and subsequent proliferation of oligodendrocytes in focal cerebral ischemia. Neuroscience 1997, 77, 849–861. [Google Scholar] [CrossRef]
- Bain, J.M.; Moore, L.; Ren, Z.; Simonishvili, S.; Levison, S.W. Vascular Endothelial Growth Factors A and C are Induced in the SVZ Following Neonatal Hypoxia-Ischemia and Exert Different Effects on Neonatal Glial Progenitors. Transl. Stroke Res. 2013, 4, 158–170. [Google Scholar] [CrossRef]
- Kim, H.J.; Chuang, D.M. HDAC inhibitors mitigate ischemia-induced oligodendrocyte damage: Potential roles of oligodendrogenesis, VEGF, and anti-inflammation. Am. J. Transl. Res. 2014, 6, 206. [Google Scholar] [PubMed]
- Zhang, L.; Chopp, M.; Zhang, R.L.; Wang, L.; Zhang, J.; Wang, Y.; Toh, Y.; Santra, M.; Lu, M.; Zhang, Z.G. Erythropoietin amplifies stroke-induced oligodendrogenesis in the rat. PLoS ONE 2010, 5, e11016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Chopp, M.; Roberts, C.; Wei, M.; Wang, X.; Liu, X.; Lu, M.; Zhang, Z.G. Sildenafil Enhances Neurogenesis and Oligodendrogenesis in Ischemic Brain of Middle-Aged Mouse. PLoS ONE 2012, 7, e48141. [Google Scholar] [CrossRef]
- Mount, C.W.; Monje, M. Wrapped to Adapt: Experience-Dependent Myelination. Neuron 2017, 95, 743–756. [Google Scholar] [CrossRef]
- Nagy, B.; Hovhannisyan, A.; Barzan, R.; Chen, T.J.; Kukley, M. Different patterns of neuronal activity trigger distinct responses of oligodendrocyte precursor cells in the corpus callosum. PLoS Biol. 2017, 15, e2001993. [Google Scholar] [CrossRef]
- Chen, T.J.; Kula, B.; Nagy, B.; Barzan, R.; Gall, A.; Ehrlich, I.; Kukley, M. In Vivo Regulation of Oligodendrocyte Precursor Cell Proliferation and Differentiation by the AMPA-Receptor Subunit GluA2. Cell Rep. 2018, 25, 852–861. [Google Scholar] [CrossRef]
- Ortiz, F.C.; Habermacher, C.; Graciarena, M.; Houry, P.Y.; Nishiyama, A.; Oumesmar, B.N.; Angulo, M.C. Neuronal activity in vivo enhances functional myelin repair. JCI Insight 2019, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef] [PubMed]
- Lobsiger, C.S.; Smith, P.M.; Buchstaller, J.; Schweitzer, B.; Franklin, R.J.M.; Suter, U.; Taylor, V. A role for oligodendrocyte-derived IGF-1 in trophic support of cortical neurons. Glia 2001, 36, 48–57. [Google Scholar] [CrossRef]
- Caroni, P.; Schwab, M.E. Antibody against myelin associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1988, 1, 85–96. [Google Scholar] [CrossRef]
- Atwal, J.K.; Pinkston-Gosse, J.; Syken, J.; Stawicki, S.; Wu, Y.; Shatz, C.; Tessier-Lavigne, M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 2008, 322, 967–970. [Google Scholar] [CrossRef]
- Wang, K.C.; Koprivica, V.; Kim, J.A.; Sivasankaran, R.; Guo, Y.; Neve, R.L.; He, Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 2002, 417, 941–944. [Google Scholar] [CrossRef]
- Kempf, A.; Tews, B.; Arzt, M.; Weinmann, O.; Obermair, F.; Pernet, V.; Zagrebelsky, M.; Delekate, A.; Iobbi, C.; Zemmar, A.; et al. The sphingolipid receptor S1PR2 is a receptor for Nogo-a repressing synaptic plasticity. PLoS Biol. 2014, 12, e1001763. [Google Scholar] [CrossRef]
- Papadopoulos, C.M.; Tsai, S.Y.; Alsbiei, T.; O’Brien, T.E.; Schwab, M.E.; Kartje, G.L. Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann. Neurol. 2002, 51, 433–441. [Google Scholar] [CrossRef]
- Seymour, A.B.; Andrews, E.M.; Tsai, S.Y.; Markus, T.M.; Bollnow, M.R.; Brenneman, M.M.; O’Brien, T.E.; Castro, A.J.; Schwab, M.E.; Kartje, G.L. Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J. Cereb. Blood Flow Metab. 2005, 25, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.M.; Tsai, S.-Y.; Cheatwood, J.L.; Bollnow, M.R.; Kolb, B.E.; Schwab, M.E.; Kartje, G.L. Dendritic Plasticity in the Adult Rat Following Middle Cerebral Artery Occlusion and Nogo-A Neutralization. Cereb. Cortex 2006, 16, 529–536. [Google Scholar] [CrossRef]
- Zhan, H.; Sun, S.J.; Cai, J.; Li, Y.Q.; Hu, C.L.; Lee, D.H.S.; So, K.F.; Li, X. The effect of an NgR1 antagonist on the neuroprotection of cortical axons after cortical infarction in rats. Neurochem. Res. 2013, 38, 1333–1340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fournier, A.E.; Gould, G.C.; Liu, B.P.; Strittmatter, S.M. Truncated soluble Nogo receptor binds Nogo-66 and blocks inhibition of axon growth by myelin. J. Neurosci. 2002, 22, 8876–8883. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, J.E.; Sivula, M.; Strittmatter, S.M. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J. Neurosci. 2004, 24, 6209–6217. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.S.; Omlor, W.; Rubio, J.C.; Chen, J.L.; Zheng, H.; Schröter, A.; Gullo, M.; Weinmann, O.; Kobayashi, K.; Helmchen, F.; et al. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science 2014, 344, 1250–1255. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Abrams, C.K.; Scherer, S.S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 2008, 35, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J. Neurosci. Res. 2010, 88, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Aoyama, M.; Kakita, H.; Hida, H.; Kato, I.; Ito, T.; Goto, T.; Hussein, M.H.; Sawamoto, K.; Togari, H.; et al. Endogenous erythropoietin from astrocyte protects the oligodendrocyte precursor cell against hypoxic and reoxygenation injury. J. Neurosci. Res. 2011, 89, 1566–1574. [Google Scholar] [CrossRef]
- Gonzalez, F.F.; Larpthaveesarp, A.; McQuillen, P.; Derugin, N.; Wendland, M.; Spadafora, R.; Ferriero, D.M. Erythropoietin Increases neurogenesis and oligodendrogliosis of subventricular zone precursor cells after neonatal stroke. Stroke 2013, 44, 753–758. [Google Scholar] [CrossRef]
- Miyamoto, N.; Maki, T.; Shindo, A.; Liang, A.C.; Maeda, M.; Egawa, N.; Itoh, K.; Lo, E.K.; Lok, J.; Ihara, M.; et al. Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J. Neurosci. 2015, 35, 14002–14008. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Cejudo, J.; Gutiérrez-Fernández, M.; Otero-Ortega, L.; Rodríguez-Frutos, B.; Fuentes, B.; Vallejo-Cremades, M.T.; Hernanz, T.N.; Cerdán, S.; Díez-Tejedor, E. Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 2015, 46, 221–228. [Google Scholar] [CrossRef]
- Rowe, D.D.; Collier, L.A.; Seifert, H.A.; Chapman, C.B.; Leonardo, C.C.; Willing, A.E.; Pennypacker, K.R. Leukemia inhibitor factor promotes functional recovery and oligodendrocyte survival in rat models of focal ischemia. Eur. J. Neurosci. 2014, 40, 3111–3119. [Google Scholar] [CrossRef]
- Arai, K.; Lo, E.H. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci. 2009, 29, 4351–4355. [Google Scholar] [CrossRef]
- Maki, T.; Morancho, A.; Segundo, P.M.S.; Hayakawa, K.; Takase, H.; Liang, A.C.; Gabriel-Salazar, M.; Medina-Gutiérrez, E.; Washida, K.; Montaner, J.; et al. Endothelial progenitor cell secretome and oligovascular repair in a mouse model of prolonged cerebral hypoperfusion. Stroke 2018, 49, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Kishida, N.; Maki, T.; Takagi, Y.; Yasuda, K.; Kinoshita, H.; Ayaki, T.; Noro, T.; Kinoshita, Y.; Ono, Y.; Kataoka, H.; et al. Role of Perivascular Oligodendrocyte Precursor Cells in Angiogenesis After Brain Ischemia. J. Am. Heart Assoc. 2019, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rust, R.; Grönnert, L.; Gantner, C.; Enzler, A.; Mulders, G.; Weber, R.Z.; Siewert, A.; Limasale, Y.D.P.; Meinhardt, A.; Maurer, M.A.; et al. Nogo-A targeted therapy promotes vascular repair and functional recovery following stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 14270–14279. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Lee, S.; Lee, W.-H.; Lee, H.-J.; Suk, K. Stimulation of glucocorticoid-induced tumor necrosis factor receptor family-related protein ligand (GITRL) induces inflammatory activation of microglia in culture. J. Neurosci. Res. 2010, 88, 2188–2196. [Google Scholar] [CrossRef]
- Takata, M.; Nakagomi, T.; Kashiwamura, S.; Nakano-Doi, A.; Saino, O.; Nakagomi, N.; Okamura, H.; Mimura, O.; Taguchi, A.; Matsuyama, T. Glucocorticoid-induced TNF receptor-triggered T cells are key modulators for survival/death of neural stem/progenitor cells induced by ischemic stroke. Cell Death Differ. 2012, 19, 756–767. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kumon, Y.; Watanabe, H.; Ohnishi, T.; Takahashi, H.; Imai, Y.; Tanaka, J. Expression of CD200 by macrophage-like cells in ischemic core of rat brain after transient middle cerebral artery occlusion. Neurosci. Lett. 2007, 418, 44–48. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.-j; Zhang, C.; Chen, R.; Li, L.; He, J.; Xie, Y.; Chen, Y. Loss of neuronal CD200 contributed to microglial activation after acute cerebral ischemia in mice. Neurosci. Lett. 2018, 678, 48–54. [Google Scholar] [CrossRef]
- Ritzel, R.M.; Al Mamun, A.; Crapser, J.; Verma, R.; Patel, A.R.; Knight, B.E.; Harris, N.; Mancini, N.; Roy-O’reilly, M.; Ganesh, B.P.; et al. CD200-CD200R1 inhibitory signaling prevents spontaneous bacterial infection and promotes resolution of neuroinflammation and recovery after stroke. J. Neuroinflamm. 2019, 16, 40. [Google Scholar] [CrossRef]
- Sun, H.; He, X.; Tao, X.; Hou, T.; Chen, M.; He, M.; Liao, H. The CD200/CD200R signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J. Neuroinflamm. 2020, 17, 171. [Google Scholar] [CrossRef]
- Li, Z.; Ye, H.; Cai, X.; Sun, W.; He, B.; Yang, Z.; Xu, P. Bone marrow-mesenchymal stem cells modulate microglial activation in the peri-infarct area in rats during the acute phase of stroke. Brain Res. Bull. 2019, 153, 324–333. [Google Scholar] [CrossRef]
- Kong, T.; Park, J.-M.; Jang, J.H.; Kim, C.-Y.; Bae, S.-H.; Choi, Y.; Jeong, Y.-H.; Kim, C.; Chang, S.W.; Kim, J.; et al. Immunomodulatory effect of CD200-positive human placenta-derived stem cells in the early phase of stroke. Exp. Mol. Med. 2018, 50, e425. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; Mcnamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Dénes, Á.D.; Ferenczi, S.; Halász, J.Z.; Rnyei, Z.K.; Kovács, K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef]
- Fumagalli, S.; Perego, C.; Ortolano, F.; De Simoni, M.G. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia 2013, 61, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dzyubenko, E.; Sanchez-Mendoza, E.H.; Sardari, M.; Silva de Carvalho, T.; Doeppner, T.R.; Kaltwasser, B.; Machado, P.; Kleinschnitz, C.; Bassetti, C.L.; et al. Postacute Delivery of GABAA α5 Antagonist Promotes Postischemic Neurological Recovery and Peri-infarct Brain Remodeling. Stroke 2018, 49, 2495–2503. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Wang, C.; Wang, Q.; Lin, Y.Z.; Ge, Y.S.; Li, D.M.; Mao, G.S. Role of fractalkine/CX3CR1 signaling pathway in the recovery of neurological function after early ischemic stroke in a rat model. Life Sci. 2017, 184, 87–94. [Google Scholar] [CrossRef]
- Tang, Z.; Gan, Y.; Liu, Q.; Yin, J.-X.; Liu, Q.; Shi, J.; Shi, F.-D. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J. Neuroinflamm. 2014, 11, 1–13. [Google Scholar] [CrossRef]
- Soriano, S.G.; Amaravadi, L.S.; Wang, Y.F.; Zhou, H.; Yu, G.X.; Tonra, J.R.; Fairchild-Huntress, V.; Fang, Q.; Dunmore, J.H.; Huszar, D.; et al. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J. Neuroimmunol. 2002, 125, 59–65. [Google Scholar] [CrossRef]
- Lauro, C.; Di Angelantonio, S.; Cipriani, R.; Sobrero, F.; Antonilli, L.; Brusadin, V.; Ragozzino, D.; Limatola, C. Activity of Adenosine Receptors Type 1 Is Required for CX 3 CL1-Mediated Neuroprotection and Neuromodulation in Hippocampal Neurons. J. Immunol. 2008, 180, 7590–7596. [Google Scholar] [CrossRef]
- Lauro, C.; Cipriani, R.; Catalano, M.; Trettel, F.; Chece, G.; Brusadin, V.; Antonilli, L.; Van Rooijen, N.; Eusebi, F.; Fredholm, B.B.; et al. Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against glu-induced death. Neuropsychopharmacology 2010, 35, 1550–1559. [Google Scholar] [CrossRef]
- Cipriani, R.; Villa, P.; Chece, G.; Lauro, C.; Paladini, A.; Micotti, E.; Perego, C.; De Simoni, M.-G.; Fredholm, B.B.; Eusebi, F.; et al. Neurobiology of Disease CX3CL1 Is Neuroprotective in Permanent Focal Cerebral Ischemia in Rodents. J. Neurosci. 2011, 31, 16327–16335. [Google Scholar] [CrossRef] [PubMed]
- Donohue, M.M.; Cain, K.; Zierath, D.; Shibata, D.; Tanzi, P.M.; Becker, K.J. Higher Plasma Fractalkine Is Associated With Better 6-Month Outcome From Ischemic Stroke. Stroke 2012, 43, 2300–2306. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Boyd, J.D.; Murphy, T.H. Longitudinal in vivo Imaging Reveals Balanced and Branch-Specific Remodeling of Mature Cortical Pyramidal Dendritic Arbors after Stroke. J. Cereb. Blood Flow Metab. 2010, 30, 783–791. [Google Scholar] [CrossRef]
- Allegra Mascaro, A.L.; Conti, E.; Lai, S.; Di Giovanna, A.P.; Spalletti, C.; Alia, C.; Panarese, A.; Scaglione, A.; Sacconi, L.; Micera, S.; et al. Combined Rehabilitation Promotes the Recovery of Structural and Functional Features of Healthy Neuronal Networks after Stroke. Cell Rep. 2019, 28, 3474–3485.e6. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chen, S.; Wu, Y.; Murphy, T.H. Prolonged Deficits in Parvalbumin Neuron Stimulation-Evoked Network Activity Despite Recovery of Dendritic Structure and Excitability in the Somatosensory Cortex following Global Ischemia in Mice. J. Neurosci. 2014, 34, 14890–14900. [Google Scholar] [CrossRef]
- Alia, C.; Spalletti, C.; Lai, S.; Panarese, A.; Lamola, G.; Bertolucci, F.; Vallone, F.; Di Garbo, A.; Chisari, C.; Micera, S.; et al. Neuroplastic Changes Following Brain Ischemia and their Contribution to Stroke Recovery: Novel Approaches in Neurorehabilitation. Front. Cell. Neurosci. 2017, 11, 76. [Google Scholar] [CrossRef]
- Povysheva, N.; Nigam, A.; Brisbin, A.K.; Johnson, J.W.; Barrionuevo, G. Oxygen–Glucose Deprivation Differentially Affects Neocortical Pyramidal Neurons and Parvalbumin-Positive Interneurons. Neuroscience 2019, 412, 72–82. [Google Scholar] [CrossRef]
- Wang, J.H. Short-term cerebral ischemia causes the dysfunction of interneurons and more excitation of pyramidal neurons in rats. Brain Res. Bull. 2003, 60, 53–58. [Google Scholar] [CrossRef]
- Hu, H.; Gan, J.; Jonas, P. Fast-spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science 2014, 345, 1255263. [Google Scholar] [CrossRef]
- Kann, O.; Papageorgiou, I.E.; Draguhn, A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J. Cereb. Blood Flow Metab. 2014, 34, 1270–1282. [Google Scholar] [CrossRef]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef]
- Huchzermeyer, C.; Berndt, N.; Holzhütter, H.G.; Kann, O. Oxygen consumption rates during three different neuronal activity states in the hippocampal CA3 network. J. Cereb. Blood Flow Metab. 2013, 33, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Hájos, N.; Ellender, T.J.; Zemankovics, R.; Mann, E.O.; Exley, R.; Cragg, S.J.; Freund, T.F.; Paulsen, O. Maintaining network activity in submerged hippocampal slices: Importance of oxygen supply. Eur. J. Neurosci. 2009, 29, 319–327. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Kulak, A.; Kraftsik, R.; Chen, Y.; Dalton, T.P.; Cuenod, M.; Do, K.Q. Redox dysregulation affects the ventral but not dorsal hippocampus: Impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J. Neurosci. 2010, 30, 2547–2558. [Google Scholar] [CrossRef]
- Spalletti, C.; Alia, C.; Lai, S.; Panarese, A.; Conti, S.; Micera, S.; Caleo, M. Combining robotic training and inactivation of the healthy hemisphere restores pre-stroke motor patterns in mice. eLife 2017, 6, e28662. [Google Scholar] [CrossRef] [PubMed]
- Frahm, C.; Haupt, C.; Witte, O. GABA neurons survive focal ischemic injury. Neuroscience 2004, 127, 341–346. [Google Scholar] [CrossRef]
- Balbi, M.; Xiao, D.; Jativa Vega, M.; Hu, H.; Vanni, M.P.; Bernier, L.P.; LeDue, J.; MacVicar, B.; Murphy, T.H. Gamma frequency activation of inhibitory neurons in the acute phase after stroke attenuates vascular and behavioral dysfunction. Cell Rep. 2021, 34, 108696. [Google Scholar] [CrossRef]
- Wang, Y.; Galeffi, F.; Wang, W.; Li, X.; Lu, L.; Sheng, H.; Hoffmann, U.; Turner, D.A.; Yang, W. Chemogenetics-mediated acute inhibition of excitatory neuronal activity improves stroke outcome. Exp. Neurol. 2020, 326, 113206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, W.; Mamtilahun, M.; Song, Y.; Ma, Y.; Qu, M.; Lu, Y.; He, X.; Zheng, J.; Fu, Z.; et al. Optogenetic inhibition of Striatal GABAergic neuronal activity improves outcomes after ischemic brain injury. Stroke 2017, 48, 3375–3383. [Google Scholar] [CrossRef]
- Schiene, K.; Bruehl, C.; Zilles, K.; Qü, M.; Hagemann, G.; Kraemer, M.; Witte, O.W. Neuronal hyperexcitability and reduction of GABA(A)-receptor expression in the surround of cerebral photothrombosis. J. Cereb. Blood Flow Metab. 1996, 16, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Hiu, T.; Farzampour, Z.; Paz, J.T.; Wang, E.H.J.; Badgely, C.; Olson, A.; Micheva, K.D.; Wang, G.; Lemmens, R.; Tran, K.V.; et al. Enhanced phasic GABA inhibition during the repair phase of stroke: A novel therapeutic target. Brain 2016, 139, 468–480. [Google Scholar] [CrossRef]
- Lazar, R.M.; Berman, M.F.; Festa, J.R.; Geller, A.E.; Matejovsky, T.G.; Marshall, R.S. GABAergic but not anti-cholinergic agents re-induce clinical deficits after stroke. J. Neurol. Sci. 2010, 292, 72–76. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef]
- Alia, C.; Spalletti, C.; Lai, S.; Panarese, A.; Micera, S.; Caleo, M. Reducing GABA A-mediated inhibition improves forelimb motor function after focal cortical stroke in mice. Sci. Rep. 2016, 6, 37823. [Google Scholar] [CrossRef]
- Jaenisch, N.; Liebmann, L.; Guenther, M.; Hübner, C.A.; Frahm, C.; Witte, O.W. Reduced tonic inhibition after stroke promotes motor performance and epileptic seizures. Sci. Rep. 2016, 6, 26173. [Google Scholar] [CrossRef]
- Chabriat, H.; Bassetti, C.L.; Marx, U.; Audoli-Inthavong, M.L.; Sors, A.; Lambert, E.; Wattez, M.; Hermann, D.M.; ALTHAUS, K.; AMARO, S.; et al. Safety and efficacy of GABAA α5 antagonist S44819 in patients with ischaemic stroke: A multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2020, 19, 226–233. [Google Scholar] [CrossRef]
- Bocchi, R.; Götz, M. Neuronal Reprogramming for Brain Repair: Challenges and Perspectives. Trends Mol. Med. 2020, 26, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D Self-Organized Microvascular Model of the Human Blood-Brain Barrier with Endothelial Cells, Pericytes and Astrocytes. Biomaterials 2018, 180, 117. [Google Scholar] [CrossRef]
- Wang, S.N.; Wang, Z.; Xu, T.Y.; Cheng, M.H.; Li, W.L.; Miao, C.Y. Cerebral Organoids Repair Ischemic Stroke Brain Injury. Transl. Stroke Res. 2020, 11, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- Wevers, N.R.; Kasi, D.G.; Gray, T.; Wilschut, K.J.; Smith, B.; van Vught, R.; Shimizu, F.; Sano, Y.; Kanda, T.; Marsh, G.; et al. A perfused human blood–brain barrier on-a-chip for high-throughput assessment of barrier function and antibody transport. Fluids Barriers CNS 2018, 15, 23. [Google Scholar] [CrossRef]
- Salman, M.M.; Marsh, G.; Kusters, I.; Delincé, M.; Di Caprio, G.; Upadhyayula, S.; de Nola, G.; Hunt, R.; Ohashi, K.G.; Gray, T.; et al. Design and Validation of a Human Brain Endothelial Microvessel-on-a-Chip Open Microfluidic Model Enabling Advanced Optical Imaging. Front. Bioeng. Biotechnol. 2020, 8, 1077. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alia, C.; Cangi, D.; Massa, V.; Salluzzo, M.; Vignozzi, L.; Caleo, M.; Spalletti, C. Cell-to-Cell Interactions Mediating Functional Recovery after Stroke. Cells 2021, 10, 3050. https://doi.org/10.3390/cells10113050
Alia C, Cangi D, Massa V, Salluzzo M, Vignozzi L, Caleo M, Spalletti C. Cell-to-Cell Interactions Mediating Functional Recovery after Stroke. Cells. 2021; 10(11):3050. https://doi.org/10.3390/cells10113050
Chicago/Turabian StyleAlia, Claudia, Daniele Cangi, Verediana Massa, Marco Salluzzo, Livia Vignozzi, Matteo Caleo, and Cristina Spalletti. 2021. "Cell-to-Cell Interactions Mediating Functional Recovery after Stroke" Cells 10, no. 11: 3050. https://doi.org/10.3390/cells10113050
APA StyleAlia, C., Cangi, D., Massa, V., Salluzzo, M., Vignozzi, L., Caleo, M., & Spalletti, C. (2021). Cell-to-Cell Interactions Mediating Functional Recovery after Stroke. Cells, 10(11), 3050. https://doi.org/10.3390/cells10113050