
Therapeutic Implications of miRNAs for Muscle-Wasting Conditions



Abstract

1. Introduction
2. The Biosynthesis of miRNAs
3. MyomiRs Regulate Crucial Events in the Muscular Niche
4. Implication of miRNAs in Muscle-Wasting Conditions
5. Duchenne Muscular Dystrophy
6. Becker Muscular Dystrophy
7. Myotonic Dystrophy
8. Limb-Girdle Muscular Dystrophy
9. Facioscapulohumeral Muscular Dystrophy
10. Cancer Cachexia
11. Sarcopenia
12. Therapeutic Options
13. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Boyer, O.; Butler-Browne, G.; Chinoy, H.; Cossu, G.; Galli, F.; Lilleker, J.B.; Magli, A.; Mouly, V.; Perlingeiro, R.C.R.; Previtali, S.C.; et al. Myogenic Cell Transplantation in Genetic and Acquired Diseases of Skeletal Muscle. Front. Genet. 2021, 12, 1226. [Google Scholar] [CrossRef]
- Cassano, M.; Quattrocelli, M.; Crippa, S.; Perini, I.; Ronzoni, F.; Sampaolesi, M. Cellular mechanisms and local progenitor activation to regulate skeletal muscle mass. J. Muscle Res. Cell Motil. 2009, 30, 243–253. [Google Scholar] [CrossRef]
- Brzeszczyńska, J.; Brzeszczyński, F.; Hamilton, D.F.; McGregor, R.; Simpson, A.H.R.W. Role of microRNA in muscle regeneration and diseases related to muscle dysfunction in atrophy, cachexia, osteoporosis, and osteoarthritis. Bone Jt. Res. 2020, 9, 798–807. [Google Scholar] [CrossRef]
- Gu, K.; Mok, L.; Chong, M.M. Regulating gene expression in animals through RNA endonucleolytic cleavage. Heliyon 2018, 4, e00908. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.-P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Giarratana, N.; Conti, F.; La Rovere, R.; Gijsbers, R.; Carai, P.; Duelen, R.; Vervliet, T.; Bultynck, G.; Ronzoni, F.; Piciotti, R.; et al. MICAL2 is essential for myogenic lineage commitment. Cell Death Dis. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Ronzoni, F.L.; Giarratana, N.; Crippa, S.; Quattrocelli, M.; Cassano, M.; Ceccarelli, G.; Benedetti, L.; Van Herck, J.; De Angelis, M.G.C.; Vitale, M.; et al. Guide Cells Support Muscle Regeneration and Affect Neuro-Muscular Junction Organization. Int. J. Mol. Sci. 2021, 22, 1939. [Google Scholar] [CrossRef] [PubMed]
- Giacomazzi, G.; Holvoet, B.; Trenson, S.; Caluwé, E.; Kravic, B.; Grosemans, H.; Cortés-Calabuig, Á.; Deroose, C.M.; Huylebroeck, D.; Hashemolhosseini, S.; et al. MicroRNAs promote skeletal muscle differentiation of mesodermal iPSC-derived progenitors. Nat. Commun. 2017, 8, 1249. [Google Scholar] [CrossRef] [PubMed]
- Ronzoni, F.; Ceccarelli, G.; Perini, I.; Benedetti, L.; Galli, D.; Mulas, F.; Balli, M.; Magenes, G.; Bellazzi, R.; De Angelis, G.C.; et al. Met-Activating Genetically Improved Chimeric Factor-1 Promotes Angiogenesis and Hypertrophy in Adult Myogenesis. Curr. Pharm. Biotechnol. 2017, 18, 309–317. [Google Scholar] [CrossRef]
- Pozzo, E.; Giarratana, N.; Sassi, G.; Elmastas, M.; Killian, T.; Wang, C.-C.; Marini, V.; Ronzoni, F.; Yustein, J.; Uyttebroeck, A.; et al. Upregulation of miR181a/miR212 Improves Myogenic Commitment in Murine Fusion-Negative Rhabdomyosarcoma. Front. Physiol. 2021, 12, 1146. [Google Scholar] [CrossRef]
- Cheung, T.H.T.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.R.; Georges, S.A.; Seay, H.; Tapscott, S.J.; McManus, M.; Goldhamer, D.J.; Swanson, M.; Harfe, B.D. Essential role for Dicer during skeletal muscle development. Dev. Biol. 2007, 311, 359–368. [Google Scholar] [CrossRef]
- Hill, M.; Tran, N. miRNA interplay: Mechanisms and consequences in cancer. Dis. Model. Mech. 2021, 14, dmm047662. [Google Scholar] [CrossRef]
- Gregory, R.I.; Yan, K.-P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear Export of MicroRNA Precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Caudy, A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Song, J.-J.; Smith, S.K.; Hannon, G.J.; Joshua-Tor, L. Crystal Structure of Argonaute and Its Implications for RISC Slicer Activity. Science 2004, 305, 1434–1437. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Hutvagner, G.; Du, T.; Xu, Z.; Aronin, N.; Zamore, P.D. Asymmetry in the Assembly of the RNAi Enzyme Complex. Cell 2003, 115, 199–208. [Google Scholar] [CrossRef]
- BioRender.com. microRNA in Cancer. 2021. Available online: https://app.biorender.com/biorender-templates (accessed on 28 October 2021).
- Koutsoulidou, A.; Photiades, M.; Kyriakides, T.C.; Georgiou, K.; Prokopi, M.; Kapnisis, K.; Łusakowska, A.; Nearchou, M.; Christou, Y.; Papadimas, G.K.; et al. Identification of exosomal muscle-specific miRNAs in serum of myotonic dystrophy patients relating to muscle disease progress. Hum. Mol. Genet. 2017, 26, 3285–3302. [Google Scholar] [CrossRef]
- Arroyo, J.; Chevillet, J.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.; Bennett, C.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Burwinkel, B. Distinct AGO1 and AGO2 associated miRNA profiles in human cells and blood plasma. RNA Biol. 2012, 9, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nature 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Röxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of Levels and Cellular Transfer of Circulating Lipoprotein-Bound MicroRNAs. Arter. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef]
- Barone, R.; Macaluso, F.; Sangiorgi, C.; Campanella, C.; Gammazza, A.M.; Moresi, V.; Coletti, D.; De Macario, E.C.; Macario, A.J.; Cappello, F.; et al. Skeletal muscle Heat shock protein 60 increases after endurance training and induces peroxisome proliferator-activated receptor gamma coactivator 1 α1 expression. Sci. Rep. 2016, 6, 19781. [Google Scholar] [CrossRef]
- Marceca, G.P.; Nigita, G.; Calore, F.; Croce, C.M. MicroRNAs in Skeletal Muscle and Hints on Their Potential Role in Muscle Wasting During Cancer Cachexia. Front. Oncol. 2020, 10, 2604. [Google Scholar] [CrossRef]
- McCarthy, J.J. MicroRNA-206: The skeletal muscle-specific myomiR. Biochim. Biophys. Acta Bioenerg. 2008, 1779, 682–691. [Google Scholar] [CrossRef]
- Sempere, L.F.; Freemantle, S.; Pitha-Rowe, I.; Moss, E.; Dmitrovsky, E.; Ambros, V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004, 5, R13. [Google Scholar] [CrossRef]
- Small, E.M.; O’Rourke, J.R.; Moresi, V.; Sutherland, L.B.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. Regulation of PI3-kinase/Akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 2010, 107, 4218–4223. [Google Scholar] [CrossRef]
- van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J.; Olson, E.N. A Family of microRNAs Encoded by Myosin Genes Governs Myosin Expression and Muscle Performance. Dev. Cell 2009, 17, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Torres, F.; Aranega, A.E.; Franco, D. Identification of regulatory elements directing miR-23a–miR-27a–miR-24-2 transcriptional regulation in response to muscle hypertrophic stimuli. Biochim. Biophys. Acta Bioenerg. 2014, 1839, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.L.; Rudnick, M.A. Molecular mechanisms regulating myogenic determination and differentiation. Front. Biosci. J. Virtual Libr. 2000, 5, D750–D767. [Google Scholar] [CrossRef]
- Rao, P.K.; Kumar, R.M.; Farkhondeh, M.; Baskerville, S.; Lodish, H.F. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 2006, 103, 8721–8726. [Google Scholar] [CrossRef]
- Goljanek-Whysall, K.; Pais, H.; Rathjen, T.; Sweetman, D.; Dalmay, T.; Münsterberg, A. Regulation of multiple target genes by miR-1/miR-206 is pivotal for C2C12 myoblast differentiation. J. Cell Sci. 2012, 125, 3590–3600. [Google Scholar] [CrossRef]
- Yin, Z.; Tong, Y.; Zhu, H.; Watsky, M.A. ClC-3 is required for LPA-activated Cl− current activity and fibroblast-to-myofibroblast differentiation. Am. J. Physiol. Physiol. 2008, 294, C535–C542. [Google Scholar] [CrossRef]
- Habas, R.; Kato, Y.; He, X. Wnt/Frizzled Activation of Rho Regulates Vertebrate Gastrulation and Requires a Novel Formin Homology Protein Daam1. Cell 2001, 107, 843–854. [Google Scholar] [CrossRef]
- Kennedy, K.A.; Porter, T.; Mehta, V.; Ryan, S.D.; Price, F.; Peshdary, V.; Karamboulas, C.; Savage, J.; Drysdale, T.A.; Li, S.C.; et al. Retinoic acid enhances skeletal muscle progenitor formation and bypasses inhibition by bone morphogenetic protein 4 but not dominant negative beta-catenin. BMC Biol. 2009, 7, 1–21. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as Alternative Hpo/Mst-like Kinases in the Hippo Kinase Cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef]
- Alteri, A.; De Vito, F.; Messina, G.; Pompili, M.; Calconi, A.; Visca, P.; Mottolese, M.; Presutti, C.; Grossi, M. Cyclin D1 is a major target of miR-206 in cell differentiation and transformation. J. Cell Biol. 2013, 12, 3781–3790. [Google Scholar] [CrossRef]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Li, L.; Sarver, A.L.; Alamgir, S.; Subramanian, S. Downregulation of microRNAs miR-1, -206 and -29 stabilizes PAX3 and CCND2 expression in rhabdomyosarcoma. Lab. Investig. J. Tech. Methods Pathol. 2012, 92, 571–583. [Google Scholar] [CrossRef]
- Winbanks, C.E.; Beyer, C.; Hagg, A.; Qian, H.; Sepulveda, P.V.; Gregorevic, P. miR-206 Represses Hypertrophy of Myogenic Cells but Not Muscle Fibers via Inhibition of HDAC4. PLoS ONE 2013, 8, e73589. [Google Scholar] [CrossRef]
- Koutsoulidou, A.; Kyriakides, T.C.; Papadimas, G.K.; Christou, Y.; Kararizou, E.; Papanicolaou, E.Z.; Phylactou, L.A. Elevated Muscle-Specific miRNAs in Serum of Myotonic Dystrophy Patients Relate to Muscle Disease Progress. PLoS ONE 2015, 10, e0125341. [Google Scholar] [CrossRef] [PubMed]
- Ivey, K.N.; Muth, A.; Arnold, J.; King, F.W.; Yeh, R.-F.; Fish, J.E.; Hsiao, E.C.; Schwartz, R.J.; Conklin, B.R.; Bernstein, H.S.; et al. MicroRNA Regulation of Cell Lineages in Mouse and Human Embryonic Stem Cells. Cell Stem Cell 2008, 2, 219–229. [Google Scholar] [CrossRef]
- Chen, J.-F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.-Z. The Role of MicroRNA-1 and MicroRNA-133 in Skeletal Muscle Proliferation and Differentiation. Nat. Genet. 2005, 38, 228–233. [Google Scholar] [CrossRef]
- Boutz, P.L.; Chawla, G.; Stoilov, P.; Black, D.L. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007, 21, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wu, X.; Ling, Z.; Yuan, L.; Cheng, Y.; Chen, J.; Xiang, C. microRNA133a TargetsFoxl2and Promotes Differentiation of C2C12 into Myogenic Progenitor Cells. DNA Cell Biol. 2015, 34, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Dai, A.; Sun, H.; Fang, T.; Zhang, Q.; Wu, S.; Jiang, Y.; Ding, L.; Yan, G.; Hu, Y. MicroRNA-133b stimulates ovarian estradiol synthesis by targeting Foxl2. FEBS Lett. 2013, 587, 2474–2482. [Google Scholar] [CrossRef]
- Cheng, Z.; Liu, F.; Wang, G.; Li, Y.; Zhang, H.; Li, F. miR-133 is a key negative regulator of CDC42–PAK pathway in gastric cancer. Cell. Signal. 2014, 26, 2667–2673. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Dong, P.; Ma, C.; Mitchelson, K.; Deng, T.; Zhang, L.; Sun, Y.; Feng, X.; Ding, Y.; Lu, X.; et al. MicroRNA-133b is a key promoter of cervical carcinoma development through the activation of the ERK and AKT1 pathways. Oncogene 2011, 31, 4067–4075. [Google Scholar] [CrossRef]
- Qiu, T.; Zhou, X.; Wang, J.; Du, Y.; Xu, J.; Huang, Z.; Zhu, W.; Shu, Y.; Liu, P. MiR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett. 2014, 588, 1168–1177. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, J.; Zhang, L.; Qu, Y.; Li, J.; Yu, B.; Yan, M.; Yu, Y.; Liu, B.; Zhu, Z. MiR-133b is frequently decreased in gastric cancer and its overexpression reduces the metastatic potential of gastric cancer cells. BMC Cancer 2014, 14, 34. [Google Scholar] [CrossRef]
- Boettger, T.; Wüst, S.; Nolte, H.; Braun, T. The miR-206/133b cluster is dispensable for development, survival and regeneration of skeletal muscle. Skelet. Muscle 2014, 4, 23. [Google Scholar] [CrossRef]
- Gan, Z.; Rumsey, J.; Hazen, B.C.; Lai, L.; Leone, T.C.; Vega, R.B.; Xie, H.; Conley, K.E.; Auwerx, J.; Smith, S.R.; et al. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J. Clin. Investig. 2013, 123, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational Suppression of Atrophic Regulators by MicroRNA-23a Integrates Resistance to Skeletal Muscle Atrophy. J. Biol. Chem. 2011, 286, 38456–38465. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.-F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar] [CrossRef]
- Zhang, A.; Li, M.; Wang, B.; Klein, J.D.; Price, S.R.; Wang, X.H. miRNA-23a/27a attenuates muscle atrophy and renal fibrosis through muscle-kidney crosstalk. J. Cachexia Sarcopenia Muscle 2018, 9, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Feron, M.; Guével, L.; Rouger, K.; Dubreil, L.; Arnaud, M.-C.; Ledevin, M.; Megeney, L.; Cherel, Y.; Sakanyan, V. PTEN Contributes to Profound PI3K/Akt Signaling Pathway Deregulation in Dystrophin-Deficient Dog Muscle. Am. J. Pathol. 2009, 174, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nature 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Dalkilic, I.; Kunkel, L.M. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Dhanapal, R.; Saraswathi, T.; Govind, R.N. Cancer cachexia. J. Oral Maxillofac. Pathol. 2011, 15, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Ziaaldini, M.M.; Marzetti, E.; Picca, A.; Murlasits, Z. Biochemical Pathways of Sarcopenia and Their Modulation by Physical Exercise: A Narrative Review. Front. Med. 2017, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Lovering, R.M.; Porter, N.C.; Bloch, R.J. The Muscular Dystrophies: From Genes to Therapies. Phys. Ther. 2005, 85, 1372–1388. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; De Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 1–21. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Mercuri, E.; Bonnemann, C.; Muntoni, F. Comprehensive overview of the clinical and genetic aspects of muscular dystrophies. Lancet 2019, 394, 2025–2038. [Google Scholar] [CrossRef]
- Alexander, M.; Casar, J.C.; Motohashi, N.; Vieira, N.M.; Eisenberg, I.; Marshall, J.L.; Gasperini, M.J.; Lek, A.; Myers, J.A.; Estrella, E.A.; et al. MicroRNA-486–dependent modulation of DOCK3/PTEN/AKT signaling pathways improves muscular dystrophy–associated symptoms. J. Clin. Investig. 2014, 124, 2651–2667. [Google Scholar] [CrossRef]
- Alexander, M.; Kawahara, G.; Motohashi, N.; Casar, J.C.; Eisenberg, I.; Myers, J.A.; Gasperini, M.J.; Estrella, E.A.; Kho, A.T.; Mitsuhashi, S.; et al. MicroRNA-199a is induced in dystrophic muscle and affects WNT signaling, cell proliferation, and myogenic differentiation. Cell Death Differ. 2013, 20, 1194–1208. [Google Scholar] [CrossRef]
- Jeanson-Leh, L.; Lameth, J.; Krimi, S.; Buisset, J.; Amor, F.; Le Guiner, C.; Barthélémy, I.; Servais, L.; Blot, S.; Voit, T.; et al. Serum Profiling Identifies Novel Muscle miRNA and Cardiomyopathy-Related miRNA Biomarkers in Golden Retriever Muscular Dystrophy Dogs and Duchenne Muscular Dystrophy Patients. Am. J. Pathol. 2014, 184, 2885–2898. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zhao, L.; Zhang, D.; Yao, X.; Zhang, H.; Wang, Y.-C.; Wang, X.-Y.; Xia, H.; Yan, J.; et al. Circulating Muscle-specific miRNAs in Duchenne Muscular Dystrophy Patients. Mol. Ther. Nucleic Acids 2014, 3, e177. [Google Scholar] [CrossRef] [PubMed]
- Zaharieva, I.T.; Calissano, M.; Scoto, M.; Preston, M.D.; Cirak, S.; Feng, L.; Collins, J.; Kole, R.; Guglieri, M.; Straub, V.; et al. Dystromirs as Serum Biomarkers for Monitoring the Disease Severity in Duchenne Muscular Dystrophy. PLoS ONE 2013, 8, e80263. [Google Scholar] [CrossRef]
- Liu, J.; Liang, X.; Zhou, D.; Lai, L.; Xiao, L.; Liu, L.; Fu, T.; Kong, Y.; Zhou, Q.; Vega, R.B.; et al. Coupling of mitochondrial function and skeletal muscle fiber type by a miR-499/Fnip1/AMPK circuit. EMBO Mol. Med. 2016, 8, 1212–1228. [Google Scholar] [CrossRef]
- Thomson, D.M. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef]
- Dong, X.; Hui, T.; Chen, J.; Yu, Z.; Ren, D.; Zou, S.; Wang, S.; Fei, E.; Jiao, H.; Lai, X. Metformin Increases Sarcolemma Integrity and Ameliorates Neuromuscular Deficits in a Murine Model of Duchenne Muscular Dystrophy. Front. Physiol. 2021, 12, 642908. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Théret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Göransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKα1 Regulates Macrophage Skewing at the Time of Resolution of Inflammation during Skeletal Muscle Regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Miura, P.; Burt, M.; Boudreault, L.; Khogali, S.; Lunde, J.A.; Renaud, J.-M.; Jasmin, B.J. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum. Mol. Genet. 2011, 20, 3478–3493. [Google Scholar] [CrossRef]
- Zhai, L.; Wu, R.; Han, W.; Zhang, Y.; Zhu, D. miR-127 enhances myogenic cell differentiation by targeting S1PR3. Cell Death Dis. 2017, 8, e2707. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, L.; Jiang, P.; Lu, L.; Chen, X.; Lan, H.Y.; Guttridge, D.C.; Sun, H.; Wang, H. Loss of miR-29 in Myoblasts Contributes to Dystrophic Muscle Pathogenesis. Mol. Ther. 2012, 20, 1222–1233. [Google Scholar] [CrossRef]
- Perkins, K.J.; Davies, K.E. Alternative utrophin mRNAs contribute to phenotypic differences between dystrophin-deficient mice and Duchenne muscular dystrophy. FEBS Lett. 2018, 592, 1856–1869. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Torcinaro, A.; Madaro, L.; Marchetti, L.; Sileno, S.; Beji, S.; Salis, C.; Proietti, D.; Imeneo, G.; Capogrossi, M.C.; et al. Role of miR-200c in Myogenic Differentiation Impairment via p66Shc: Implication in Skeletal Muscle Regeneration of Dystrophic mdx Mice. Oxidative Med. Cell. Longev. 2018, 2018, 4814696. [Google Scholar] [CrossRef]
- Zanotti, S.; Gibertini, S.; Curcio, M.; Savadori, P.; Pasanisi, B.; Morandi, L.; Cornelio, F.; Mantegazza, R.; Mora, M. Opposing roles of miR-21 and miR-29 in the progression of fibrosis in Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 1451–1464. [Google Scholar] [CrossRef]
- Morgoulis, D.; Berenstein, P.; Cazacu, S.; Kazimirsky, G.; Dori, A.; Barnea, E.R.; Brodie, C. sPIF promotes myoblast differentiation and utrophin expression while inhibiting fibrosis in Duchenne muscular dystrophy via the H19/miR-675/let-7 and miR-21 pathways. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P. Dystrophinopathies. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 4th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 1103–1111. [Google Scholar]
- Marozzo, R.; Pegoraro, V.; Angelini, C. MiRNAs, Myostatin, and Muscle MRI Imaging as Biomarkers of Clinical Features in Becker Muscular Dystrophy. Diagnostics 2020, 10, 713. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a Key Modulator of Skeletal Muscle Development and Disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef]
- Singh, G.B.; Cowan, D.B.; Wang, D.-Z. Tiny Regulators of Massive Tissue: MicroRNAs in Skeletal Muscle Development, Myopathies, and Cancer Cachexia. Front. Oncol. 2020, 10, 2647. [Google Scholar] [CrossRef]
- Eisenberg, I.; Alexander, M.S.; Kunkel, L.M. miRNAS in normal and diseased skeletal muscle. J. Cell. Mol. Med. 2008, 13, 2–11. [Google Scholar] [CrossRef]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar] [CrossRef]
- Bachinski, L.L.; Udd, B.; Meola, G.; Sansone, V.; Bassez, G.; Eymard, B.; Thornton, C.A.; Moxley, R.T.; Harper, P.S.; Rogers, M.T.; et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: A single shared haplotype indicates an ancestral founder effect. Am. J. Hum. Genet. 2003, 73, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.-P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Fu, Y.; Pizzuti, A.; Fenwick, R.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; De Jong, P. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P.W. Myotonic Dystrophy Type 2 Caused by a CCTG Expansion in Intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic Dystrophy Mutation: An Unstable CTG Repeat in the 3′ Untranslated region of the Gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Heatwole, C.R.; Johnson, N.E. Myotonic Dystrophy: From Bench to Bedside. Semin. Neurol. 2012, 32, 246–254. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.-I.; Hashido, K. Three novel serum biomarkers, miR-1, miR-133a, and miR-206 for Limb-girdle muscular dystrophy, Facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ. Health Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef]
- Roberts, T.C.; Godfrey, C.; McClorey, G.; Vader, P.; Briggs, D.; Gardiner, C.; Aoki, Y.; Sargent, I.; Morgan, J.E.; Wood, M.J. Extracellular microRNAs are dynamic non-vesicular biomarkers of muscle turnover. Nucleic Acids Res. 2013, 41, 9500–9513. [Google Scholar] [CrossRef]
- Strafella, C.; Campoli, G.; Galota, R.M.; Caputo, V.; Pagliaroli, G.; Carboni, S.; Zampatti, S.; Peconi, C.; Mela, J.; Sancricca, C.; et al. Limb-Girdle Muscular Dystrophies (LGMDs): The Clinical Application of NGS Analysis, a Family Case Report. Front. Neurol. 2019, 10, 619. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: The yield and the pitfalls. Muscle Nerve 2015, 52, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Fanin, M. Calpainopathy. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2017; pp. 1–30. [Google Scholar]
- Mori-Yoshimura, M.; Segawa, K.; Minami, N.; Oya, Y.; Komaki, H.; Nonaka, I.; Nishino, I.; Murata, M. Cardiopulmonary dysfunction in patients with limb-girdle muscular dystrophy 2A. Muscle Nerve 2016, 55, 465–469. [Google Scholar] [CrossRef]
- Okere, A.; Reddy, S.S.; Gupta, S.; Shinnar, M. A Cardiomyopathy in a Patient with Limb Girdle Muscular Dystrophy Type 2A. Circ. Hear. Fail. 2013, 6, e12–e13. [Google Scholar] [CrossRef]
- Pegoraro, V.; Angelini, C. Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies. Genes 2021, 12, 85. [Google Scholar] [CrossRef]
- Statland, J.M.; Tawil, R. Facioscapulohumeral Muscular Dystrophy. Continuum 2016, 22, 1916–1931. [Google Scholar] [PubMed]
- Statland, J.; Tawil, R. Facioscapulohumeral muscular dystrophy. Neurol. Clin. 2014, 32, 721–728. [Google Scholar] [CrossRef] [PubMed]
- van der Maarel, S.M.; Tawil, R.; Tapscott, S.J. Facioscapulohumeral muscular dystrophy and DUX4: Breaking the silence. Trends Mol. Med. 2011, 17, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Portilho, D.M.; Alves, M.R.; Kratassiouk, G.; Roche, S.; Magdinier, F.; De Santana, E.C.; Polesskaya, A.; Harel-Bellan, A.; Mouly, V.; Savino, W.; et al. miRNA Expression in Control and FSHD Fetal Human Muscle Biopsies. PLoS ONE 2015, 10, e0116853. [Google Scholar] [CrossRef] [PubMed]
- Arashiro, P.; Eisenberg, I.; Kho, A.T.; Cerqueira, A.M.P.; Canovas, M.; Silva, H.C.A.; Pavanello, R.C.M.; Verjovski-Almeida, S.; Kunkel, L.M.; Zatz, M. Transcriptional regulation differs in affected facioscapulohumeral muscular dystrophy patients compared to asymptomatic related carriers. Proc. Natl. Acad. Sci. USA 2009, 106, 6220–6225. [Google Scholar] [CrossRef]
- Greco, S.; Perfetti, A.; Fasanaro, P.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Deregulated MicroRNAs in Myotonic Dystrophy Type 2. PLoS ONE 2012, 7, e39732. [Google Scholar] [CrossRef]
- Dmitriev, P.; Barat, A.; Polesskaya, A.; O’Connell, M.J.; Robert, T.; Dessen, P.; Walsh, T.A.; Lazar, V.; Turki, A.; Carnac, G.; et al. Simultaneous miRNA and mRNA transcriptome profiling of human myoblasts reveals a novel set of myogenic differentiation-associated miRNAs and their target genes. BMC Genom. 2013, 14, 265. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.J.; Durick, K.; Weiner, J.A.; Chun, J.; Taylor, S.S. D-AKAP2, a novel protein kinase A anchoring protein with a putative RGS domain. Proc. Natl. Acad. Sci. USA 1997, 94, 11184–11189. [Google Scholar] [CrossRef]
- McNab, F.W.; Rajsbaum, R.; Stoye, J.; O’Garra, A. Tripartite-motif proteins and innate immune regulation. Curr. Opin. Immunol. 2011, 23, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; da Silva, S.P.; da Costa, R.M.; Medeiros, R. The Emerging Role of MicroRNAs and Other Non-Coding RNAs in Cancer Cachexia. Cancers 2020, 12, 1004. [Google Scholar] [CrossRef]
- Lee, D.E.; Brown, J.L.; Rosa-Caldwell, M.E.; Blackwell, T.A.; Perry Jr, R.A.; Brown, L.A.; Khatri, B.; Seo, D.; Bottje, W.G.; Washington, T.A.; et al. Cancer cachexia-induced muscle atrophy: Evidence for alterations in microRNAs important for muscle size. Physiol. Genom. 2017, 49, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Liu, Z. MicroRNA-147 suppresses proliferation, invasion and migration through the AKT/mTOR signaling pathway in breast cancer. Oncol. Lett. 2016, 11, 405–410. [Google Scholar] [CrossRef]
- Li, P.; Xue, W.-J.; Feng, Y.; Mao, Q.-S. MicroRNA-205 functions as a tumor suppressor in colorectal cancer by targeting cAMP responsive element binding protein 1 (CREB1). Am. J. Transl. Res. 2015, 7, 2053–2059. [Google Scholar]
- Jang, S.J.; Choi, I.-S.; Park, G.; Moon, D.-S.; Choi, J.-S.; Nam, M.-H.; Yoon, S.-Y.; Choi, C.H.; Kang, S.-H. MicroRNA-205-5p is upregulated in myelodysplastic syndromes and induces cell proliferation via PTEN suppression. Leuk. Res. 2016, 47, 172–177. [Google Scholar] [CrossRef]
- Narasimhan, A.; Ghosh, S.; Stretch, C.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Small RNAome profiling from human skeletal muscle: Novel miRNAs and their targets associated with cancer cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 405–416. [Google Scholar] [CrossRef] [PubMed]
- van de Worp, W.R.; Schols, A.M.; Dingemans, A.C.; Kamp, C.M.O.D.; Degens, J.H.; Kelders, M.C.; Coort, S.; Woodruff, H.C.; Kratassiouk, G.; Harel-Bellan, A.; et al. Identification of microRNAs in skeletal muscle associated with lung cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 11, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Camargo, R.G.; Ribeiro, H.Q.T.; Geraldo, M.V.; Matos-Neto, E.; Neves, R.X.; Carnevali, L.C.; Donatto, F.F.; Alcântara, P.S.M.; Ottoch, J.P.; Seelaender, M. Cancer Cachexia and MicroRNAs. Mediat. Inflamm. 2015, 2015, 367561. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.; Sun, P.Y.; Davies, K.J.; Grune, T. Sarcopenia—Molecular mechanisms and open questions. Ageing Res. Rev. 2020, 65, 101200. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; De Feudis, M.; Angelino, E.; Belli, R.; Teixeira, M.A.; Zaggia, I.; Tamiso, E.; Raiteri, T.; Scircoli, A.; Ronzoni, F.L.; et al. Both ghrelin deletion and unacylated ghrelin overexpression preserve muscles in aging mice. Aging 2020, 12, 13939–13957. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.M.; Cohn, R.D. TGFβ signaling its role in fibrosis formation and myopathies. Curr. Opin. Rheumatol. 2012, 24, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Yanai, K.; Kaneko, S.; Ishii, H.; Aomatsu, A.; Ito, K.; Hirai, K.; Ookawara, S.; Ishibashi, K.; Morishita, Y. MicroRNAs in Sarcopenia: A Systematic Review. Front. Med. 2020, 7, 180. [Google Scholar] [CrossRef]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef]
- Yin, J.; Qian, Z.; Chen, Y.; Li, Y.; Zhou, X. MicroRNA regulatory networks in the pathogenesis of sarcopenia. J. Cell. Mol. Med. 2020, 24, 4900–4912. [Google Scholar] [CrossRef]
- Ge, Y.; Sun, Y.; Chen, J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol. 2011, 192, 69–81. [Google Scholar] [CrossRef]
- Jia, L.; Li, Y.F.; Wu, G.F.; Song, Z.Y.; Lu, H.Z.; Song, C.C.; Zhang, Q.L.; Zhu, J.Y.; Yang, G.S.; Shi, X.E. MiRNA-199a-3p regulates C2C12 myoblast differentiation through IGF-1/AKT/mTOR signal pathway. Int. J. Mol. Sci. 2013, 15, 296–308. [Google Scholar] [CrossRef]
- Li, G.; Luo, W.; Abdalla, B.A.; Ouyang, H.; Yu, J.; Hu, F.; Nie, Q.; Zhang, X. miRNA-223 upregulated by MYOD inhibits myoblast proliferation by repressing IGF2 and facilitates myoblast differentiation by inhibiting ZEB1. Cell Death Dis. 2017, 8, e3094. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tan, J.; Qi, Q.; Yang, L.; Wang, Y.; Zhang, C.; Hu, L.; Chen, H.; Fang, X. miR-487b-3p Suppresses the Proliferation and Differentiation of Myoblasts by Targeting IRS1 in Skeletal Muscle Myogenesis. Int. J. Biol. Sci. 2018, 14, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, R.; Workeneh, B.; Dong, Y.; Wang, X.; Hu, Z. Transcription factor FoxO1, the dominant mediator of muscle wasting in chronic kidney disease, is inhibited by microRNA-486. Kidney Int. 2012, 82, 401–411. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 Delays ALS Progression and Promotes Regeneration of Neuromuscular Synapses in Mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Bithell, A.; Soldati, C.; Buckley, N.J.; Stanton, L.W. MiR-375 is Essential for Human Spinal Motor Neuron Development and May Be Involved in Motor Neuron Degeneration. STEM CELLS 2015, 34, 124–134. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef]
- Kaifer, K.A.; Villalón, E.; O’Brien, B.S.; Sison, S.L.; Smith, C.; Simon, M.E.; Marquez, J.; O’Day, S.; Hopkins, A.E.; Neff, R.; et al. AAV9-mediated delivery of miR-23a reduces disease severity in Smn2B/−SMA model mice. Hum. Mol. Genet. 2019, 28, 3199–3210. [Google Scholar] [CrossRef]
- Snieckute, G.; Baltaci, O.; Liu, H.; Li, L.; Hu, Z.; Pocock, R. mir-234 controls neuropeptide release at the Caenorhabditis elegans neuromuscular junction. Mol. Cell. Neurosci. 2019, 98, 70–81. [Google Scholar] [CrossRef]
- Natarajan, A.; Lemos, D.R.; Rossi, F.M. Fibro/adipogenic progenitors: A double-edged sword in skeletal muscle regeneration. Cell Cycle 2010, 9, 2045–2046. [Google Scholar] [CrossRef][Green Version]
- Uezumi, A.; Ikemoto-Uezumi, M.; Tsuchida, K. Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front. Physiol. 2014, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Li, P.; Ling, H.; Xu, Z.; Yi, B.; Zhu, S. MiR-499/PRDM16 axis modulates the adipogenic differentiation of mouse skeletal muscle satellite cells. Hum. Cell 2018, 31, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, M.; Guan, J.; Li, P.; Wang, H.; Guo, Y.; Shuai, S.; Li, X. MicroRNAs miR-27a and miR-143 Regulate Porcine Adipocyte Lipid Metabolism. Int. J. Mol. Sci. 2011, 12, 7950–7959. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.; Roubenoff, R. Recent advances in the biology and therapy of muscle wasting. Ann. N. Y. Acad. Sci. 2010, 1211, 25–36. [Google Scholar] [CrossRef]
- Morvan, F.; Rondeau, J.M.; Zou, C.; Minetti, G.; Scheufler, C.; Scharenberg, M.; Jacobi, C.; Brebbia, P.; Ritter, V.; Toussaint, G.; et al. Blockade of activin type II receptors with a dual anti-ActRIIA/IIB antibody is critical to promote maximal skeletal muscle hypertrophy. Proc. Natl. Acad. Sci. USA 2017, 114, 12448–12453. [Google Scholar] [CrossRef]
- Suh, J.; Lee, Y.S. Myostatin Inhibitors: Panacea or Predicament for Musculoskeletal Disorders? J. Bone Metab. 2020, 27, 151–165. [Google Scholar] [CrossRef]
- Kemp, G.J.; Birrell, F.; Clegg, P.D.; Cuthbertson, D.J.; De Vito, G.; Van Dieën, J.H.; Del Din, S.; Eastell, R.; Garnero, P.; Goljanek–Whysall, K.; et al. Developing a toolkit for the assessment and monitoring of musculoskeletal ageing. Age Ageing 2018, 47, iv1–iv19. [Google Scholar] [CrossRef]
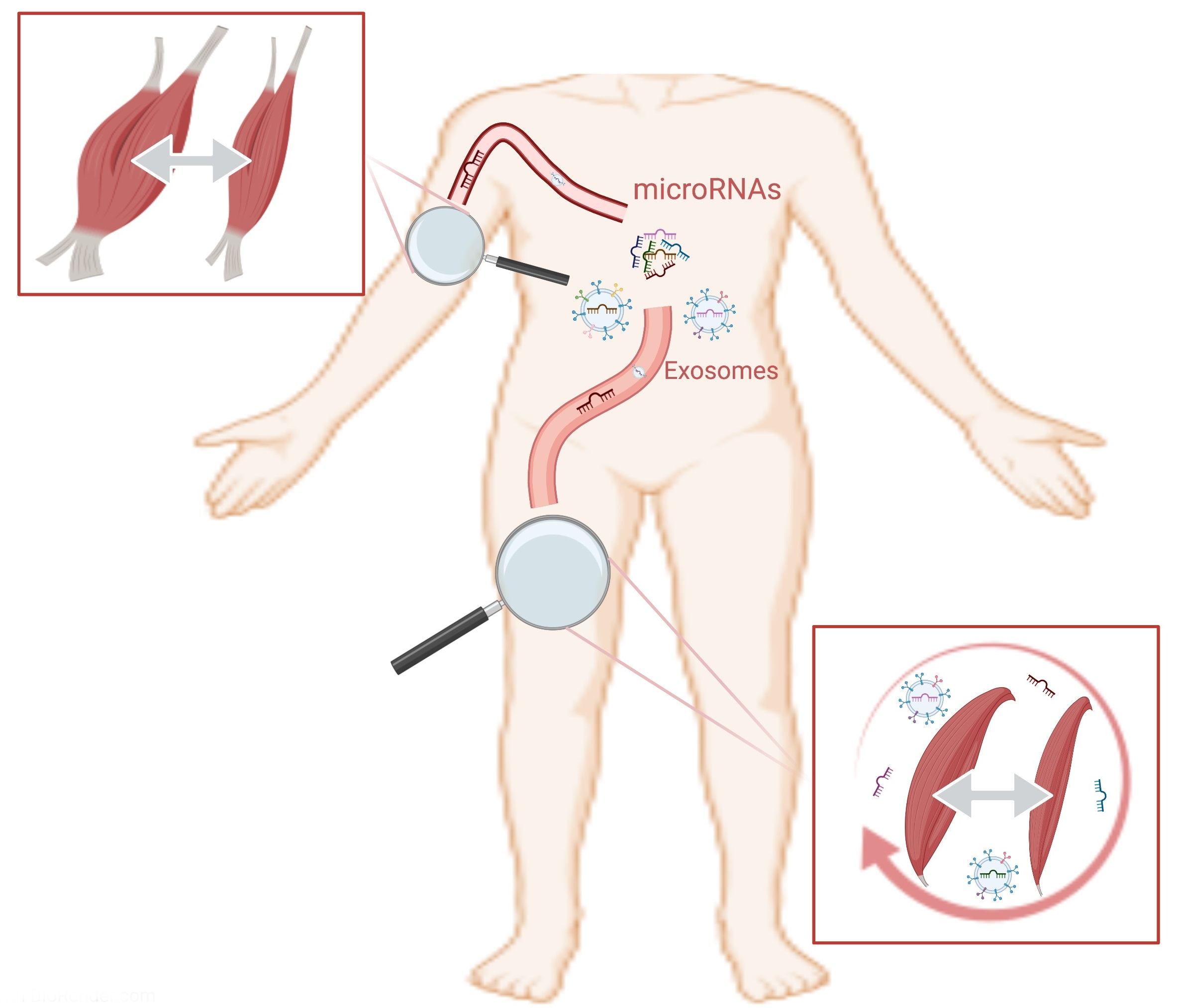
- Siracusa, J.; Koulmann, N.; Banzet, S. Circulating myomiRs: A new class of biomarkers to monitor skeletal muscle in physiology and medicine. J. Cachexia Sarcopenia Muscle 2017, 9, 20–27. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.-S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Miravirsen works against hepatitis C virus. BMJ 2013, 346, f2069. [Google Scholar] [CrossRef] [PubMed]
- van der Ree, M.H.; Van Der Meer, A.J.; Van Nuenen, A.C.; De Bruijne, J.; Ottosen, S.; Janssen, H.L.; Kootstra, N.A.; Reesink, H.W. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment. Pharmacol. Ther. 2015, 43, 102–113. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
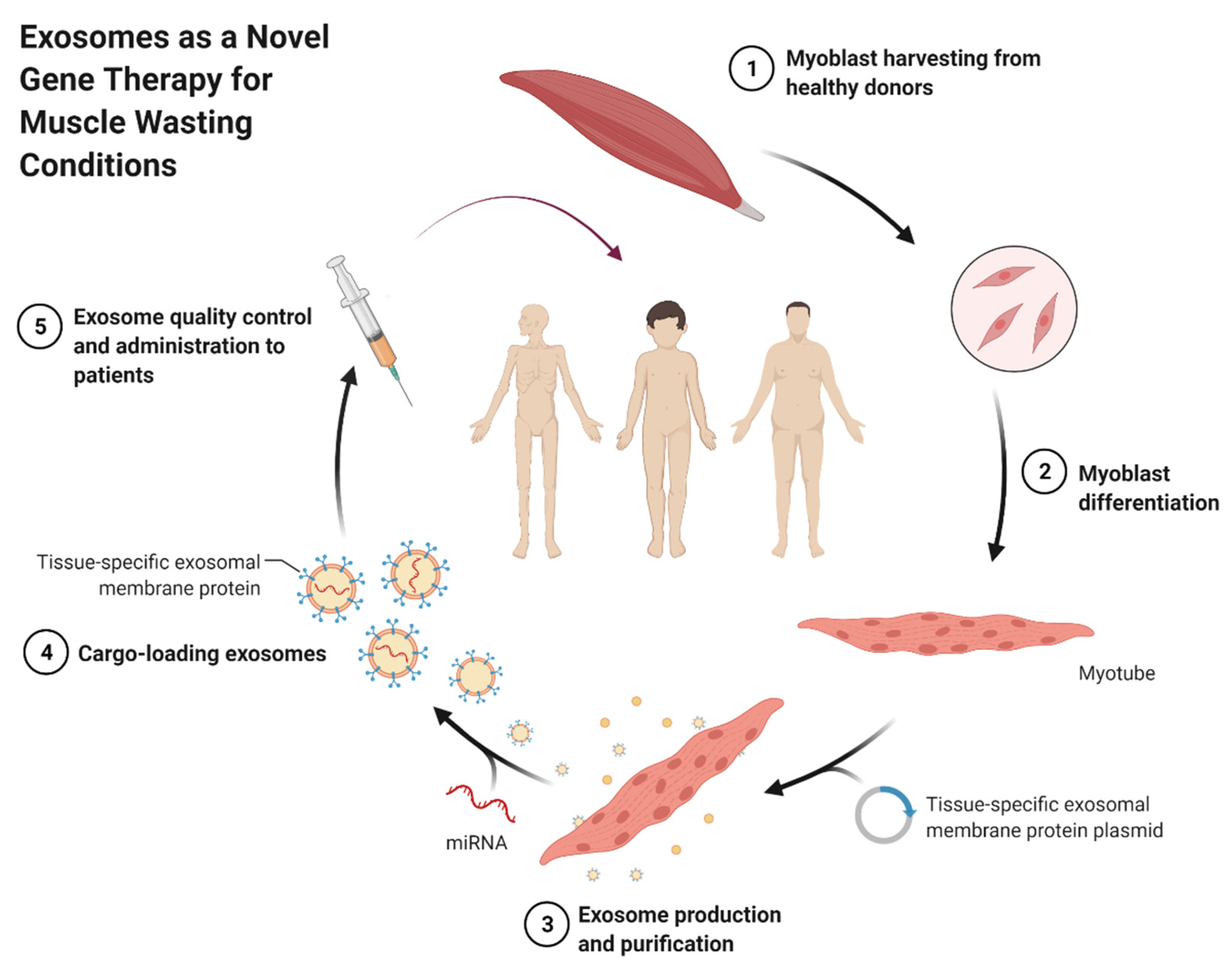
- BioRender.com. Self-Derived Exosomes as a Novel Gene Therapy. 2021. Available online: https://app.biorender.com/biorender-templates/t-5fc96e5428df8200ae6faf29-self-derived-exosomes-as-a-novel-gene-therapy (accessed on 28 October 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MyomiR | Expression Pattern | Prominent Targets |
|---|---|---|
| miR-1-1 | Skeletal muscle and heart | PAX3/7, POLA1, CCDN1/2, YY1, CX43, HDAC4, MEOX2, RARB, BAF47, BAF60A, FZD7, CNN3, SFRP1, NOTCH3, HAND2, DII-1, HES1, FRS2, myocardin |
| miR-1-2 | Skeletal muscle and heart | - |
| miR-133a-1 | Skeletal muscle and heart | FGFR1, PP2AC, CCN1, RUNX2, BAF60B, PRDM16, SRF, nPTB, IGF-1R, UCP2, FOXL2, FGFR1, PP2AC, ESFR, SNAI1, cyclin D2, SP1 |
| miR-133a-2 | Skeletal muscle and heart | - |
| miR-206 | Skeletal muscle (Type I fibers) | PAX3/7, POLA1, CCDN1/2, YY1, CX43, HDAC4, MEOX2, RARB, BAF47, BAF60A, FZD7, UTM, FSTL1, nPTB |
| miR-208a | Skeletal muscle (mass regulator), heart | MSTN, MYH7, MYH7B, THRAP1 |
| miR-208b | Skeletal muscle (type I fibers), heart (low expression) | SOX6, MYH6 |
| miR-486 | Skeletal muscle and heart | PAX7, PTEN, FOXO1A |
| miR-499 | Skeletal muscle (type I fibers), heart | SOX6, MEF2C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yedigaryan, L.; Sampaolesi, M. Therapeutic Implications of miRNAs for Muscle-Wasting Conditions. Cells 2021, 10, 3035. https://doi.org/10.3390/cells10113035
Yedigaryan L, Sampaolesi M. Therapeutic Implications of miRNAs for Muscle-Wasting Conditions. Cells. 2021; 10(11):3035. https://doi.org/10.3390/cells10113035
Chicago/Turabian StyleYedigaryan, Laura, and Maurilio Sampaolesi. 2021. "Therapeutic Implications of miRNAs for Muscle-Wasting Conditions" Cells 10, no. 11: 3035. https://doi.org/10.3390/cells10113035
APA StyleYedigaryan, L., & Sampaolesi, M. (2021). Therapeutic Implications of miRNAs for Muscle-Wasting Conditions. Cells, 10(11), 3035. https://doi.org/10.3390/cells10113035