
Platelet Function, Role in Thrombosis, Inflammation, and Consequences in Chronic Myeloproliferative Disorders


Abstract
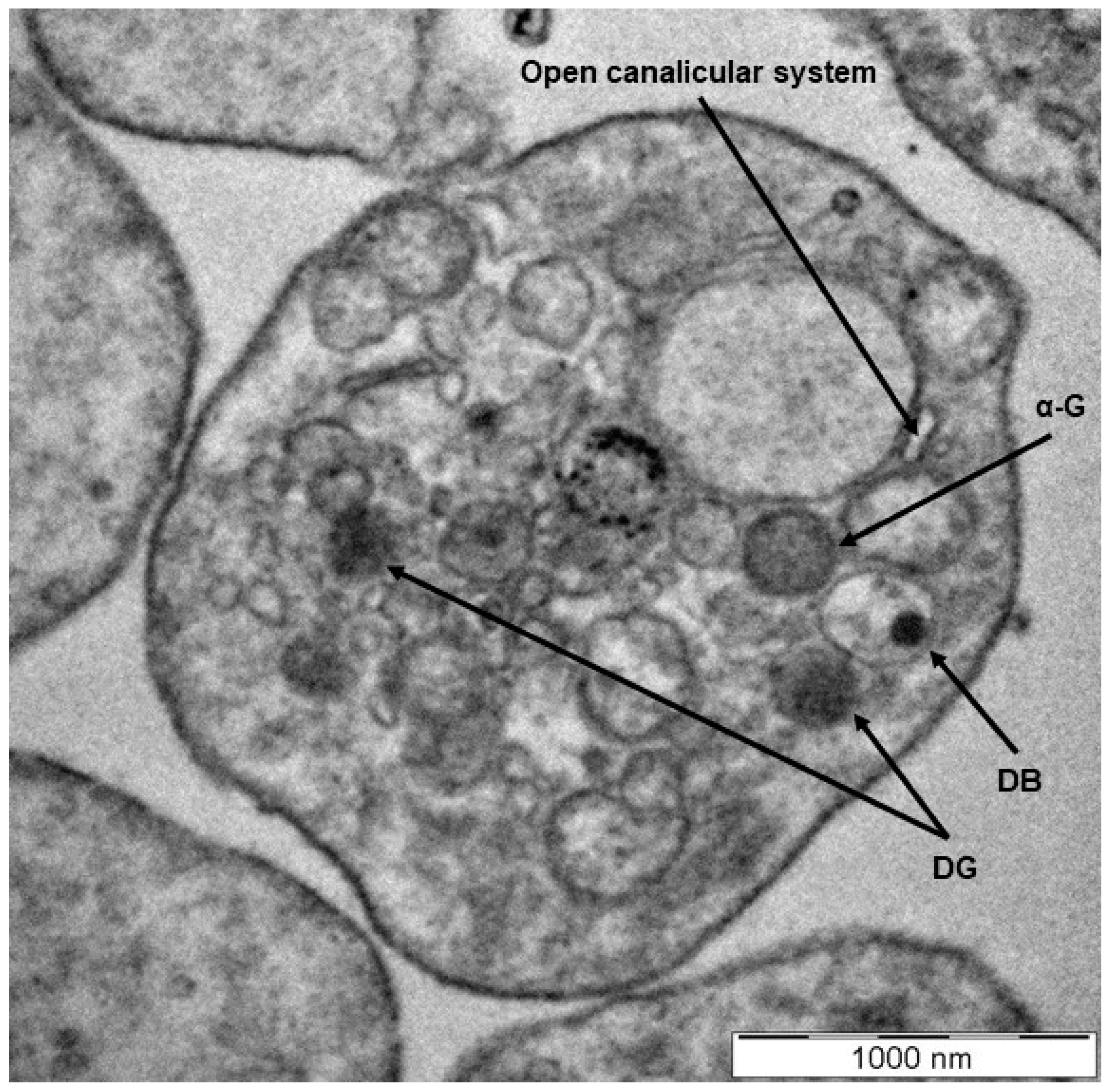
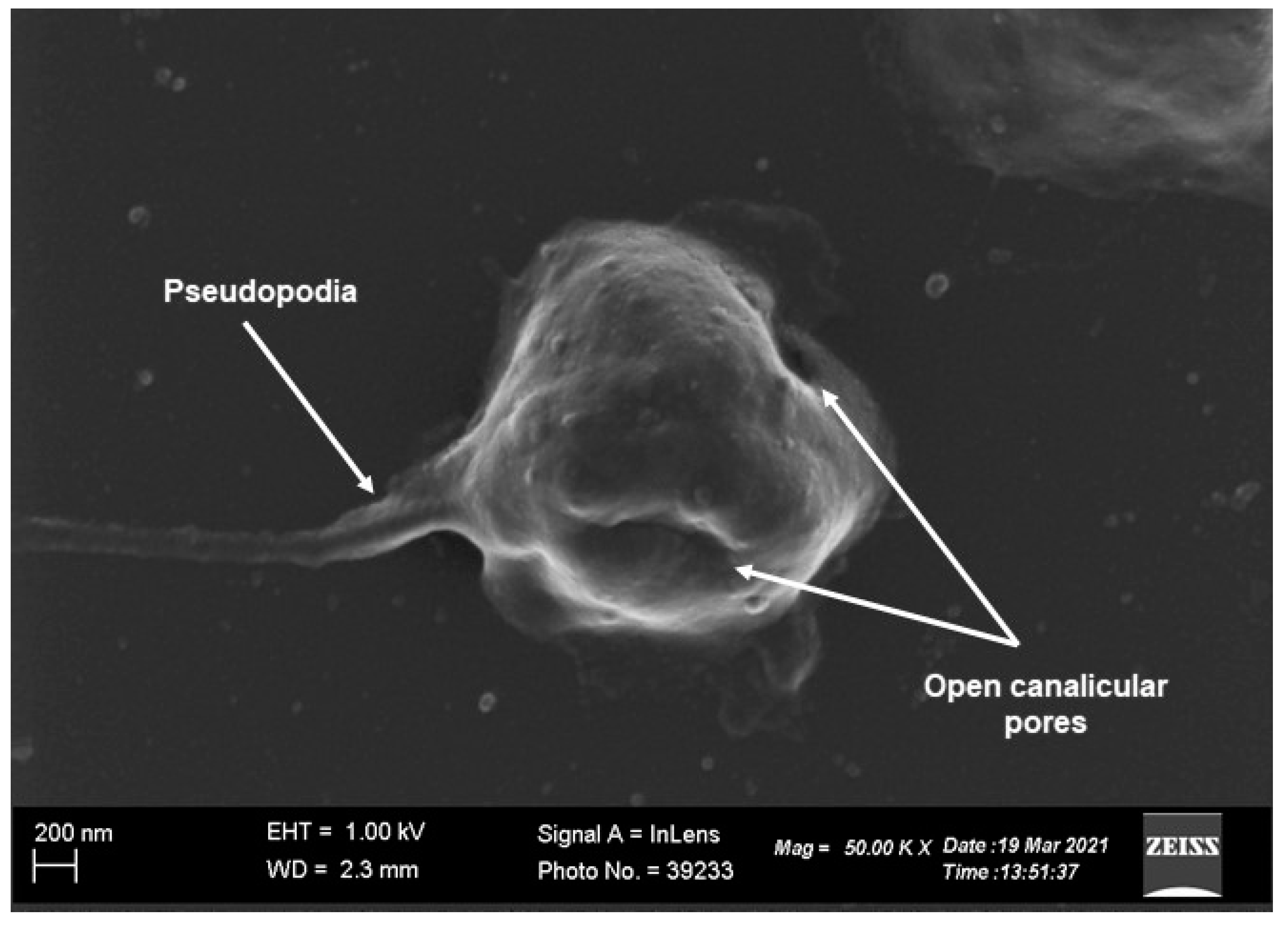
:1. Platelet Morphology and Function
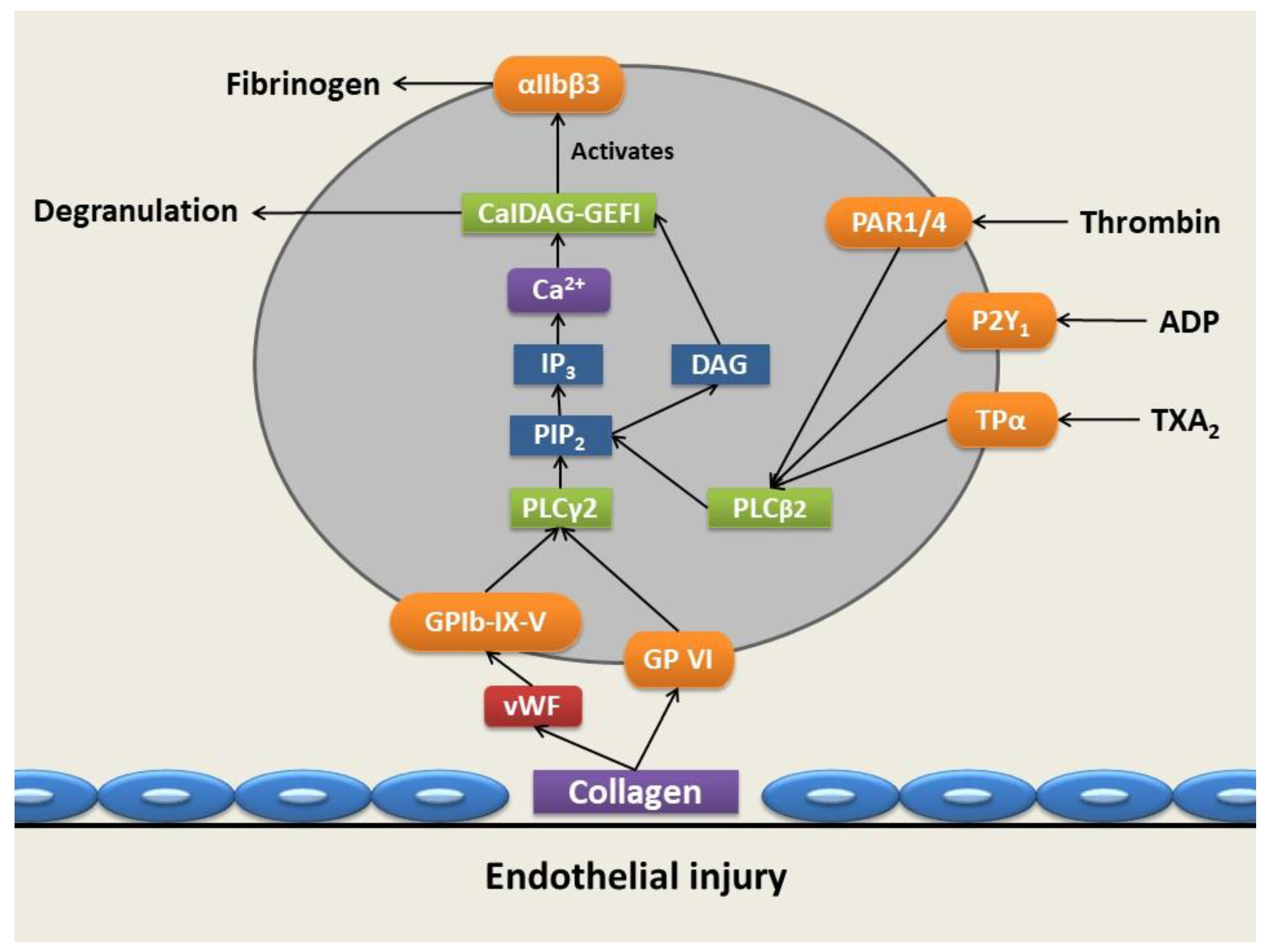
2. Platelet Granule Constituents and Membrane Receptors
3. Platelets’ Role in Inflammation and Cancer
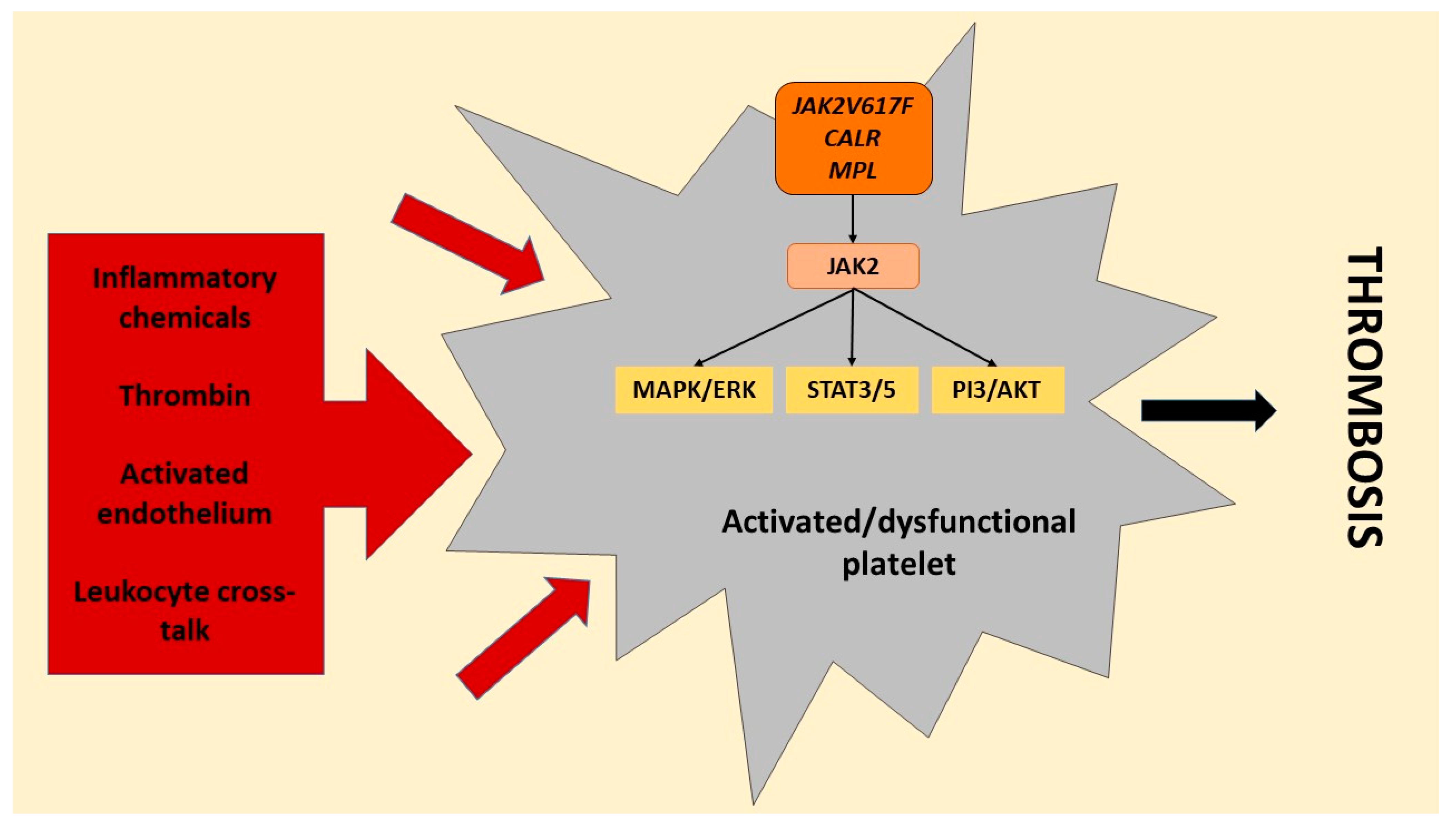
4. Platelets’ Role in Myeloproliferative Disorders
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Latger-Cannard, V.; Fenneteau, O.; Salignac, S.; Lecompte, T.P.; Schlegel, N. Platelet morphology analysis. Haemostasis 2013, 992, 207–225. [Google Scholar]
- van der Meijden, P.E.; Heemskerk, J.W. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Holinstat, M. Normal platelet function. Cancer Metastasis Rev. 2017, 36, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E. Ultrastructural changes in platelet membranes due to cigarette smoking. Ultrastruct. Pathol. 2012, 36, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Buys, A.V.; Pretorius, E. Comparing different preparation methods to study human fibrin fibers and platelets using TEM. Microsc. Res. Tech. 2012, 75, 801–806. [Google Scholar] [CrossRef]
- Repsold, L. Angiogenic, Apoptotic and Autophagic Profiling of Chronic Myeloid Leukaemia Patients’ Platelets ex vivo before and after Treatment with Imatinib. 2021; Unpublished. [Google Scholar]
- Repsold, L.; Pretorius, E.; Joubert, A.M. An estrogen analogue and promising anticancer agent refrains from inducing morphological damage and reactive oxygen species generation in erythrocytes, fibrin and platelets: A pilot study. Cancer Cell Int. 2014, 14, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Plooy, J.N.; Buys, A.; Duim, W.; Pretorius, E. Comparison of platelet ultrastructure and elastic properties in thrombo-embolic ischemic stroke and smoking using atomic force and scanning electron microscopy. PLoS ONE 2013, 8, e69774. [Google Scholar]
- Rumbaut, R.E.; Thiagarajan, P. Chapter 2: General characteristics of platelets. In Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- White, J.G.; Gerrard, J.M. The ultrastructure of defective human platelets. Mol. Cell. Biochem. 1978, 21, 109–128. [Google Scholar] [CrossRef]
- Zufferey, A.; Fontana, P.; Reny, J.L.; Nolli, S.; Sanchez, J.C. Platelet proteomics. Mass Spectrom. Rev. 2012, 31, 331–351. [Google Scholar] [CrossRef]
- Ware, J.A.; Coller, B.S. Platelet morphology, biochemistry, and function. Williams Hematol. 1995, 5, 1161–1201. [Google Scholar]
- Lewandrowski, U.; Wortelkamp, S.; Lohrig, K.; Zahedi, R.P.; Wolters, D.A.; Walter, U.; Sickmann, A. Platelet membrane proteomics: A novel repository for functional research. Blood 2009, 114, e10–e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troxler, M.; Dickinson, K.; Homer-Vanniasinkam, S. Platelet function and antiplatelet therapy. Br. J. Surg. 2007, 94, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Whiteheart, S.W. Platelet granules: Surprise packages. Blood 2011, 118, 1190–1191. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet α-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Rendu, F.; Brohard-Bohn, B. The platelet release reaction: Granules’ constituents, secretion and functions. Platelets 2001, 12, 261–273. [Google Scholar] [CrossRef]
- Reed, G.L. Platelet secretory mechanisms. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers, Inc.: New York, NY, USA, 2004; Volume 30, pp. 441–450. [Google Scholar]
- King, S.M.; Reed, G.L. Development of platelet secretory granules. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2002; Volume 13, pp. 293–302. [Google Scholar]
- McNicol, A.; Israels, S.J. Platelet dense granules: Structure, function and implications for haemostasis. Thromb. Res. 1999, 95, 1–18. [Google Scholar] [CrossRef]
- Kauskot, A.; Hoylaerts, M.F. Platelet receptors. Antiplatelet Agents 2012, 210, 23–57. [Google Scholar]
- Fong, K.P.; Barry, C.; Tran, A.N.; Traxler, E.A.; Wannemacher, K.M.; Tang, H.Y.; Speicher, K.D.; Blair, I.A.; Speicher, D.W.; Grosser, T.; et al. Deciphering the human platelet sheddome. Blood 2011, 117, e15–e26. [Google Scholar] [CrossRef] [Green Version]
- Michelson, A.D. Platelets; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Kunicki, T.J. Platelet membrane glycoproteins and their function: An overview. Ann. Hematol. 1989, 59, 30–34. [Google Scholar] [CrossRef]
- Nurden, A.T. Platelet membrane glycoproteins: A historical review. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: Stuttgart, Germany, 2014; Volume 40, pp. 577–584. [Google Scholar]
- Andrews, R.K.; Berndt, M.C. Platelet physiology and thrombosis. Thromb. Res. 2004, 114, 447–453. [Google Scholar] [CrossRef]
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at work in primary hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef]
- Broos, K.; De Meyer, S.F.; Feys, H.B.; Vanhoorelbeke, K.; Deckmyn, H. Blood platelet biochemistry. Thromb. Res. 2012, 129, 245–249. [Google Scholar] [CrossRef]
- Nurden, A.T. Platelets, inflammation and tissue regeneration. Thromb. Haemost. 2011, 105, S13–S33. [Google Scholar] [CrossRef] [PubMed]
- George, J.N.; Pickett, E.B.; Saucerman, S.; McEver, R.P.; Kunicki, T.J.; Kieffer, N.; Newman, P.J. Platelet surface glycoproteins. Studies on resting and activated platelets and platelet membrane microparticles in normal subjects, and observations in patients during adult respiratory distress syndrome and cardiac surgery. J. Clin. Investig. 1986, 78, 340–348. [Google Scholar] [CrossRef]
- Park, K.; Mao, F.W.; Park, H. Morphological characterization of surface-induced platelet activation. Biomaterials 1990, 11, 24–31. [Google Scholar] [CrossRef]
- Goubran, H.A.; Burnouf, T.; Radosevic, M.; El-Ekiaby, M. The platelet–cancer loop. Eur. J. Intern. Med. 2013, 24, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.A.; Stakiw, J.; Radosevic, M.; Burnouf, T. Platelet-Cancer Interactions. Semin. Thromb. Hemost. 2014, 40, 296–305. [Google Scholar] [PubMed]
- Estevez, B.; Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Mangin, P.; Ohlmann, P.; Eckly, A.; Cazenave, J.P.; Lanza, F.; Gachet, C. The P2Y1 receptor plays an essential role in the platelet shape change induced by collagen when TxA2 formation is prevented. J. Thromb. Haemost. 2004, 2, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Saboor, M.; Ayub, Q.; Samina Ilyas, M. Platelet receptors; an instrumental of platelet physiology. Pak. J. Med. Sci. 2013, 29, 891. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.T.; Corken, A.; Ware, J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood J. Am. Soc. Hematol. 2015, 126, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezger, M.; Nording, H.; Sauter, R.; Graf, T.; Heim, C.; von Bubnoff, N.; Ensminger, S.M.; Langer, H.F. Platelets and immune responses during Thromboinflammation. Front. Immunol. 2019, 10, 1731. [Google Scholar] [CrossRef] [PubMed]
- Schattner, M.; Jenne, C.N.; Negrotto, S.; Ho-Tin-Noe, B. Platelets and Immune Responses during Thromboinflammation. Front. Immunol. 2020, 11, 1079. [Google Scholar] [CrossRef]
- Meikle, C.K.; Kelly, C.A.; Garg, P.; Wuescher, L.M.; Ali, R.A.; Worth, R.G. Cancer and thrombosis: The platelet perspective. Front. Cell Dev. Biol. 2017, 4, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, A.K.; Cedervall, J. The pro-inflammatory role of platelets in cancer. Platelets 2018, 29, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Acedo, A.L.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Platelets, thrombo-inflammation, and cancer: Collaborating with the enemy. Front. Immunol. 2019, 10, 1805. [Google Scholar] [CrossRef] [Green Version]
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46. [Google Scholar] [CrossRef] [Green Version]
- Elaskalani, O.; Berndt, M.C.; Falasca, M.; Metharom, P. Targeting platelets for the treatment of cancer. Cancers 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Anders, H.J.; Gudermann, T.; Mammadova-Bach, E. Platelet-cancer interplay: Molecular mechanisms and new therapeutic avenues. Front. Oncol. 2021, 11, 665534. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges for aspirin and other antiplatelet agents. Blood J. Am. Soc. Hematol. 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Mufti, G.J.; Flandrin, G.; Schaefer, H.E.; Sandberg, A.A.; Kanfer, E.J.; Bryon, P.A. An atlas of malignant haematology: Cytology, histology, and cytogenetics. Hématologie 1997, 3, 478–480. [Google Scholar]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic myeloid leukemia: A model disease of the past, present and future. Cells 2021, 10, 117. [Google Scholar] [CrossRef]
- Akay, O.M.; Mutlu, F.; Gülbas, Z. Platelet dysfunction in patients with chronic myeloid leukemia: Does imatinib mesylate improve it? Turk. J. Hematol. 2016, 33, 127–130. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Han, X.; Kantarjian, H.; Cortes, J. Tyrosine kinase inhibitor-induced platelet dysfunction in patients with chronic myeloid leukemia. Blood 2009, 114, 261–263. [Google Scholar] [CrossRef] [Green Version]
- Marin Oyarzún, C.P.; Heller, P.G. Platelets as mediators of thromboinflammation in chronic myeloproliferative neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Pardanani, A. Myeloproliferative neoplasms: A contemporary review. JAMA Oncol. 2015, 1, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, B.S. Myeloproliferative neoplasms. Diagn. Histopathol. 2021, 27, 373–379. [Google Scholar] [CrossRef]
- Harrison, C.N.; Lee, J.S. Myeloproliferative neoplasms. Medicine 2017, 45, 275–279. [Google Scholar] [CrossRef]
- Gerrard, J.M.; McNicol, A. Platelet storage pool deficiency, leukemia, and myelodysplastic syndromes. Leuk. Lymphoma 1992, 8, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T. The biology of the platelet with special reference to inflammation, wound healing and immunity. Front. Biosci. 2018, 23, 726–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors Released from α-Granules | |
|---|---|
| Adhesion proteins | Fibrinogen Fibronectin Thrombospondin Vitronectin von Willebrand factor |
| Chemokines | Connective tissue activating peptide III CXC chemokine ligand (CXCL)-1, 2, 3, 4, 5, 6, 7, 8, 12 and 16 Macrophage inflammatory protein 1α Neutrophil-activating peptide 2 Platelet factor 4 Regulated upon activation, normal T cell expressed and presumably secreted (RANTES) β-Thromboglobulin |
| Coagulation factors | Factor V Factor VII Factor XI Factor XIII Kininogens Plasminogen Protein S |
| Growth factors | Angiopoietin-1 and 2 Basic fibroblast growth factor Endothelial cell growth factor Epidermal growth factor Hepatocyte growth factor Insulin-like growth factor 1 Interleukin-2, 3, 4, 6, 7, and 8 Interleukin-β Platelet-derived growth factor Transforming growth factor β Vascular endothelial growth factor A & C |
| Immunoglobulins | Immunoglobulin A (IgA) Immunoglobulin E (IgE) Immunoglobulin G (IgG) Immunoglobulin M (IgM) |
| Protease inhibitors | C1-inhibitor Plasminogen activator inhibitor-1 Platelet inhibitor of factor XIα Platelet-derived collagenase inhibitor Protease nexin-II/amyloid β-protein precursor Tissue factor pathway inhibitor α-1-proteinase inhibitor α2-antiplasmin α2-antitrypsin α2-macroglobulin |
| Proteoglycans | Histidine-rich glycoprotein Neutrophil-activating peptide 2 Platelet basic protein Serglycin |
| Other | Albumin Glycoprotein Iα/Multimerin |
| Factors Released from Dense Granules | |
|---|---|
| Amines | Adrenaline Dopamine Histamine Noradrenaline Serotonin |
| Bivalent cations | Calcium (Ca2+) Magnesium Polyphosphates Pyrophosphate |
| Nucleotides | Adenosine 5′-diphosphate Adenosine 5′-triphosphate Guanosine 5′-diphosphate Guanosine 5′-triphosphate |
| Factors Present in Lysosomes | |
|---|---|
| Acid Proteases | Acid phosphatase Arylsulphatase Carboxypeptidases A & B Cathepsins D & E Collagenase Proline carboxypeptidase |
| Glycohydrolases | Heparinase α-D-glucosidase α-D-mannosidase α-L-arabinosidase α-L-fucosidase β-D-fucosidase β-D-galactosidase β-D-glucoronidase β-D-glucosidase β-glycerophosphatase β-N-acetyl-D-hexosaminidase |
| Receptors Present on Platelet Membrane | |
|---|---|
| C-type lectin receptor family (selectins) | Cluster of differentiation 72 (CD72) Cluster of differentiation 93 (CD93) C-type lectin 2 (CLEC-2) P-selectin (CD62P) |
| Immunoglobulin receptors | Cluster of differentiation 23 (CD23/FcεRI) Cluster of differentiation 32 (CD32/FcγRIIA) Cluster of differentiation 47 (CD47) Endothelial cell-selective adhesion molecule (ESAM) G6B Glycoprotein VI (GPVI) Intercellular adhesion molecule 2 (ICAM-2) Junction adhesion molecule 1 (JAM-1) Junction adhesion molecule 3 (JAM-3) Platelet and T cell activation antigen 1 (PTA-1) Platelet endothelial cell adhesion molecule (PECAM-1) TREM-like transcript 1 (TLT-1) |
| Integrins (glycoproteins) | α2β1 (GPIa-IIa—collagen-binding receptor) α5β1 (fibronectin receptor) α6β1 (laminin binding cell adhesion receptor) αIIbβ3 (GPIIb/IIIa (CD41/CD61)) αLβ2 (leukocyte function-associated antigen 1) αvβ3 (vitronectin receptor) |
| Leucine-rich repeats receptors | GPIb-IX-V complex Toll-like receptors (TLR) 1, 2, 4 and 6 Matrix metalloproteinases (MMP) 1, 2, 3, 9 and 14 |
| Lipid receptors | Lysophosphatidic acid receptor (LPL-R) Platelet-activating factor (PAF) receptor Sphingosine-1-phosphate receptors |
| Other membrane receptors | C3-specific binding protein (CD46) Cluster of differentiation 100 (CD100) Cluster of differentiation 40 ligand (CD40L) Collectin receptor (C1q) Extracellular matrix metalloproteinase inducer (EMMPRIN) (CD147) Galectin receptors Glutamate receptors GPIIIb (CD36) Liver X receptors (LXR) Lysosomal-associated membrane protein 1 (LAMP-1) Lysosomal-associated membrane protein 2 (LAMP-2) P2X purinoceptor 1 (P2 × 1) (ion channel receptor) Peroxisome proliferator-activated receptor γ (PPARγ) P-selectin glycoprotein ligand 1 (PSGL-1) Semaphorin 3A Tight junction receptors Tumor necrosis factor (TNF) |
| Tetraspanins | Cluster of differentiation 53 (CD53) Cluster of differentiation 9 (CD9) Lysosomal membrane-associated glycoprotein-3 (LAMP-3) (CD63) |
| G protein-coupled receptors | A2a-adenosine C-C chemokine receptor 1 (CCR1) C-C chemokine receptor 3 (CCR3) C-C chemokine receptor 4 (CCR4) C-X-C chemokine receptor 1 (CXCR1) C-X-C chemokine receptor 4 (CXCR4) Dopamine receptors P2Y1 and P2Y12 (guanosine triphosphate-coupled protein receptor) Platelet factor 4 Prostaglandin D2 receptor (DP2), Prostaglandin E1 receptor (EP), Prostaglandin E2 receptor (EP), Prostaglandin F2 receptor (FP), Prostaglandin I2 receptor (IP) Protease-activated receptor (PAR) 1, 2, 3 and 4 Serotonin (5-HT2A) TXA2 receptor (TP) V1a vasopressin β2-adrenergic Β-Thromoglobulin |
| Tyrosine kinase receptors | c-MPL (thrombopoietin receptor) (CD110) Insulin receptors Platelet-derived growth factor receptors Tyrosine kinase with immunoglobulin and epidermal growth factor homology-1 receptors (Tie-1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Repsold, L.; Joubert, A.M. Platelet Function, Role in Thrombosis, Inflammation, and Consequences in Chronic Myeloproliferative Disorders. Cells 2021, 10, 3034. https://doi.org/10.3390/cells10113034
Repsold L, Joubert AM. Platelet Function, Role in Thrombosis, Inflammation, and Consequences in Chronic Myeloproliferative Disorders. Cells. 2021; 10(11):3034. https://doi.org/10.3390/cells10113034
Chicago/Turabian StyleRepsold, Lisa, and Anna Margaretha Joubert. 2021. "Platelet Function, Role in Thrombosis, Inflammation, and Consequences in Chronic Myeloproliferative Disorders" Cells 10, no. 11: 3034. https://doi.org/10.3390/cells10113034
APA StyleRepsold, L., & Joubert, A. M. (2021). Platelet Function, Role in Thrombosis, Inflammation, and Consequences in Chronic Myeloproliferative Disorders. Cells, 10(11), 3034. https://doi.org/10.3390/cells10113034