
Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs

 ,
,
Abstract
:1. Introduction
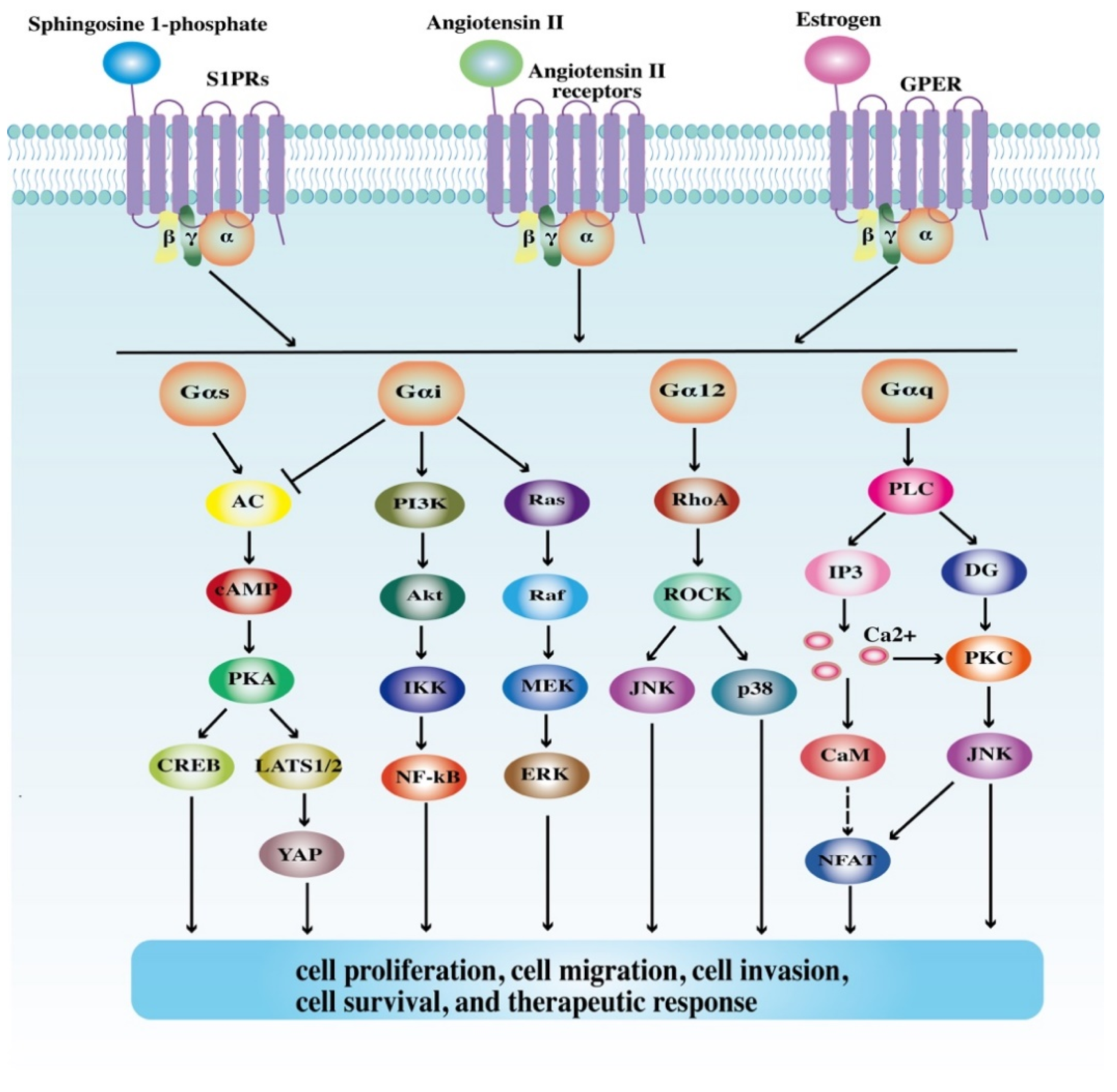
2. Structures and Signaling Pathways of GPCRs
3. Roles of GPCRs in GI Cancers
3.1. Colorectal Cancer
3.1.1. Sphingosine 1-Phosphate Receptors
3.1.2. Angiotensin II Receptors
3.1.3. Estrogen-Related GPCRs
3.1.4. Other GPCRs
3.2. Gastric Cancer
3.2.1. Sphingosine 1-Phosphate Receptors
3.2.2. Angiotensin II Receptors
3.2.3. Estrogen-Related GPCRs
3.2.4. Other GPCRs
3.3. Esophageal Cancer
3.3.1. Sphingosine 1-Phosphate Receptors
3.3.2. Angiotensin II Receptors
3.3.3. Estrogen-Related GPCRs
3.3.4. Other GPCRs
4. Targeting GPCRs in GI Cancer Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. Available online: https://gco.iarc.fr/today/home (accessed on 17 October 2021).
- Global Cancer Observatory: Cancer Tomorrow. Lyon, France: International Agency for Research on Cancer. Available online: https://gco.iarc.fr/tomorrow/en (accessed on 17 October 2021).
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e315. [Google Scholar] [CrossRef]
- Sukocheva, O.A.; Furuya, H.; Ng, M.L.; Friedemann, M.; Menschikowski, M.; Tarasov, V.V.; Chubarev, V.N.; Klochkov, S.G.; Neganova, M.E.; Mangoni, A.A.; et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol. Ther. 2020, 207, 107464. [Google Scholar] [CrossRef]
- Soond, S.M.; Zamyatnin, A.A., Jr. Targeting G protein-coupled receptors in cancer therapy. Adv. Cancer Res. 2020, 145, 49–97. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Lorito, N.; Smiriglia, A.; Morandi, A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer 2020, 7, 198–213. [Google Scholar] [CrossRef]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X.; et al. Emerging role of lipid metabolism alterations in Cancer stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 118. [Google Scholar] [CrossRef] [PubMed]
- Germain, N.; Dhayer, M.; Boileau, M.; Fovez, Q.; Kluza, J.; Marchetti, P. Lipid Metabolism and Resistance to Anticancer Treatment. Biology 2020, 9, 474. [Google Scholar] [CrossRef] [PubMed]
- Perini, M.V.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the benefits of renin-angiotensin system inhibitors as cancer treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Ager, E.I.; Neo, J.; Christophi, C. Regulation of colorectal cancer cell epithelial to mesenchymal transition by the renin angiotensin system. J. Gastroenterol. Hepatol. 2016, 31, 1773–1782. [Google Scholar] [CrossRef]
- Abancens, M.; Bustos, V.; Harvey, H.; McBryan, J.; Harvey, B.J. Sexual Dimorphism in Colon Cancer. Front. Oncol. 2020, 10, 607909. [Google Scholar] [CrossRef] [PubMed]
- Wesołowska, M.; Pawlik, P.; Jagodziński, P.P. The clinicopathologic significance of estrogen receptors in human gastric carcinoma. Biomed. Pharmacother. 2016, 83, 314–322. [Google Scholar] [CrossRef]
- Arnold, M.; Soerjomataram, I.; Ferlay, J.; Forman, D. Global incidence of oesophageal cancer by histological subtype in 2012. Gut 2015, 64, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Chen, Z.; Jiang, G.; Zhou, Y.; Yang, X.; Huang, H.; Liu, H.; Du, J.; Wang, H. Epigenetic down regulation of G protein-coupled estrogen receptor (GPER) functions as a tumor suppressor in colorectal cancer. Mol. Cancer 2017, 16, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Xu, Z.; Sun, J.; Lv, H.; Wang, Y.; Ni, Y.; Chen, S.; Hu, C.; Wang, L.; Chen, W.; et al. Cisplatin resistance in gastric cancer cells is involved with GPR30-mediated epithelial-mesenchymal transition. J. Cell. Mol. Med. 2020, 24, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, R.J. A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. 2013, 52, 6366–6378. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mu, J.; Zhu, M.; Mukherjee, A.; Zhang, H. Transient Receptor Potential Channels and Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 180. [Google Scholar] [CrossRef] [Green Version]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; et al. The Concise Guide to Pharmacology 2017/18: G protein-coupled receptors. Br. J. Pharmacol. 2017, 174 (Suppl. 1), S17–S129. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Mukherjee, A.; Varghese, A.P.; Yang, X.L.; Chen, S.; Zhang, H. Roles of G protein-coupled receptors in inflammatory bowel disease. World J. Gastroenterol. 2020, 26, 1242–1261. [Google Scholar] [CrossRef]
- Lohse, M.J. Dimerization in GPCR mobility and signaling. Curr. Opin. Pharmacol. 2010, 10, 53–58. [Google Scholar] [CrossRef]
- AbdAlla, S.; Lother, H.; Langer, A.; el Faramawy, Y.; Quitterer, U. Factor XIIIA transglutaminase crosslinks AT1 receptor dimers of monocytes at the onset of atherosclerosis. Cell 2004, 119, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Sun, L.; Jiao, Y.; Lee, L.T.O. The Role of G Protein-coupled Receptor Kinases in Cancer. Int. J. Biol. Sci. 2018, 14, 189–203. [Google Scholar] [CrossRef] [Green Version]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, Y.; Luo, C.; Zhang, H. Early detection of ulcerative colitis-associated colorectal cancer. Gastroenterol. Rep. 2018, 6, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Zhang, H. The Role of Proinflammatory Pathways in the Pathogenesis of Colitis-Associated Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 5126048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Petti, L.; Rizzo, G.; Rubbino, F.; Elangovan, S.; Colombo, P.; Restelli, S.; Piontini, A.; Arena, V.; Carvello, M.; Romano, B.; et al. Unveiling role of sphingosine-1-phosphate receptor 2 as a brake of epithelial stem cell proliferation and a tumor suppressor in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 253. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Luo, D.D.; Wan, S.B.; Qu, X.J. S1PR2 inhibitors potently reverse 5-FU resistance by downregulating DPD expression in colorectal cancer. Pharm. Res. 2020, 155, 104717. [Google Scholar] [CrossRef]
- Gu, X.; Jiang, Y.; Xue, W.; Song, C.; Wang, Y.; Liu, Y.; Cui, B. SPNS2 promotes the malignancy of colorectal cancer cells via regulating Akt and ERK pathway. Clin. Exp. Pharmacol. Physiol. 2019, 46, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Olesch, C.; Sirait-Fischer, E.; Berkefeld, M.; Fink, A.F.; Susen, R.M.; Ritter, B.; Michels, B.E.; Steinhilber, D.; Greten, F.R.; Savai, R.; et al. S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J. Clin. Investig. 2020, 130, 5461–5476. [Google Scholar] [CrossRef]
- Zhou, H.; Yin, X.; Bai, F.; Liu, W.; Jiang, S.; Zhao, J. The Role and Mechanism of S1PR5 in Colon Cancer. Cancer Manag. Res. 2020, 12, 4759–4775. [Google Scholar] [CrossRef] [PubMed]
- Neo, J.H.; Malcontenti-Wilson, C.; Muralidharan, V.; Christophi, C. Effect of ACE inhibitors and angiotensin II receptor antagonists in a mouse model of colorectal cancer liver metastases. J. Gastroenterol. Hepatol. 2007, 22, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Chen, I.X.; Tong, R.; Ng, M.R.; Martin, J.D.; Naxerova, K.; Wu, M.W.; Huang, P.; Boucher, Y.; Kohane, D.S.; et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 10674–10680. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yaguchi, T.; Ohmura, G.; Kobayashi, A.; Kawamura, N.; Iwata, T.; Kiniwa, Y.; Okuyama, R.; Kawakami, Y. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2018, 109, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Lappano, R.; De Marco, P.; Pupo, M.; Vivacqua, A.; Sisci, D.; Abonante, S.; Iacopetta, D.; Cappello, A.R.; Dolce, V.; et al. G protein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17β-estradiol in cancer cells and cancer-associated fibroblasts. J. Biol. Chem. 2012, 287, 43234–43245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilligan, L.C.; Rahman, H.P.; Hewitt, A.M.; Sitch, A.J.; Gondal, A.; Arvaniti, A.; Taylor, A.E.; Read, M.L.; Morton, D.G.; Foster, P.A. Estrogen Activation by Steroid Sulfatase Increases Colorectal Cancer Proliferation via GPER. J. Clin. Endocrinol. Metab. 2017, 102, 4435–4447. [Google Scholar] [CrossRef]
- Bustos, V.; Nolan, Á.M.; Nijhuis, A.; Harvey, H.; Parker, A.; Poulsom, R.; McBryan, J.; Thomas, W.; Silver, A.; Harvey, B.J. GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget 2017, 8, 84258–84275. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, S.; Chun, E.; Bae, S.; Brennan, C.A.; Gallini Comeau, C.A.; Lang, J.K.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Fuller, M.H.; et al. Expression of Free Fatty Acid Receptor 2 by Dendritic Cells Prevents Their Expression of Interleukin 27 and Is Required for Maintenance of Mucosal Barrier and Immune Response Against Colorectal Tumors in Mice. Gastroenterology 2020, 158, 1359–1372.e1359. [Google Scholar] [CrossRef]
- Hatanaka, H.; Tsukui, M.; Takada, S.; Kurashina, K.; Choi, Y.L.; Soda, M.; Yamashita, Y.; Haruta, H.; Hamada, T.; Ueno, T.; et al. Identification of transforming activity of free fatty acid receptor 2 by retroviral expression screening. Cancer Sci. 2010, 101, 54–59. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Y.; Jiang, H.; Robbins, G.T.; Nie, D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int. J. Cancer 2011, 128, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, Z.; Hu, Y.; Dong, S. Butyrate-induced GPR41 activation inhibits histone acetylation and cell growth. J. Genet. Genom. 2012, 39, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Ramanathan, V.; Quante, M.; Baik, G.H.; Yang, X.; Wang, S.S.; Tu, S.; Gordon, S.A.; Pritchard, D.M.; Varro, A.; et al. Inactivating cholecystokinin-2 receptor inhibits progastrin-dependent colonic crypt fission, proliferation, and colorectal cancer in mice. J. Clin. Investig. 2009, 119, 2691–2701. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Westphalen, C.B.; Hayakawa, Y.; Worthley, D.L.; Asfaha, S.; Yang, X.; Chen, X.; Si, Y.; Wang, H.; Tailor, Y.; et al. Progastrin stimulates colonic cell proliferation via CCK2R- and β-arrestin-dependent suppression of BMP2. Gastroenterology 2013, 145, 820–830.e810. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Feng, Y.; Sun, Y.; Ji, D.; Qian, W.; Zhang, Z.; Wang, Q.; Zhang, Y.; Zhang, C.; Sun, Y. GPR56 promotes proliferation of colorectal cancer cells and enhances metastasis via epithelial-mesenchymal transition through PI3K/AKT signaling activation. Oncol. Rep. 2018, 40, 1885–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Chatterjee, T.; Godoy, C.; Wu, L.; Liu, Q.J.; Carmon, K.S. GPR56 Drives Colorectal Tumor Growth and Promotes Drug Resistance through Upregulation of MDR1 Expression via a RhoA-Mediated Mechanism. Mol. Cancer Res. 2019, 17, 2196–2207. [Google Scholar] [CrossRef] [Green Version]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef]
- Zheng, X.; Li, W.; Ren, L.; Liu, J.; Pang, X.; Chen, X.; Kang, D.; Wang, J.; Du, G. The sphingosine kinase-1/sphingosine-1-phosphate axis in cancer: Potential target for anticancer therapy. Pharmacol. Ther. 2019, 195, 85–99. [Google Scholar] [CrossRef]
- Kawamori, T.; Osta, W.; Johnson, K.R.; Pettus, B.J.; Bielawski, J.; Tanaka, T.; Wargovich, M.J.; Reddy, B.S.; Hannun, Y.A.; Obeid, L.M.; et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006, 20, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell. Signal. 2017, 34, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Sanchez, T.; Milne, G.L.; Morrow, J.D.; Hla, T.; Ferrer, F. S1P/S1P2 signaling induces cyclooxygenase-2 expression in Wilms tumor. J. Urol. 2009, 181, 1347–1352. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Zheng, Y.; Bleu, T. Sphingosine 1-phosphate-induced cell proliferation, survival, and related signaling events mediated by G protein-coupled receptors Edg3 and Edg5. J. Biol. Chem. 2000, 275, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Young, N.; Van Brocklyn, J.R. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.M.; Li, L.; Jing, B.Q.; Zhao, Y.S.; Wang, C.L.; Feng, L.; Xie, Y.E. Effect of S1P5 on proliferation and migration of human esophageal cancer cells. World J. Gastroenterol. 2010, 16, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Bilotta, A.J.; Cong, Y. Gut microbiota metabolite regulation of host defenses at mucosal surfaces: Implication in precision medicine. Precis. Clin. Med. 2019, 2, 110–119. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar]
- Kawabata, A.; Baoum, A.; Ohta, N.; Jacquez, S.; Seo, G.M.; Berkland, C.; Tamura, M. Intratracheal administration of a nanoparticle-based therapy with the angiotensin II type 2 receptor gene attenuates lung cancer growth. Cancer Res. 2012, 72, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Makar, G.A.; Holmes, J.H.; Yang, Y.X. Angiotensin-converting enzyme inhibitor therapy and colorectal cancer risk. J. Natl. Cancer Inst. 2014, 106, djt374. [Google Scholar] [CrossRef] [Green Version]
- Chiang, Y.Y.; Chen, K.B.; Tsai, T.H.; Tsai, W.C. Lowered cancer risk with ACE inhibitors/ARBs: A population-based cohort study. J. Clin. Hypertens. 2014, 16, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yi, C.H.; Ya, K.G. Renin-angiotensin system inhibitor use and colorectal cancer risk and mortality: A dose-response meta analysis. J. Renin-Angiotensin-Aldosterone Syst. 2020, 21, 1470320319895646. [Google Scholar] [CrossRef]
- Friis, S.; Sørensen, H.T.; Mellemkjaer, L.; McLaughlin, J.K.; Nielsen, G.L.; Blot, W.J.; Olsen, J.H. Angiotensin-converting enzyme inhibitors and the risk of cancer: A population-based cohort study in Denmark. Cancer 2001, 92, 2462–2470. [Google Scholar] [CrossRef]
- Htoo, P.T.; Stürmer, T.; Jonsson-Funk, M.; Pate, V.; Simpson, R.J., Jr.; Lund, J.L. Renin-Angiotensin-Aldosterone System-based Antihypertensive Agents and the Risk of Colorectal Cancer Among Medicare Beneficiaries. Epidemiol. Camb. Mass. 2019, 30, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Dierssen-Sotos, T.; Gómez-Acebo, I.; Palazuelos, C.; Rodriguez-Moranta, F.; Pérez-Gómez, B.; Fernández Vazquez, J.P.; Amiano, P.; Barricarte, A.; Mirón-Pozo, B.; Tardon, A.; et al. Relationship between drugs affecting the renin-angiotensin system and colorectal cancer: The MCC-Spain study. Prev. Med. 2017, 99, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, I.M.; Wickremesekera, A.C.; Wickremesekera, S.K.; Davis, P.F.; Tan, S.T. Therapeutic Targeting of Cancer Stem Cells via Modulation of the Renin-Angiotensin System. Front. Oncol. 2019, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef]
- Chen, D.; Wang, C.Y. Targeting cancer stem cells in squamous cell carcinoma. Precis. Clin. Med. 2019, 2, 152–165. [Google Scholar] [CrossRef]
- Gao, W.; Chen, L.; Ma, Z.; Du, Z.; Zhao, Z.; Hu, Z.; Li, Q. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology 2013, 145, 636–646.e635. [Google Scholar] [CrossRef] [PubMed]
- Humphries, H.N.; Wickremesekera, S.K.; Marsh, R.W.; Brasch, H.D.; Mehrotra, S.; Tan, S.T.; Itinteang, T. Characterization of Cancer Stem Cells in Colon Adenocarcinoma Metastasis to the Liver. Front. Surg. 2017, 4, 76. [Google Scholar] [CrossRef] [PubMed]
- Murphy, N.; Xu, L.; Zervoudakis, A.; Xue, X.; Kabat, G.; Rohan, T.E.; Wassertheil-Smoller, S.; O’Sullivan, M.J.; Thomson, C.; Messina, C.; et al. Reproductive and menstrual factors and colorectal cancer incidence in the Women’s Health Initiative Observational Study. Br. J. Cancer 2017, 116, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, N.N.; Wu, L.; Gong, T.T.; Wang, Y.L.; Lin, B.; Wu, Q.J. Nonlinear reduction in risk for colorectal cancer by oral contraceptive use: A meta-analysis of epidemiological studies. Cancer Causes Control 2015, 26, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Troisi, R.; Bjørge, T.; Gissler, M.; Grotmol, T.; Kitahara, C.M.; Myrtveit Saether, S.M.; Ording, A.G.; Sköld, C.; Sørensen, H.T.; Trabert, B.; et al. The role of pregnancy, perinatal factors and hormones in maternal cancer risk: A review of the evidence. J. Intern. Med. 2018, 283, 430–445. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Zhang, Y.; Wang, L.; Huang, Y.; Yu, Q.; Guo, P.; Li, K. Risk of colorectal cancer with hysterectomy and oophorectomy: A systematic review and meta-analysis. Int. J. Surg. 2016, 34, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zervoudakis, A.; Strickler, H.D.; Park, Y.; Xue, X.; Hollenbeck, A.; Schatzkin, A.; Gunter, M.J. Reproductive history and risk of colorectal cancer in postmenopausal women. J. Natl. Cancer Inst. 2011, 103, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Stevanato Filho, P.R.; Aguiar Júnior, S.; Begnami, M.D.; Ferreira, F.O.; Nakagawa, W.T.; Spencer, R.; Bezerra, T.S.; Boggiss, P.E.; Lopes, A. Estrogen Receptor β as a Prognostic Marker of Tumor Progression in Colorectal Cancer with Familial Adenomatous Polyposis and Sporadic Polyps. Pathol. Oncol. Res. 2018, 24, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Hugerth, L.W.; Hases, L.; Saxena, A.; Seifert, M.; Thomas, Q.; Gustafsson, J.; Engstrand, L.; Williams, C. Colitis-induced colorectal cancer and intestinal epithelial estrogen receptor beta impact gut microbiota diversity. Int. J. Cancer 2019, 144, 3086–3098. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free Fatty Acid Receptors in Health and Disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, G.; Lv, J.; Yang, M.; Wang, M.; Zhu, M.; Wang, T.; Yan, C.; Yu, C.; Ding, Y.; Li, G.; et al. Genetic risk, incident gastric cancer, and healthy lifestyle: A meta-analysis of genome-wide association studies and prospective cohort study. Lancet. Oncol. 2020, 21, 1378–1386. [Google Scholar] [CrossRef]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, H.; Gong, W.; Liu, A. Up-regulation and tumor-promoting role of SPHK1 were attenuated by miR-330-3p in gastric cancer. IUBMB Life 2018, 70, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, H.; Kitayama, J.; Shida, D.; Yamaguchi, H.; Mori, K.; Osada, M.; Aoki, S.; Yatomi, Y.; Takuwa, Y.; Nagawa, H. Sphingosine 1-phosphate receptor expression profile in human gastric cancer cells: Differential regulation on the migration and proliferation. J. Surg. Res. 2006, 130, 80–87. [Google Scholar] [CrossRef]
- Shida, D.; Kitayama, J.; Yamaguchi, H.; Yamashita, H.; Mori, K.; Watanabe, T.; Yatomi, Y.; Nagawa, H. Sphingosine 1-phosphate transactivates c-Met as well as epidermal growth factor receptor (EGFR) in human gastric cancer cells. FEBS Lett. 2004, 577, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Carl-McGrath, S.; Ebert, M.P.; Lendeckel, U.; Röcken, C. Expression of the local angiotensin II system in gastric cancer may facilitate lymphatic invasion and nodal spread. Cancer Biol. Ther. 2007, 6, 1218–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.M.; Guo, A.B.; Sun, J.F.; Chen, X.L.; Yin, Z.Y. Angiotensin II promotes the progression of human gastric cancer. Mol. Med. Rep. 2014, 9, 1056–1060. [Google Scholar] [CrossRef]
- Sugimoto, M.; Ohno, T.; Yamaoka, Y. Expression of angiotensin II type 1 and type 2 receptor mRNAs in the gastric mucosa of Helicobacter pylori-infected Mongolian gerbils. J. Gastroenterol. 2011, 46, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Wu, Y.L.; Zhong, J.; Jiang, F.X.; Tian, X.L.; Yu, L.F. Angiotensin II type 1 receptor antagonist suppress angiogenesis and growth of gastric cancer xenografts. Dig. Dis. Sci. 2008, 53, 1206–1210. [Google Scholar] [CrossRef]
- Huang, W.; Yu, L.F.; Zhong, J.; Qiao, M.M.; Jiang, F.X.; Du, F.; Tian, X.L.; Wu, Y.L. Angiotensin II type 1 receptor expression in human gastric cancer and induces MMP2 and MMP9 expression in MKN-28 cells. Dig. Dis. Sci. 2008, 53, 163–168. [Google Scholar] [CrossRef]
- Kinoshita, J.; Fushida, S.; Harada, S.; Yagi, Y.; Fujita, H.; Kinami, S.; Ninomiya, I.; Fujimura, T.; Kayahara, M.; Yashiro, M.; et al. Local angiotensin II-generation in human gastric cancer: Correlation with tumor progression through the activation of ERK1/2, NF-kappaB and survivin. Int. J. Oncol. 2009, 34, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Fushida, S.; Harada, S.; Tsukada, T.; Kinoshita, J.; Oyama, K.; Tajima, H.; Ninomiya, I.; Fujimura, T.; Ohta, T. The angiotensin II type 1 receptor blocker candesartan suppresses proliferation and fibrosis in gastric cancer. Cancer Lett. 2014, 355, 46–53. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, T.W.; Park, G.L.; Hwang, Y.S.; Cho, H.J.; Kim, J.T.; Lee, H.G. G protein-coupled estrogen receptor-1 agonist induces chemotherapeutic effect via ER stress signaling in gastric cancer. BMB Rep. 2019, 52, 647–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, E.; Xia, X.; Jiang, C.; Li, Z.; Yang, Z.; Zheng, C.; Wang, X.; Du, S.; Miao, J.; Wang, F.; et al. GPER1 Silencing Suppresses the Proliferation, Migration, and Invasion of Gastric Cancer Cells by Inhibiting PI3K/AKT-Mediated EMT. Front. Cell Dev. Biol. 2020, 8, 591239. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, Y.; Dong, S.; Ge, C.; Xiao, Y.; Li, R.; Ma, X.; Xue, Y.; Zhang, Q.; Lv, J.; et al. Association of CXCR1 and 2 expressions with gastric cancer metastasis in ex vivo and tumor cell invasion in vitro. Cytokine 2014, 69, 6–13. [Google Scholar] [CrossRef]
- Zhao, R.; Wan, Q.; Wang, Y.; Wu, Y.; Xiao, S.; Li, Q.; Shen, X.; Zhuang, W.; Zhou, Y.; Xia, L.; et al. M1-like TAMs are required for the efficacy of PD-L1/PD-1 blockades in gastric cancer. Oncoimmunology 2020, 10, 1862520. [Google Scholar] [CrossRef]
- Tang, C.; Lei, X.; Xiong, L.; Hu, Z.; Tang, B. HMGA1B/2 transcriptionally activated-POU1F1 facilitates gastric carcinoma metastasis via CXCL12/CXCR4 axis-mediated macrophage polarization. Cell Death Dis. 2021, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Long, Q.; Zhuang, K.; Han, K.; Zhang, X.; Guo, H.; Lu, X. Retinoblastoma tumor suppressor gene 1 enhances 5-Fluorouracil chemosensitivity through SDF-1/CXCR4 axis by regulating autophagy in gastric cancer. Pathol. Res. Pract. 2021, 224, 153532. [Google Scholar] [CrossRef]
- Takiguchi, G.; Nishita, M.; Kurita, K.; Kakeji, Y.; Minami, Y. Wnt5a-Ror2 signaling in mesenchymal stem cells promotes proliferation of gastric cancer cells by activating CXCL16-CXCR6 axis. Cancer Sci. 2016, 107, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Xin, Q.; Zhang, N.; Yu, H.B.; Zhang, Q.; Cui, Y.F.; Zhang, C.S.; Ma, Z.; Yang, Y.; Liu, W. CXCR7/CXCL12 axis is involved in lymph node and liver metastasis of gastric carcinoma. World J. Gastroenterol. 2017, 23, 3053–3065. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wang, H.; Kim, W.; Liu, Y.; Deng, H.; Liu, H.; Jiang, Z.; Niu, Z.; Sheng, W.; Nápoles, O.C.; et al. Hormonal Suppression of Stem Cells Inhibits Symmetric Cell Division and Gastric Tumorigenesis. Cell Stem Cell 2020, 26, 739–754.e738. [Google Scholar] [CrossRef]
- Grojean, M.; Schwarz, M.A.; Schwarz, J.R.; Hassan, S.; von Holzen, U.; Zhang, C.; Schwarz, R.E.; Awasthi, N. Targeted dual inhibition of c-Met/VEGFR2 signalling by foretinib improves antitumour effects of nanoparticle paclitaxel in gastric cancer models. J. Cell. Mol. Med. 2021, 25, 4950–4961. [Google Scholar] [CrossRef] [PubMed]
- Shida, D.; Kitayama, J.; Yamaguchi, H.; Yamashita, H.; Mori, K.; Watanabe, T.; Nagawa, H. Lysophospholipids transactivate HER2/neu (erbB-2) in human gastric cancer cells. Biochem. Biophys. Res. Commun. 2005, 327, 907–914. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Alderton, F.; Rakhit, S.; Kong, K.C.; Palmer, T.; Sambi, B.; Pyne, S.; Pyne, N.J. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J. Biol. Chem. 2001, 276, 28578–28585. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Q.; Lin, C.Y.; Huang, Y.L.; Wang, S.W.; Wang, Y.; Huang, B.F.; Lai, Y.W.; Weng, S.L.; Fong, Y.C.; Tang, C.H.; et al. Sphingosine-1-phosphate promotes PDGF-dependent endothelial progenitor cell angiogenesis in human chondrosarcoma cells. Aging 2019, 11, 11040–11053. [Google Scholar] [CrossRef] [PubMed]
- Lepannetier, S.; Zanou, N.; Yerna, X.; Emeriau, N.; Dufour, I.; Masquelier, J.; Muccioli, G.; Tajeddine, N.; Gailly, P. Sphingosine-1-phosphate-activated TRPC1 channel controls chemotaxis of glioblastoma cells. Cell Calcium 2016, 60, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yu, C.P.; Xia, J.T.; Zhang, L.; Weng, G.X.; Zheng, H.Q.; Kong, Q.L.; Hu, L.J.; Zeng, M.S.; Zeng, Y.X.; et al. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin. Cancer Res. 2009, 15, 1393–1399. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wu, Z.; Yu, C.; He, W.; Zheng, H.; He, Y.; Jian, W.; Chen, L.; Zhang, L.; Li, W. miR-124 inhibits cell proliferation in gastric cancer through down-regulation of SPHK1. J. Pathol. 2012, 227, 470–480. [Google Scholar] [CrossRef]
- Zeng, Z.; Mukherjee, A.; Zhang, H. From Genetics to Epigenetics, Roles of Epigenetics in Inflammatory Bowel Disease. Front. Genet. 2019, 10, 1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshaker, H.; Thrower, H.; Pchejetski, D. Sphingosine Kinase 1 in Breast Cancer-A New Molecular Marker and a Therapy Target. Front. Oncol. 2020, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Yan, Y.; Tian, F.; Wu, D.; Huang, Y. Prognostic value of estrogen receptor α and estrogen receptor β in gastric cancer based on a meta-analysis and The Cancer Genome Atlas (TCGA) datasets. Int. J. Surg. 2018, 53, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Hallersund, P.; Elfvin, A.; Helander, H.F.; Fändriks, L. The expression of renin-angiotensin system components in the human gastric mucosa. J. Renin-Angiotensin-Aldosterone Syst. 2011, 12, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Juillerat-Jeanneret, L. The Other Angiotensin II Receptor: AT(2)R as a Therapeutic Target. J. Med. Chem. 2020, 63, 1978–1995. [Google Scholar] [CrossRef]
- Karnik, S.S.; Unal, H.; Kemp, J.R.; Tirupula, K.C.; Eguchi, S.; Vanderheyden, P.M.; Thomas, W.G. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli. Pharmacol. Rev. 2015, 67, 754–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Butler, L.M.; Wu, A.H.; Koh, W.P.; Jin, A.; Wang, R.; Yuan, J.M. Reproductive factors, hormone use and gastric cancer risk: The Singapore Chinese Health Study. Int. J. Cancer 2016, 138, 2837–2845. [Google Scholar] [CrossRef]
- Tian, S.; Zhan, N.; Li, R.; Dong, W. Downregulation of G Protein-Coupled Estrogen Receptor (GPER) is Associated with Reduced Prognosis in Patients with Gastric Cancer. Med. Sci. Monit. 2019, 25, 3115–3126. [Google Scholar] [CrossRef]
- Hata, M.; Kinoshita, H.; Hayakawa, Y.; Konishi, M.; Tsuboi, M.; Oya, Y.; Kurokawa, K.; Hayata, Y.; Nakagawa, H.; Tateishi, K.; et al. GPR30-Expressing Gastric Chief Cells Do Not Dedifferentiate But Are Eliminated via PDK-Dependent Cell Competition During Development of Metaplasia. Gastroenterology 2020, 158, 1650–1666.e1615. [Google Scholar] [CrossRef]
- Nam, K.T.; Lee, H.J.; Sousa, J.F.; Weis, V.G.; O’Neal, R.L.; Finke, P.E.; Romero-Gallo, J.; Shi, G.; Mills, J.C.; Peek, R.M., Jr.; et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010, 139, 2028–2037.e2029. [Google Scholar] [CrossRef] [Green Version]
- Burclaff, J.; Willet, S.G.; Sáenz, J.B.; Mills, J.C. Proliferation and Differentiation of Gastric Mucous Neck and Chief Cells During Homeostasis and Injury-induced Metaplasia. Gastroenterology 2020, 158, 598–609.e595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Teng, R.; Xu, C.; Wang, Q.; Guo, J.; Xu, C.; Li, Z.; Xie, S.; Shen, J.; Wang, L. Overexpression of ERα inhibits proliferation and invasion of MKN28 gastric cancer cells by suppressing β-catenin. Oncol. Rep. 2013, 30, 1622–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Deng, H.; Zou, F.; Fu, Z.; Chen, Y.; Wang, Z.; Liu, L. ER-α36-mediated gastric cancer cell proliferation via the c-Src pathway. Oncol. Lett. 2013, 6, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Zhen, H.; Zou, F.; Wang, X.; Chen, Y.; Liu, L. Involvement of the Akt signaling pathway in ER-α36/GRP94-mediated signaling in gastric cancer. Oncol. Lett. 2014, 8, 2077–2080. [Google Scholar] [CrossRef] [Green Version]
- Ryu, W.S.; Kim, J.H.; Jang, Y.J.; Park, S.S.; Um, J.W.; Park, S.H.; Kim, S.J.; Mok, Y.J.; Kim, C.S. Expression of estrogen receptors in gastric cancer and their clinical significance. J. Surg. Oncol. 2012, 106, 456–461. [Google Scholar] [CrossRef]
- Chandanos, E.; Lindblad, M.; Rubio, C.A.; Jia, C.; Warner, M.; Gustafsson, J.A.; Lagergren, J. Tamoxifen exposure in relation to gastric adenocarcinoma development. Eur. J. Cancer 2008, 44, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Sunakawa, Y.; Cao, S.; Berger, M.D.; Matsusaka, S.; Yang, D.; Zhang, W.; Ning, Y.; Parekh, A.; Stremitzer, S.; Mendez, A.; et al. Estrogen receptor-beta genetic variations and overall survival in patients with locally advanced gastric cancer. Pharm. J 2017, 17, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Jin, J.; Zhou, L.; Wu, L.; Cao, Y.; Yan, H.; Huang, Q.; Wang, L.; Zou, X. Suppression of estrogen receptor-beta promotes gastric cancer cell apoptosis with induction of autophagy. Am. J. Transl. Res. 2020, 12, 4397–4409. [Google Scholar] [PubMed]
- Kumral, Z.N.; Memi, G.; Ercan, F.; Yeğen, B.C. Estrogen alleviates acetic acid-induced gastric or colonic damage via both ERα- and ERβ-mediated and direct antioxidant mechanisms in rats. Inflammation 2014, 37, 694–705. [Google Scholar] [CrossRef]
- Chen, C.; Gong, X.; Yang, X.; Shang, X.; Du, Q.; Liao, Q.; Xie, R.; Chen, Y.; Xu, J. The roles of estrogen and estrogen receptors in gastrointestinal disease. Oncol. Lett. 2019, 18, 5673–5680. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Li, K.; Maskey, N.; Xu, Z.; Yu, F.; Peng, C.; Li, Y.; Yang, G. Overexpression of the chemokine receptor CXCR3 and its correlation with favorable prognosis in gastric cancer. Hum. Pathol. 2015, 46, 1872–1880. [Google Scholar] [CrossRef]
- Wang, T.C.; Dangler, C.A.; Chen, D.; Goldenring, J.R.; Koh, T.; Raychowdhury, R.; Coffey, R.J.; Ito, S.; Varro, A.; Dockray, G.J.; et al. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 2000, 118, 36–47. [Google Scholar] [CrossRef]
- Zavros, Y.; Rieder, G.; Ferguson, A.; Samuelson, L.C.; Merchant, J.L. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology 2002, 122, 119–133. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Enzinger, P.C.; Mayer, R.J. Esophageal cancer. N. Engl. J. Med. 2003, 349, 2241–2252. [Google Scholar] [CrossRef] [Green Version]
- Noone, A.M.; Cronin, K.A.; Altekruse, S.F.; Howlader, N.; Lewis, D.R.; Petkov, V.I.; Penberthy, L. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol. Biomark. Prev. 2017, 26, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhi, Y.; Song, H.; Zong, M.; Yi, J.; Mao, G.; Chen, L.; Huang, G. S1PR1 promotes proliferation and inhibits apoptosis of esophageal squamous cell carcinoma through activating STAT3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Li, X.; Hylemon, P.B.; Zhou, H. Conjugated Bile Acids Promote Invasive Growth of Esophageal Adenocarcinoma Cells and Cancer Stem Cell Expansion via Sphingosine 1-Phosphate Receptor 2-Mediated Yes-Associated Protein Activation. Am. J. Pathol. 2018, 188, 2042–2058. [Google Scholar] [CrossRef]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.J.; Xing, H.; Wang, Y.X.; Zhang, X.; Zhan, Q.M.; Geng, M.Y.; Ding, J.; Meng, L.H. PI3Kα inhibitors sensitize esophageal squamous cell carcinoma to radiation by abrogating survival signals in tumor cells and tumor microenvironment. Cancer Lett. 2019, 459, 145–155. [Google Scholar] [CrossRef]
- Chen, Y.H.; Huang, C.H.; Lu, H.I.; Chen, C.H.; Huang, W.T.; Hsieh, M.J.; Rau, K.M.; Chang, A.Y.; Lin, W.C.; Li, S.H. Prognostic impact of renin-angiotensin system blockade in esophageal squamous cell carcinoma. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 1185–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Chiyo, T.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Nakahara, M.; Kobayashi, N.; Nishiyama, N.; Yachida, T.; et al. Telmisartan Inhibits Cell Proliferation and Tumor Growth of Esophageal Squamous Cell Carcinoma by Inducing S-Phase Arrest In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 3197. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, S.; Morishita, A.; Ogawa, K.; Tadokoro, T.; Chiyo, T.; Kato, K.; Kobara, H.; Mori, H.; Iwama, H.; Masaki, T. The angiotensin II type 1 receptor antagonist telmisartan inhibits cell proliferation and tumor growth of esophageal adenocarcinoma via the AMPKα/mTOR pathway in vitro and in vivo. Oncotarget 2017, 8, 8536–8549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Deng, L.; Lai, Y.; Liu, Z. Over expression of GPR30, indicating poor prognosis and promoting proliferation, upregulates Beclin-1 expression via p38MAPK signaling in esophageal squamous cell carcinoma progression. Int. J. Clin. Exp. Pathol. 2018, 11, 3426–3435. [Google Scholar] [PubMed]
- Jiang, P.; De Li, S.; Li, Z.G.; Zhu, Y.C.; Yi, X.J.; Li, S.M. The expression of protease-activated receptors in esophageal carcinoma cells: The relationship between changes in gene expression and cell proliferation, apoptosis in vitro and growing ability in vivo. Cancer Cell Int. 2018, 18, 81. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Deng, X.; Zhang, Q.; Liu, H.; Wang, N.; Liu, Z.; Dai, E.; Deng, Q. PAR-2 promotes invasion and migration of esophageal cancer cells by activating MEK/ERK and PI3K/Akt signaling pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 787–797. [Google Scholar]
- Chen, J.; Xie, L.; Zheng, Y.; Liu, C. Effects of silenced PAR-2 on cell proliferation, invasion and metastasis of esophageal cancer. Oncol. Lett. 2017, 14, 4115–4121. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; An, S.; Wang, D.; Ji, H.; Guo, X.; Wang, Z. Activation of PAR4 Upregulates p16 through Inhibition of DNMT1 and HDAC2 Expression via MAPK Signals in Esophageal Squamous Cell Carcinoma Cells. J. Immunol. Res. 2018, 2018, 4735752. [Google Scholar] [CrossRef]
- Cui, Z.; Li, D.; Liu, J.; Zhang, Y.; Xu, H.; Yin, H.; Li, H.; Wang, G.; Cai, H.; Zhang, L.; et al. G-protein-coupled receptor 120 regulates the development and progression of human esophageal cancer. Oncol. Rep. 2018, 40, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Haigh, C.R.; Attwood, S.E.; Thompson, D.G.; Jankowski, J.A.; Kirton, C.M.; Pritchard, D.M.; Varro, A.; Dimaline, R. Gastrin induces proliferation in Barrett’s metaplasia through activation of the CCK2 receptor. Gastroenterology 2003, 124, 615–625. [Google Scholar] [CrossRef]
- Abdalla, S.I.; Lao-Sirieix, P.; Novelli, M.R.; Lovat, L.B.; Sanderson, I.R.; Fitzgerald, R.C. Gastrin-induced cyclooxygenase-2 expression in Barrett’s carcinogenesis. Clin. Cancer Res. 2004, 10, 4784–4792. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Tao, Y.F.; Zhou, Z.; Cao, B.R.; Wu, S.Y.; Zhang, Y.L.; Hu, S.Y.; Zhao, W.L.; Wang, J.; Lou, G.L.; et al. An novel role of sphingosine kinase-1 (SPHK1) in the invasion and metastasis of esophageal carcinoma. J. Transl. Med. 2011, 9, 157. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, M.; Ichikawa, H.; Nagahashi, M.; Hanyu, T.; Ishikawa, T.; Kano, Y.; Muneoka, Y.; Wakai, T. Phospho-Sphingosine Kinase 1 Expression in Lymphatic Spread of Esophageal Squamous Cell Carcinoma. J. Surg. Res. 2019, 234, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Kusaczuk, M. Tauroursodeoxycholate-Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Rees, J.R.; Onwuegbusi, B.A.; Save, V.E.; Alderson, D.; Fitzgerald, R.C. In vivo and in vitro evidence for transforming growth factor-beta1-mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res. 2006, 66, 9583–9590. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Nisar, S.; Maacha, S.; Carneiro-Lobo, T.C.; Akhtar, S.; Siveen, K.S.; Wani, N.A.; Rizwan, A.; Bagga, P.; Singh, M.; et al. Cytokine-chemokine network driven metastasis in esophageal cancer; promising avenue for targeted therapy. Mol. Cancer 2021, 20, 2. [Google Scholar] [CrossRef]
- Tang, H.; Fu, S.; Zhai, S.; Song, Y.; Asgari, M.M.; Han, J. Use of antihypertensive drugs and risk of keratinocyte carcinoma: A meta-analysis of observational studies. Pharmacoepidemiol. Drug Saf. 2018, 27, 279–288. [Google Scholar] [CrossRef]
- Bratlie, S.O.; Wallenius, V.; Edebo, A.; Fändriks, L.; Casselbrant, A. Proteomic Approach to the Potential Role of Angiotensin II in Barrett Dysplasia. Proteomics. Clin. Appl. 2019, 13, e1800102. [Google Scholar] [CrossRef]
- Zeng, F.M.; He, J.Z.; Wang, S.H.; Liu, D.K.; Xu, X.E.; Wu, J.Y.; Li, E.M.; Xu, L.Y. A Novel Three-Gene Model Predicts Prognosis and Therapeutic Sensitivity in Esophageal Squamous Cell Carcinoma. BioMed Res. Int. 2019, 2019, 9828637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Lu, H.I.; Lo, C.M.; Huang, C.C.; Hsiao, C.C.; Li, S.H. The clinical impact of angiotensin-(1-7)/mitochondrial assembly receptor axis in esophageal squamous cell carcinoma patients receiving curative esophagectomy. J. Formos. Med. Assoc. 2020, 119, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.B.; Wild, C.P.; Forman, D. A systematic review and meta-analysis of the sex ratio for Barrett’s esophagus, erosive reflux disease, and nonerosive reflux disease. Am. J. Epidemiol. 2005, 162, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Bohanes, P.; Yang, D.; Chhibar, R.S.; Labonte, M.J.; Winder, T.; Ning, Y.; Gerger, A.; Benhaim, L.; Paez, D.; Wakatsuki, T.; et al. Influence of sex on the survival of patients with esophageal cancer. J. Clin. Oncol. 2012, 30, 2265–2272. [Google Scholar] [CrossRef]
- Wang, Q.M.; Qi, Y.J.; Jiang, Q.; Ma, Y.F.; Wang, L.D. Relevance of serum estradiol and estrogen receptor beta expression from a high-incidence area for esophageal squamous cell carcinoma in China. Med. Oncol. 2011, 28, 188–193. [Google Scholar] [CrossRef]
- Wang, C.; Wang, P.; Liu, J.C.; Zhao, Z.A.; Guo, R.; Li, Y.; Liu, Y.S.; Li, S.G.; Zhao, Z.G. Interaction of Estradiol and Endoplasmic Reticulum Stress in the Development of Esophageal Carcinoma. Front. Endocrinol. 2020, 11, 410. [Google Scholar] [CrossRef]
- Masaka, T.; Iijima, K.; Endo, H.; Asanuma, K.; Ara, N.; Ishiyama, F.; Asano, N.; Koike, T.; Imatani, A.; Shimosegawa, T. Gender differences in oesophageal mucosal injury in a reflux oesophagitis model of rats. Gut 2013, 62, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, B.; Padget, M.R.; Schlom, J.; Hodge, J.W. Exploiting off-target effects of estrogen deprivation to sensitize estrogen receptor negative breast cancer to immune killing. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, H.T.; Li, S.; Li, K.; Lin, N.; Fan, Q.X.; Zheng, Y.L. Prognostic value of protease-activated receptor 2 expression in oesophageal squamous cell carcinoma. J. Int. Med. Res. 2010, 38, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e492. [Google Scholar] [CrossRef] [Green Version]
- Fabisiak, A.; Bartoszek, A.; Talar, M.; Binienda, A.; Dziedziczak, K.; Krajewska, J.B.; Mosińska, P.; Niewinna, K.; Tarasiuk, A.; Mokrowiecka, A.; et al. Expression of FFAR3 and FFAR4 Is Increased in Gastroesophageal Reflux Disease. J. Clin. Med. 2020, 9, 4111. [Google Scholar] [CrossRef]
- Violin, J.D.; Crombie, A.L.; Soergel, D.G.; Lark, M.W. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014, 35, 308–316. [Google Scholar] [CrossRef]
- Ryba, D.M.; Li, J.; Cowan, C.L.; Russell, B.; Wolska, B.M.; Solaro, R.J. Long-Term Biased β-Arrestin Signaling Improves Cardiac Structure and Function in Dilated Cardiomyopathy. Circulation 2017, 135, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Shen, H.; Oprea, I.; Worrall, C.; Stefanescu, R.; Girnita, A.; Girnita, L. β-Arrestin-biased agonism as the central mechanism of action for insulin-like growth factor 1 receptor-targeting antibodies in Ewing’s sarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 20620–20625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; Van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.; Vermeire, S.; Ghosh, S.; Liu, W.J.; Petersen, A.; Charles, L.; Huang, V.; Usiskin, K.; et al. Long-Term Efficacy And Safety Of Ozanimod In Moderate-To-Severe Ulcerative Colitis: Results From The Open-Label Extension Of The Randomized, Phase 2 Touchstone Study. J. Crohns Colitis 2021, 15, 1120–1129. [Google Scholar] [CrossRef]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy With FOLFIRINOX in Combination With Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Park, K.H.; Oh, S.C.; Seo, J.H.; Kim, J.S.; Shin, S.W.; Kim, Y.H. How does inhibition of the renin-angiotensin system affect the prognosis of advanced gastric cancer patients receiving platinum-based chemotherapy? Oncology 2012, 83, 354–360. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, J.C.; Vanoni, S.; Zeng, C.; Waggoner, L.; Yang, Y.; Wu, D.; Uddin, J.; Karns, R.; Kottyan, L.; Mukkada, V.; et al. 17β-Estradiol protects the esophageal epithelium from IL-13-induced barrier dysfunction and remodeling. J. Allergy Clin. Immunol. 2019, 143, 2131–2146. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Späth, S.S.; Marjani, S.L.; Zhang, W.; Pan, X. Characterization of cancer genomic heterogeneity by next-generation sequencing advances precision medicine in cancer treatment. Precis. Clin. Med. 2018, 1, 29–48. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| GPCRs | Roles | References |
|---|---|---|
| Sphingosine 1-phosphate receptors | ||
| S1PR1 | Aggravates intestinal inflammation and promotes colitis-associated colorectal tumorigenesis | [30] |
| S1PR2 | Inhibits tumorigenesis and reverses 5-FU chemoresistance | [31,32] |
| S1PR3 | Promotes tumorigenesis | [33] |
| S1PR4 | Limits CD8+ T cell expansion, inhibits cancer proliferation, and reduces chemotherapy success | [34] |
| S1PR5 | Promotes CRC growth, migration, and invasion | [35] |
| Angiotensin II receptors | ||
| AT1R | Promotes tumorigenesis and regulates cancer immunotherapy | [11,36,37,38] |
| AT2R | Increases the expression levels of E-cadherin | [11] |
| Estrogen-related GPCRs | ||
| GPER | Shows bidirectional effects on tumorigenesis | [15,39,40,41] |
| Other GPCRs | ||
| FFAR2 | Shows bidirectional effects on tumorigenesis | [42,43,44] |
| GPR109A | Inhibits colon inflammation and tumorigenesis | |
| FFAR3 | Enhances cell proliferation and inhibits apoptosis | [45] |
| CCK2R | Promotes tumorigenesis | [46,47] |
| GPR56 | Enhances EMT and promotes chemoresistance | [48,49] |
| GPCRs | Roles | References |
|---|---|---|
| Sphingosine 1-phosphate receptors | ||
| S1PR1 | Promotes cell proliferation and invasion | [86] |
| S1PR2 | Shows bidirectional effects on cell migration | [87,88] |
| S1PR3 | Promotes cell migration | [87] |
| Angiotensin II receptors | ||
| AT1R | Promotes tumorigenesis and aggravates gastric inflammation | [89,90,91,92,93,94,95] |
| AT2R | Promotes tumorigenesis and aggravates gastric inflammation | [89,91] |
| Estrogen-related GPCRs | ||
| GPER | Shows bidirectional effects on tumorigenesis and regulates chemoresistance | [16,96,97] |
| Other GPCRs | ||
| CXCR1 | Promotes cell migration and invasion | [98] |
| CXCR2 | Promotes cell migration and invasion | [98] |
| CXCR3 | Improves therapeutic efficacy of PD-L1/PD-1 | [99] |
| CXCR4 | Promotes metastasis and increases 5-FU chemosensitivity | [100,101] |
| CXCR6 | Promotes cell proliferation and migration | [102] |
| CXCR7 | Promotes cell proliferation and migration | [103] |
| CCK2R | Promotes tumorigenesis | [104] |
| GPCRs | Roles | References |
|---|---|---|
| Sphingosine 1-phosphate receptors | ||
| S1PR1 | Promotes cell proliferation and inhibits apoptosis | [139] |
| S1PR2 | Promotes tumorigenesis | [140,141] |
| S1PR3 | Promotes Akt phosphorylation, and regulates radiation resistance | [142] |
| S1PR5 | Inhibits cell proliferation and migration | [58] |
| Angiotensin II receptors | ||
| AT1R | Promotes cell proliferation and angiogenesis | [143,144,145] |
| Estrogen-related GPCRs | ||
| GPER | Promotes cell proliferation | [146] |
| Other GPCRs | ||
| PAR1 | Promotes cell proliferation | [147] |
| PAR2 | Promotes cell invasion and migration | [148,149] |
| PAR4 | Inhibits cell proliferation | [150] |
| GPR120 | Promotes EMT and cancer progression | [151] |
| CCK2R | Promotes cell proliferation | [152,153] |
| Compound | Targets | Indications | Clinical Phase |
|---|---|---|---|
| Fingolimod | S1PR1, S1PR3, S1PR4, and S1PR5 agonist | Multiple sclerosis | Approved |
| Ozanimod | S1PR1 and S1PR5 agonist | Ulcerative colitis | Phase II |
| Siponimod | S1PR1 and S1PR5 agonist | Multiple sclerosis | Approved |
| Ponesimod | S1PR1 agonist | Multiple sclerosis | Approved |
| Etrasimod | S1PR1, S1PR4, and S1PR5 agonist | Ulcerative colitis and Crohn’s disease | Phase II and Phase III |
| Amiselimod | S1PR1 antagonist | Ulcerative colitis | Phase II |
| LNS8801 | GPER agonist | Solid tumor and adult lymphoma | Phase I and Phase II |
| Plerixafor | CXCR4 SMI | Advanced colorectal adenocarcinomas, pancreatic cancer, and ovarian cancer | Phase I |
| Motixafortide | CXCR4 antagonist | Metastatic pancreatic adenocarcinoma | Phase II |
| SX-682 | CXCR1 and CXCR2 SMI | Metastatic colon adenocarcinoma | Phase I and Phase II |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, Z.; Ma, C.; Chen, K.; Jiang, M.; Vasu, R.; Liu, R.; Zhao, Y.; Zhang, H. Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs. Cells 2021, 10, 2988. https://doi.org/10.3390/cells10112988
Zeng Z, Ma C, Chen K, Jiang M, Vasu R, Liu R, Zhao Y, Zhang H. Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs. Cells. 2021; 10(11):2988. https://doi.org/10.3390/cells10112988
Chicago/Turabian StyleZeng, Zhen, Chunxiang Ma, Kexin Chen, Mingshan Jiang, Reshma Vasu, Rui Liu, Yinglan Zhao, and Hu Zhang. 2021. "Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs" Cells 10, no. 11: 2988. https://doi.org/10.3390/cells10112988
APA StyleZeng, Z., Ma, C., Chen, K., Jiang, M., Vasu, R., Liu, R., Zhao, Y., & Zhang, H. (2021). Roles of G Protein-Coupled Receptors (GPCRs) in Gastrointestinal Cancers: Focus on Sphingosine 1-Shosphate Receptors, Angiotensin II Receptors, and Estrogen-Related GPCRs. Cells, 10(11), 2988. https://doi.org/10.3390/cells10112988