
Neuroprotective Effects of Fingolimod in a Cellular Model of Optic Neuritis

 , , , and
, , , and
Abstract
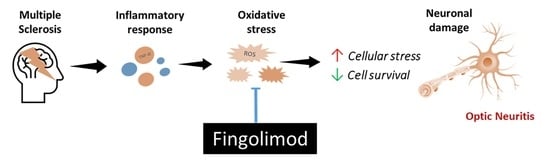
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. The In Vitro Model of Optic Neuritis
2.3. Treatments with Fingolimod
2.4. Cell Viability
2.5. Western Blot Analysis
2.6. Immunofluorescence Staining
2.7. Cellular ROS Formation Using DCF Assay
2.8. Mitochondrial ROS Measurement
2.9. H2O2 Treatment and LDH Assay
2.10. Statistical Analysis
3. Results
3.1. Fingolimod Treatment Reduces TNFα-Induced Neuronal Injury
3.2. Fingolimod Attenuates Cellular Stress and Survival Signaling
3.3. Effect of Fingolimod on Neuronal Cell Death
3.4. Effect of Fingolimod on TNFα-Induced Neuronal Damage
3.5. Effect of Fingolimod on ROS Formation
3.6. Effect of Fingolimod on Mitochondrial Dynamics and ROS Formation
3.7. Fingolimod Attenuates H2O2-Induced Cellular Damage and Stress Signaling in R28 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Used | Catalogue No. | Company | Dilution |
|---|---|---|---|
| Phospho-P38 MAP Kinase | 4511S | Cell Signaling | 1:1000 |
| Total P38 MAP Kinase | 9212S | Cell Signaling | 1:1000 |
| Phospho-Akt | 4060S | Cell Signaling | 1:1000 |
| Total Akt | 9272S | Cell Signaling | 1:1000 |
| Bcl-xL | 2764S | Cell Signaling | 1:1000 |
| Cleaved caspase-3 | 9664S | Cell Signaling | 1:1000 |
| β-Actin | A1978-200UL | Sigma | 1:10,000 |
| Tuj1 | MAB1195 | R&D Systems | 1:500 |
| Neuron Specific Enolase | NSE | Aves Lab | 1:500 |
| Mitofusin-2 | 74792 | Cell Signaling | 1:1000 |
| p-DRP-1 | 4494 | Cell Signaling | 1:1000 |
| OPA-1 | 80471S | Cell Signaling | 1:1000 |
References
- Filippi, M.; Rocca, M.A. Multiple sclerosis: New measures to monitor the disease. Lancet Neurol. 2013, 12, 12–13. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef]
- Zwibel, H.L.; Smrtka, J. Improving quality of life in multiple sclerosis: An unmet need. Am. J. Manag. Care 2011, 17 (Suppl. 5), S139–S145. [Google Scholar] [PubMed]
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.; Raininko, R.; Fagius, J. Bilateral and recurrent optic neuritis in multiple sclerosis. Acta Neurol. Scand. 2011, 123, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, T.L.; Frederiksen, J.L.; Bronnum-Hansen, H.; Petersen, H.C. Optic neuritis as onset manifestation of multiple sclerosis: A nationwide, long-term survey. Neurology 1999, 53, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.D.; Ishikawa, H.; Galetta, K.M.; Sakai, R.E.; Feller, D.J.; Henderson, S.B.; Wilson, J.A.; Maguire, M.G.; Galetta, S.L.; Frohman, E.; et al. Ganglion cell loss in relation to visual disability in multiple sclerosis. Ophthalmology 2012, 119, 1250–1257. [Google Scholar] [CrossRef]
- Trip, S.A.; Schlottmann, P.G.; Jones, S.J.; Altmann, D.R.; Garway-Heath, D.F.; Thompson, A.J.; Plant, G.T.; Miller, D.H. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann. Neurol. 2005, 58, 383–391. [Google Scholar] [CrossRef]
- Salter, A.R.; Tyry, T.; Vollmer, T.; Cutter, G.R.; Marrie, R.A. “Seeing” in NARCOMS: A look at vision-related quality of life in the NARCOMS registry. Mult. Scler. 2013, 19, 953–960. [Google Scholar] [CrossRef]
- Adachi, K.; Chiba, K. FTY720 story. Its discovery and the following accelerated development of sphingosine 1-phosphate receptor agonists as immunomodulators based on reverse pharmacology. Perspect. Med. Chem. 2007, 1, 11–23. [Google Scholar] [CrossRef]
- Park, S.J.; Im, D.S. Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol. Ther. (Seoul) 2017, 25, 80–90. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef]
- Dyckman, A.J. Modulators of Sphingosine-1-phosphate Pathway Biology: Recent Advances of Sphingosine-1-phosphate Receptor 1 (S1P1) Agonists and Future Perspectives. J. Med. Chem. 2017, 60, 5267–5289. [Google Scholar] [CrossRef] [PubMed]
- Miron, V.E.; Schubart, A.; Antel, J.P. Central nervous system-directed effects of FTY720 (fingolimod). J. Neurol. Sci. 2008, 274, 13–17. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Graler, M.H.; Goetzl, E.J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004, 18, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Chiba, K.; Yanagawa, Y.; Masubuchi, Y.; Kataoka, H.; Kawaguchi, T.; Ohtsuki, M.; Hoshino, Y. FTY720, a novel immunosuppressant, induces sequestration of circulating mature lymphocytes by acceleration of lymphocyte homing in rats. I. FTY720 selectively decreases the number of circulating mature lymphocytes by acceleration of lymphocyte homing. J. Immunol. 1998, 160, 5037–5044. [Google Scholar]
- Zhao, P.; Yang, X.; Yang, L.; Li, M.; Wood, K.; Liu, Q.; Zhu, X. Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Martin-Montanez, E.; Pavia, J.; Valverde, N.; Boraldi, F.; Lara, E.; Oliver, B.; Hurtado-Guerrero, I.; Fernandez, O.; Garcia-Fernandez, M. The S1P mimetic fingolimod phosphate regulates mitochondrial oxidative stress in neuronal cells. Free Radic. Biol. Med. 2019, 137, 116–130. [Google Scholar] [CrossRef]
- Ren, M.; Han, M.; Wei, X.; Guo, Y.; Shi, H.; Zhang, X.; Perez, R.G.; Lou, H. FTY720 Attenuates 6-OHDA-Associated Dopaminergic Degeneration in Cellular and Mouse Parkinsonian Models. Neurochem. Res. 2017, 42, 686–696. [Google Scholar] [CrossRef]
- Vidal-Martinez, G.; Vargas-Medrano, J.; Gil-Tommee, C.; Medina, D.; Garza, N.T.; Yang, B.; Segura-Ulate, I.; Dominguez, S.J.; Perez, R.G. FTY720/Fingolimod Reduces Synucleinopathy and Improves Gut Motility in A53T Mice: Contributions of pro-brain-derived neurotrophic factor (pro-bdnf) and mature bdnf. J. Biol. Chem. 2016, 291, 20811–20821. [Google Scholar] [CrossRef]
- Aytan, N.; Choi, J.K.; Carreras, I.; Brinkmann, V.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 24939. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Tao, W.; Liu, M. Systematic review and meta-analysis of the efficacy of sphingosine-1-phosphate (S1P) receptor agonist FTY720 (fingolimod) in animal models of stroke. Int. J. Neurosci. 2013, 123, 163–169. [Google Scholar] [CrossRef]
- Yang, T.; Zha, Z.; Yang, X.; Kang, Y.; Wang, X.; Tong, Y.; Zhao, X.; Wang, L.; Fan, Y. Neuroprotective Effects of Fingolimod Supplement on the Retina and Optic Nerve in the Mouse Model of Experimental Autoimmune Encephalomyelitis. Front. Neurosci. 2021, 15, 663541. [Google Scholar] [CrossRef] [PubMed]
- Seigel, G.M. Review: R28 retinal precursor cells: The first 20 years. Mol. Vis. 2014, 20, 301–306. [Google Scholar] [PubMed]
- Seigel, G.M.; Mutchler, A.L.; Imperato, E.L. Expression of glial markers in a retinal precursor cell line. Mol. Vis. 1996, 2, 9233983. [Google Scholar]
- Seigel, G.M.; Sun, W.; Wang, J.; Hershberger, D.H.; Campbell, L.M.; Salvi, R.J. Neuronal gene expression and function in the growth-stimulated R28 retinal precursor cell line. Curr. Eye Res. 2004, 28, 257–269. [Google Scholar] [CrossRef]
- Boriushkin, E.; Wang, J.J.; Li, J.; Jing, G.; Seigel, G.M.; Zhang, S.X. Identification of p58IPK as a novel neuroprotective factor for retinal neurons. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1374–1386. [Google Scholar] [CrossRef]
- Kong, D.; Gong, L.; Arnold, E.; Shanmugam, S.; Fort, P.E.; Gardner, T.W.; Abcouwer, S.F. Insulin-like growth factor 1 rescues R28 retinal neurons from apoptotic death through ERK-mediated BimEL phosphorylation independent of Akt. Exp. Eye Res. 2016, 151, 82–95. [Google Scholar] [CrossRef]
- Goc, A.; Kochuparambil, S.T.; Al-Husein, B.; Al-Azayzih, A.; Mohammad, S.; Somanath, P.R. Simultaneous modulation of the intrinsic and extrinsic pathways by simvastatin in mediating prostate cancer cell apoptosis. BMC Cancer 2012, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Hong, S.; Iizuka, Y.; Kim, C.Y.; Seong, G.J. The neuroprotective effect of maltol against oxidative stress on rat retinal neuronal cells. Korean J. Ophthalmol. 2015, 29, 58–65. [Google Scholar] [CrossRef]
- Geisert, E.E., Jr.; Frankfurter, A. The neuronal response to injury as visualized by immunostaining of class III beta-tubulin in the rat. Neurosci. Lett. 1989, 102, 137–141. [Google Scholar] [CrossRef]
- Iwanaga, T.; Takahashi, Y.; Fujita, T. Immunohistochemistry of neuron-specific and glia-specific proteins. Arch. Histol. Cytol. 1989, 52, 13–24. [Google Scholar] [CrossRef]
- Palani, C.D.; Fouda, A.Y.; Liu, F.; Xu, Z.; Mohamed, E.; Giri, S.; Smith, S.B.; Caldwell, R.B.; Narayanan, S.P. Deletion of Arginase 2 Ameliorates Retinal Neurodegeneration in a Mouse Model of Multiple Sclerosis. Mol. Neurobiol. 2019, 56, 8589–8602. [Google Scholar] [CrossRef] [PubMed]
- Candadai, A.A.; Liu, F.; Fouda, A.Y.; Alfarhan, M.; Palani, C.D.; Xu, Z.; Caldwell, R.B.; Narayanan, S.P. Deletion of arginase 2 attenuates neuroinflammation in an experimental model of optic neuritis. PLoS ONE 2021, 16, e0247901. [Google Scholar] [CrossRef]
- Long, J.; Wang, Q.; He, H.; Sui, X.; Lin, G.; Wang, S.; Yang, J.; You, P.; Luo, Y.; Wang, Y. NLRP3 inflammasome activation is involved in trimethyltin-induced neuroinflammation. Brain Res. 2019, 1718, 186–193. [Google Scholar] [CrossRef]
- Miyagi, A.; Kawashiri, T.; Shimizu, S.; Shigematsu, N.; Kobayashi, D.; Shimazoe, T. Dimethyl Fumarate Attenuates Oxaliplatin-Induced Peripheral Neuropathy without Affecting the Anti-tumor Activity of Oxaliplatin in Rodents. Biol. Pharm. Bull. 2019, 42, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Clarner, T.; Janssen, K.; Nellessen, L.; Stangel, M.; Skripuletz, T.; Krauspe, B.; Hess, F.M.; Denecke, B.; Beutner, C.; Linnartz-Gerlach, B.; et al. CXCL10 triggers early microglial activation in the cuprizone model. J. Immunol. 2015, 194, 3400–3413. [Google Scholar] [CrossRef] [PubMed]
- Gobel, K.; Ruck, T.; Meuth, S.G. Cytokine signaling in multiple sclerosis: Lost in translation. Mult. Scler. 2018, 24, 432–439. [Google Scholar] [CrossRef]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef]
- Lei, Q.; Tan, J.; Yi, S.; Wu, N.; Wang, Y.; Wu, H. Mitochonic acid 5 activates the MAPK-ERK-yap signaling pathways to protect mouse microglial BV-2 cells against TNFalpha-induced apoptosis via increased Bnip3-related mitophagy. Cell Mol. Biol. Lett. 2018, 23, 14. [Google Scholar] [CrossRef]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef]
- Pang, Y.; Qin, M.; Hu, P.; Ji, K.; Xiao, R.; Sun, N.; Pan, X.; Zhang, X. Resveratrol protects retinal ganglion cells against ischemia induced damage by increasing Opa1 expression. Int. J. Mol. Med. 2020, 46, 1707–1720. [Google Scholar] [CrossRef]
- Mathew, B.; Chennakesavalu, M.; Sharma, M.; Torres, L.A.; Stelman, C.R.; Tran, S.; Patel, R.; Burg, N.; Salkovski, M.; Kadzielawa, K.; et al. Autophagy and post-ischemic conditioning in retinal ischemia. Autophagy 2020, 17, 1479–1499. [Google Scholar] [CrossRef]
- Tan, J.; Digicaylioglu, M.; Wang, S.X.J.; Dresselhuis, J.; Dedhar, S.; Mills, J. Insulin attenuates apoptosis in neuronal cells by an integrin-linked kinase-dependent mechanism. Heliyon 2019, 5, e02294. [Google Scholar] [CrossRef] [PubMed]
- Kale, N. Optic neuritis as an early sign of multiple sclerosis. Eye Brain 2016, 8, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxidative Med. Cell Longev. 2017, 2017, 2817252. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. The role of oxidative stress in the pathogenesis of multiple sclerosis: The need for effective antioxidant therapy. J. Neurol. 2004, 251, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.A.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol. Med. 2010, 2, 504–515. [Google Scholar] [CrossRef]
- Azuchi, Y.; Kimura, A.; Guo, X.; Akiyama, G.; Noro, T.; Harada, C.; Nishigaki, A.; Namekata, K.; Harada, T. Valproic acid and ASK1 deficiency ameliorate optic neuritis and neurodegeneration in an animal model of multiple sclerosis. Neurosci. Lett. 2017, 639, 82–87. [Google Scholar] [CrossRef]
- Guo, X.; Harada, C.; Namekata, K.; Kimura, A.; Mitamura, Y.; Yoshida, H.; Matsumoto, Y.; Harada, T. Spermidine alleviates severity of murine experimental autoimmune encephalomyelitis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2696–2703. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Namekata, K.; Kimura, A.; Noro, T.; Azuchi, Y.; Semba, K.; Harada, C.; Yoshida, H.; Mitamura, Y.; Harada, T. Brimonidine suppresses loss of retinal neurons and visual function in a murine model of optic neuritis. Neurosci. Lett. 2015, 592, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Yevgi, R.; Demir, R. Oxidative stress activity of fingolimod in multiple sclerosis. Clin. Neurol. Neurosurg. 2021, 202, 106500. [Google Scholar] [CrossRef]
- Ryba, D.M.; Warren, C.M.; Karam, C.N.; Davis, R.T., 3rd; Chowdhury, S.A.K.; Alvarez, M.G.; McCann, M.; Liew, C.W.; Wieczorek, D.F.; Varga, P.; et al. Sphingosine-1-Phosphate Receptor Modulator, FTY720, Improves Diastolic Dysfunction and Partially Reverses Atrial Remodeling in a Tm-E180G Mouse Model Linked to Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2019, 12, e005835. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Medrano, J.; Segura-Ulate, I.; Yang, B.; Chinnasamy, R.; Arterburn, J.B.; Perez, R.G. FTY720-Mitoxy reduces toxicity associated with MSA-like alpha-synuclein and oxidative stress by increasing trophic factor expression and myelin protein in OLN-93 oligodendroglia cell cultures. Neuropharmacology 2019, 158, 107701. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, X.; Gao, J.; Liang, S.; Hao, Y.; Sun, C.; Xia, W.; Cao, Y.; Wu, L. Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci. 2017, 173, 43–54. [Google Scholar] [CrossRef]
- Ng, X.; Sadeghian, M.; Heales, S.; Hargreaves, I.P. Assessment of Mitochondrial Dysfunction in Experimental Autoimmune Encephalomyelitis (EAE) Models of Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4975. [Google Scholar] [CrossRef]
- Mancini, A.; Tantucci, M.; Mazzocchetti, P.; de Iure, A.; Durante, V.; Macchioni, L.; Giampa, C.; Alvino, A.; Gaetani, L.; Costa, C.; et al. Microglial activation and the nitric oxide/cGMP/PKG pathway underlie enhanced neuronal vulnerability to mitochondrial dysfunction in experimental multiple sclerosis. Neurobiol. Dis. 2018, 113, 97–108. [Google Scholar] [CrossRef]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R.; et al. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep. 2016, 6, 33249. [Google Scholar] [CrossRef]
- Chang, J.Y.; Yu, F.; Shi, L.; Ko, M.L.; Ko, G.Y. Melatonin Affects Mitochondrial Fission/Fusion Dynamics in the Diabetic Retina. J. Diabetes Res. 2019, 2019, 8463125. [Google Scholar] [CrossRef] [PubMed]
- Nivison, M.P.; Ericson, N.G.; Green, V.M.; Bielas, J.H.; Campbell, J.S.; Horner, P.J. Age-related accumulation of phosphorylated mitofusin 2 protein in retinal ganglion cells correlates with glaucoma progression. Exp. Neurol. 2017, 296, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Martin-Montanez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.Y.; Fernandez, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal Metabolism and Neuroprotection: Neuroprotective Effect of Fingolimod on Menadione-Induced Mitochondrial Damage. Cells 2020, 10, 34. [Google Scholar] [CrossRef]
- Gimenez-Molina, Y.; Garcia-Martinez, V.; Villanueva, J.; Davletov, B.; Gutierrez, L.M. Multiple sclerosis drug FTY-720 toxicity is mediated by the heterotypic fusion of organelles in neuroendocrine cells. Sci. Rep. 2019, 9, 18471. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Martinez, G.; Segura-Ulate, I.; Yang, B.; Diaz-Pacheco, V.; Barragan, J.A.; De-Leon Esquivel, J.; Chaparro, S.A.; Vargas-Medrano, J.; Perez, R.G. FTY720-Mitoxy reduces synucleinopathy and neuroinflammation, restores behavior and mitochondria function, and increases GDNF expression in Multiple System Atrophy mouse models. Exp. Neurol. 2020, 325, 113120. [Google Scholar] [CrossRef]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Park, S.Y. FTY720 protects neuronal cells from damage induced by human prion protein by inactivating the JNK pathway. Int. J. Mol. Med. 2013, 32, 1387–1393. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candadai, A.A.; Liu, F.; Verma, A.; Adil, M.S.; Alfarhan, M.; Fagan, S.C.; Somanath, P.R.; Narayanan, S.P. Neuroprotective Effects of Fingolimod in a Cellular Model of Optic Neuritis. Cells 2021, 10, 2938. https://doi.org/10.3390/cells10112938
Candadai AA, Liu F, Verma A, Adil MS, Alfarhan M, Fagan SC, Somanath PR, Narayanan SP. Neuroprotective Effects of Fingolimod in a Cellular Model of Optic Neuritis. Cells. 2021; 10(11):2938. https://doi.org/10.3390/cells10112938
Chicago/Turabian StyleCandadai, Amritha A., Fang Liu, Arti Verma, Mir S. Adil, Moaddey Alfarhan, Susan C. Fagan, Payaningal R. Somanath, and S. Priya Narayanan. 2021. "Neuroprotective Effects of Fingolimod in a Cellular Model of Optic Neuritis" Cells 10, no. 11: 2938. https://doi.org/10.3390/cells10112938
APA StyleCandadai, A. A., Liu, F., Verma, A., Adil, M. S., Alfarhan, M., Fagan, S. C., Somanath, P. R., & Narayanan, S. P. (2021). Neuroprotective Effects of Fingolimod in a Cellular Model of Optic Neuritis. Cells, 10(11), 2938. https://doi.org/10.3390/cells10112938