
Combined Assessment of the Tumor–Stroma Ratio and Tumor Immune Cell Infiltrate for Immune Checkpoint Inhibitor Therapy Response Prediction in Colon Cancer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. CIBERSORTx Digital Cytometry
2.3. Tumor–Stroma Ratio
2.4. Definition of Stromal Categories
2.5. Microsatellite Instability, Tumor Mutational Burden, and Single Nucleotide Variants
2.6. MIRACLE and TIDE Prediction Scores
2.7. Gene Set Enrichment Analysis
2.8. Statistical Analysis
3. Results
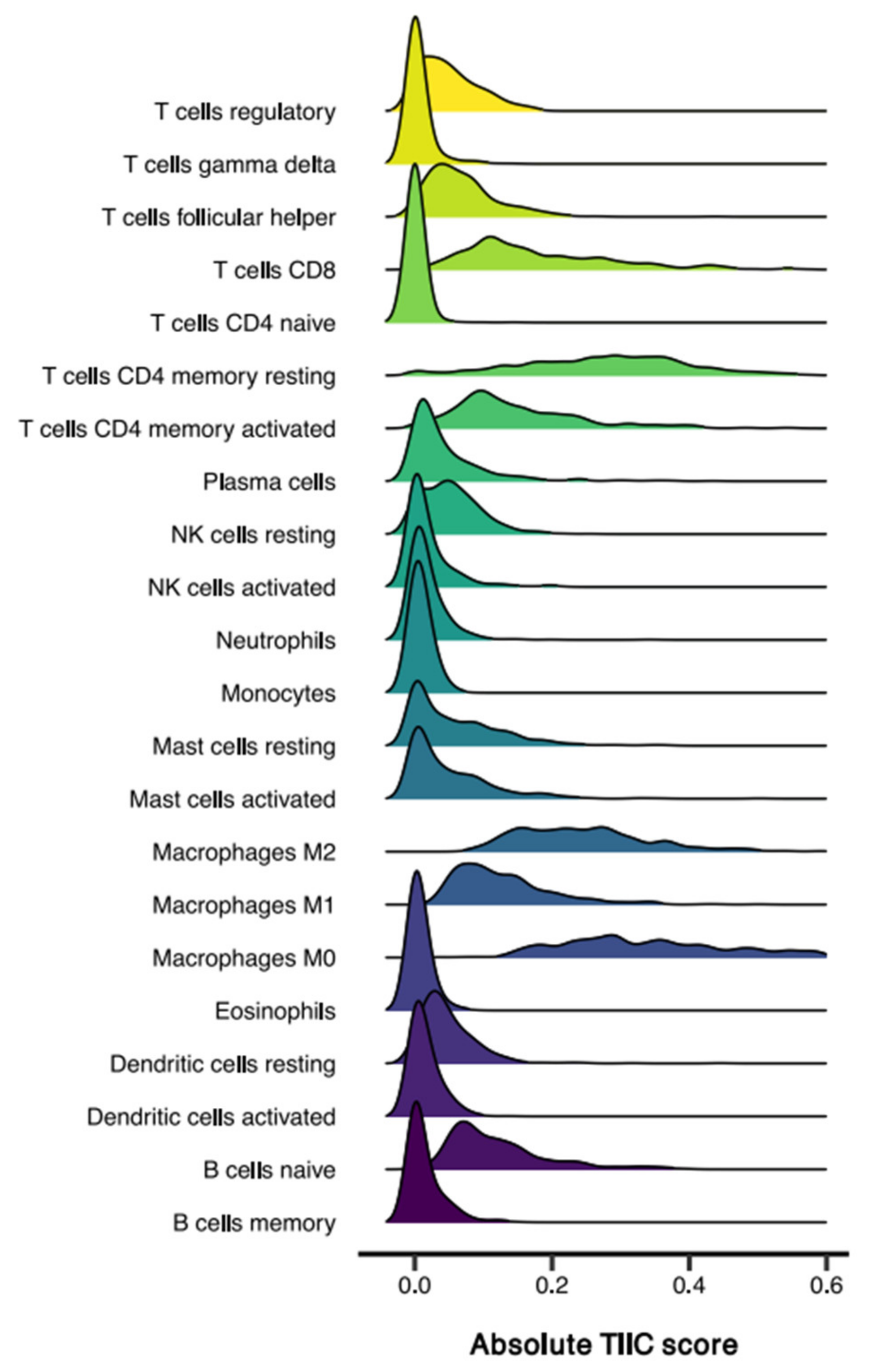
3.1. Sample Characteristics and TIIC Composition
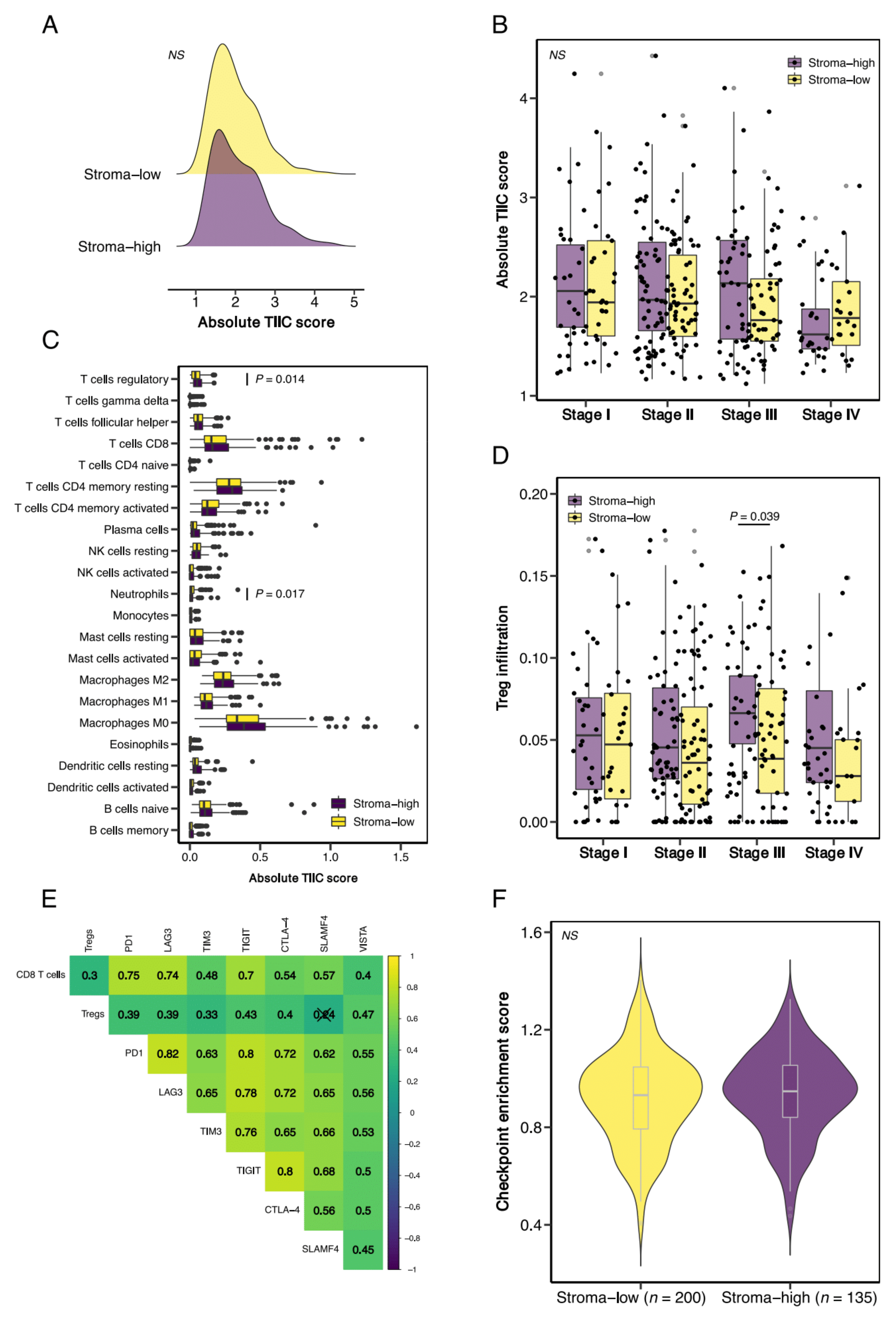
3.2. Tumor Microsatellite Status Relates to Immune Cell Infiltration but Is Not Associated with Stromal Content
3.3. Stroma-High Tumors Demonstrate Increased Infiltration of T Regulatory Cells but Are Not Associated with Increased Expression of T Cell Exhaustion Markers
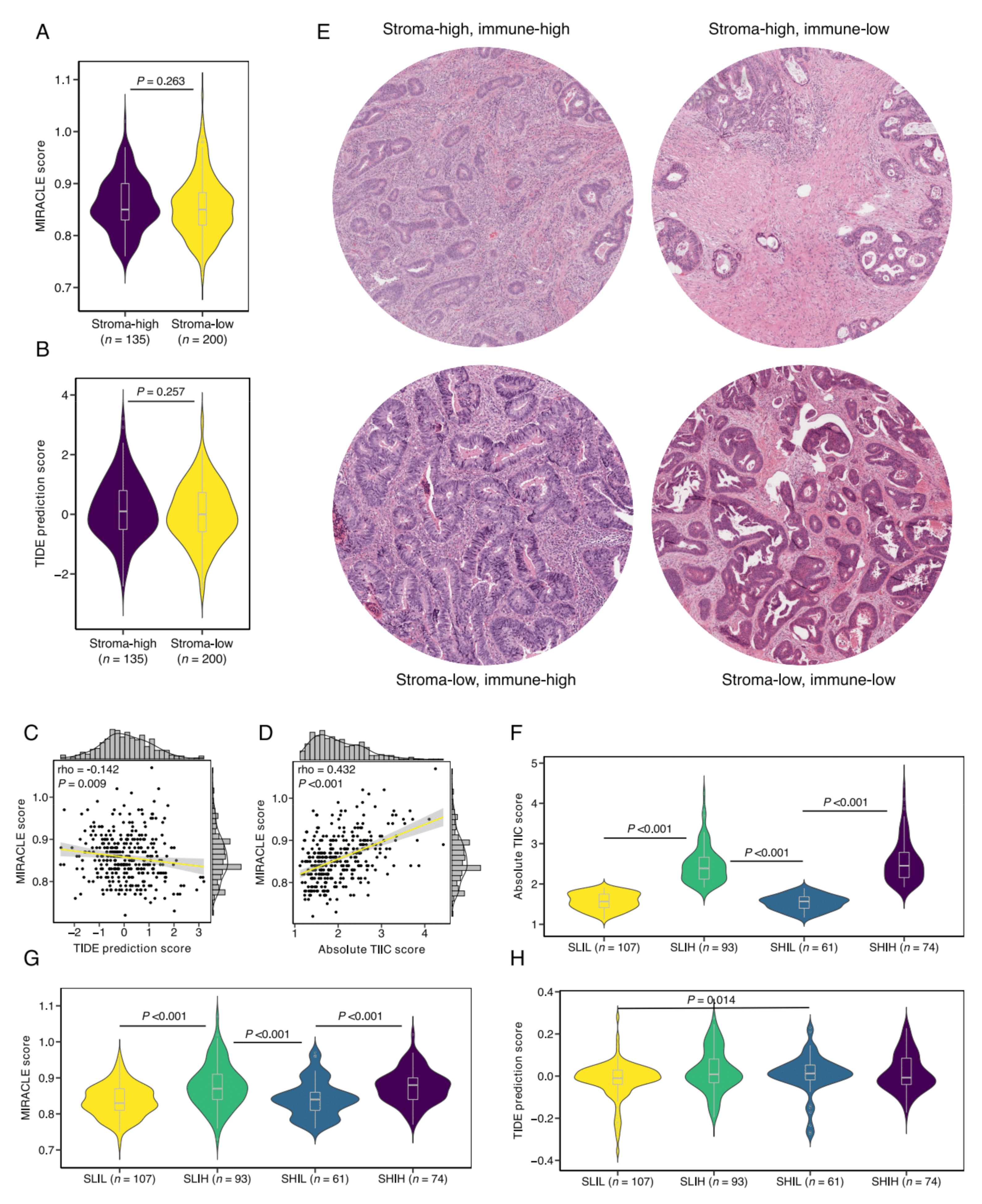
3.4. Defining Stromal Categories Based on Stromal Content and Immune Cell Infiltrate That Are Predictive of Response to ICI Therapy
3.5. SLIH Tumors Are Associated with Current Biomarkers for ICI Therapy Response Prediction
3.6. Validation in an External Cohort
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Mesker, W.E.; Junggeburt, J.M.C.; Szuhai, K.; de Heer, P.; Morreau, H.; Tanke, H.J.; Tollenaar, R.A.E.M. The Carcinoma–Stromal Ratio of Colon Carcinoma Is an Independent Factor for Survival Compared to Lymph Node Status and Tumor Stage. Anal. Cell. Pathol. 2007, 29, 387–398. [Google Scholar] [CrossRef]
- Smit, M.A.; Philipsen, M.W.; Postmus, P.E.; Putter, H.; Tollenaar, R.A.; Cohen, D.; Mesker, W.E. The prognostic value of the tumor-stroma ratio in squamous cell lung cancer, a cohort study. Cancer Treat. Res. Commun. 2020, 25, 100247. [Google Scholar] [CrossRef]
- Vangangelt, K.M.; Tollenaar, L.S.; Van Pelt, G.W.; De Kruijf, E.M.; Dekker, T.; Kuppen, P.J.; Tollenaar, R.A.; Mesker, W.E. The prognostic value of tumor-stroma ratio in tumor-positive axillary lymph nodes of breast cancer patients. Int. J. Cancer 2018, 143, 3194–3200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunder, S.M.; Perez-Lopez, R.; De Kok, B.M.; Raciti, M.V.; Van Pelt, G.W.; Dienstmann, R.; Garcia-Ruiz, A.; Meijer, C.A.; Gelderblom, H.; Tollenaar, R.A.; et al. Correlation of the tumour-stroma ratio with diffusion weighted MRI in rectal cancer. Eur. J. Radiol. 2020, 133, 109345. [Google Scholar] [CrossRef]
- Zong, L.; Zhang, Q.; Kong, Y.; Yang, F.; Zhou, Y.; Yu, S.; Wu, M.; Chen, J.; Zhang, Y.; Xiang, Y. The tumor-stroma ratio is an independent predictor of survival in patients with 2018 FIGO stage IIIC squamous cell carcinoma of the cervix following primary radical surgery. Gynecol. Oncol. 2019, 156, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yuan, S.L.; Han, Z.Z.; Huang, J.; Cui, L.; Jiang, C.Q.; Zhang, Y. Prognostic significance of the tumor-stroma ratio in gallbladder cancer. Neoplasma 2017, 64, 588–593. [Google Scholar] [CrossRef]
- Karpathiou, G.; Vieville, M.; Gavid, M.; Camy, F.; Dumollard, J.M.; Magné, N.; Froudarakis, M.; Prades, J.M.; Peoc’H, M. Prognostic significance of tumor budding, tumor-stroma ratio, cell nests size, and stroma type in laryngeal and pharyngeal squamous cell carcinomas. Head Neck 2019, 41, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Aurello, P.; Berardi, G.; Giulitti, D.; Palumbo, A.; Tierno, S.M.; Nigri, G.; D’Angelo, F.; Pilozzi, E.; Ramacciato, G. Tumor-Stroma Ratio is an independent predictor for overall survival and disease free survival in gastric cancer patients. Surgeon 2017, 15, 329–335. [Google Scholar] [CrossRef]
- Huijbers, A.; Tollenaar, R.A.E.M.; Pelt, G.W.V.; Zeestraten, E.C.M.; Dutton, S.; McConkey, C.C.; Domingo, E.; Smit, V.T.H.B.M.; Midgley, R.; Warren, B.F.; et al. The proportion of tumor-stroma as a strong prognosticator for stage II and III colon cancer patients: Validation in the VICTOR trial. Ann. Oncol. 2012, 24, 179–185. [Google Scholar] [CrossRef]
- van Pelt, G.; Krol, J.; Lips, I.; Peters, F.; van Klaveren, D.; Boonstra, J.; de Steur, W.; Tollenaar, R.; Sarasqueta, A.F.; Mesker, W.; et al. The value of tumor-stroma ratio as predictor of pathologic response after neoadjuvant chemoradiotherapy in esophageal cancer. Clin. Transl. Radiat. Oncol. 2019, 20, 39–44. [Google Scholar] [CrossRef]
- Hagenaars, S.C.; de Groot, S.; Cohen, D.; Dekker, T.J.A.; Charehbili, A.; Kranenbarg, E.M.; Carpentier, M.D.; Pijl, H.; Putter, H.; Tollenaar, R.A.E.M.; et al. Tumor-stroma ratio is associated with Miller-Payne score and pathological response to neoadjuvant chemotherapy in HER2 -negative early breast cancer. Int. J. Cancer 2021, 149, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhu, Y.; Lin, H.; Zhang, S.; Li, S.; Huang, Y.; Liu, C.; Qu, J.; Liang, C.; Zhao, K.; et al. The value of the tumour-stroma ratio for predicting neoadjuvant chemoradiotherapy response in locally advanced rectal cancer: A case control study. BMC Cancer 2021, 21, 1–11. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravensbergen, C.J.; Kuruc, M.; Polack, M.; Crobach, S.; Putter, H.; Gelderblom, H.; Roy, D.; Tollenaar, R.A.E.M.; Mesker, W.E. A Stromal-Epithelial gene signature ratio predicts colon cancer outcome and relates to the histologic tumour-stroma ratio. Br. J. Cancer 2021. submitted. [Google Scholar]
- Sandberg, T.P.; Stuart, M.P.M.E.; Oosting, J.; Tollenaar, R.A.E.M.; Sier, C.F.M.; Mesker, W.E. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 2019, 19, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Deng, C.-X. Effect of Stromal Cells in Tumor Microenvironment on Metastasis Initiation. Int. J. Biol. Sci. 2018, 14, 2083–2093. [Google Scholar] [CrossRef]
- Vangangelt, K.M.H.; Van Pelt, G.W.; Engels, C.C.; Putter, H.; Liefers, G.J.; Smit, V.T.H.B.M.; Tollenaar, R.A.E.M.; Kuppen, P.J.K.; Mesker, W.E. Prognostic value of tumor-stroma ratio combined with the immune status of tumors in invasive breast carcinoma. Breast Cancer Res. Treat. 2017, 168, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujam, F.J.A.; Edwards, J.; Mohammed, Z.M.A.; Going, J.; McMillan, D. The relationship between the tumour stroma percentage, clinicopathological characteristics and outcome in patients with operable ductal breast cancer. Br. J. Cancer 2014, 111, 157–165. [Google Scholar] [CrossRef]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Haslam, A.; Prasad, V. Estimation of the Percentage of US Patients with Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- McGrail, D.; Pilié, P.; Rashid, N.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef]
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer immunotherapy: It’s time to better predict patients’ response. Br. J. Cancer 2021, 125, 927–938. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steen, C.B.; Liu, C.L.; Alizadeh, A.A.; Newman, A.M. Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods Mol. Biol. 2020, 2117, 135–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Van Pelt, G.W.; Kjær-Frifeldt, S.; van Krieken, J.; Al Dieri, R.; Morreau, H.; Tollenaar, R.A.E.M.; Sørensen, F.B.; Mesker, W.E. Scoring the tumor-stroma ratio in colon cancer: Procedure and recommendations. Virchows Arch. 2018, 473, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Smit, M.; Van Pelt, G.; Roodvoets, A.; Kranenbarg, E.M.-K.; Putter, H.; Tollenaar, R.; Van Krieken, J.H.; Mesker, W.; Aslam, M.S.; Muddegowda, P. Uniform Noting for International Application of the Tumor-Stroma Ratio as an Easy Diagnostic Tool: Protocol for a Multicenter Prospective Cohort Study. JMIR Res. Protoc. 2019, 8, e13464. [Google Scholar] [CrossRef]
- A Smit, M.; van Pelt, G.W.; MC Dequeker, E.; Al Dieri, R.; Tollenaar, R.A.; van Krieken, J.H.J.; E Mesker, W.; UNITED Group. e-Learning for Instruction and to Improve Reproducibility of Scoring Tumor-Stroma Ratio in Colon Carcinoma: Performance and Reproducibility Assessment in the UNITED Study. JMIR Form. Res. 2021, 5, e19408. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Network, T.C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Thorsson, V.; Gibbs, D.; Brown, S.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.; Plaisier, C.L.; Eddy, J.A.; et al. Erratum: The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden–High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef] [PubMed]
- Turan, T.; Kongpachith, S.; Halliwill, K.; Roelands, J.; Hendrickx, W.; Marincola, F.M.; Hudson, T.J.; Jacob, H.J.; Bedognetti, D.; Samayoa, J.; et al. A balance score between immune stimulatory and suppressive microenvironments identifies mediators of tumour immunity and predicts pan-cancer survival. Br. J. Cancer 2020, 124, 760–769. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGready, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Kim, H.; Kwon, H.J.; Han, Y.B.; Park, S.Y.; Kim, E.S.; Kim, S.H.; Kim, Y.J.; Lee, J.S.; Chung, J.-H. Increased CD3+ T cells with a low FOXP3+/CD8+ T cell ratio can predict anti-PD-1 therapeutic response in non-small cell lung cancer patients. Mod. Pathol. 2018, 32, 367–375. [Google Scholar] [CrossRef]
- Sandberg, T.P.; Oosting, J.; Van Pelt, G.W.; Mesker, W.E.; Tollenaar, R.A.E.M.; Morreau, H. Erratum: Molecular profiling of colorectal tumors stratified by the histological tumor-stroma ratio—Increased expression of galectin-1 in tumors with high stromal content. Oncotarget 2018, 9, 31502–31515. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Berg, J.G.V.D.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef] [PubMed]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865.e7. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravensbergen, C.J.; Polack, M.; Roelands, J.; Crobach, S.; Putter, H.; Gelderblom, H.; Tollenaar, R.A.E.M.; Mesker, W.E. Combined Assessment of the Tumor–Stroma Ratio and Tumor Immune Cell Infiltrate for Immune Checkpoint Inhibitor Therapy Response Prediction in Colon Cancer. Cells 2021, 10, 2935. https://doi.org/10.3390/cells10112935
Ravensbergen CJ, Polack M, Roelands J, Crobach S, Putter H, Gelderblom H, Tollenaar RAEM, Mesker WE. Combined Assessment of the Tumor–Stroma Ratio and Tumor Immune Cell Infiltrate for Immune Checkpoint Inhibitor Therapy Response Prediction in Colon Cancer. Cells. 2021; 10(11):2935. https://doi.org/10.3390/cells10112935
Chicago/Turabian StyleRavensbergen, Cor J., Meaghan Polack, Jessica Roelands, Stijn Crobach, Hein Putter, Hans Gelderblom, Rob A. E. M. Tollenaar, and Wilma E. Mesker. 2021. "Combined Assessment of the Tumor–Stroma Ratio and Tumor Immune Cell Infiltrate for Immune Checkpoint Inhibitor Therapy Response Prediction in Colon Cancer" Cells 10, no. 11: 2935. https://doi.org/10.3390/cells10112935
APA StyleRavensbergen, C. J., Polack, M., Roelands, J., Crobach, S., Putter, H., Gelderblom, H., Tollenaar, R. A. E. M., & Mesker, W. E. (2021). Combined Assessment of the Tumor–Stroma Ratio and Tumor Immune Cell Infiltrate for Immune Checkpoint Inhibitor Therapy Response Prediction in Colon Cancer. Cells, 10(11), 2935. https://doi.org/10.3390/cells10112935