
CFTR Protein: Not Just a Chloride Channel?


Abstract
:1. Introduction
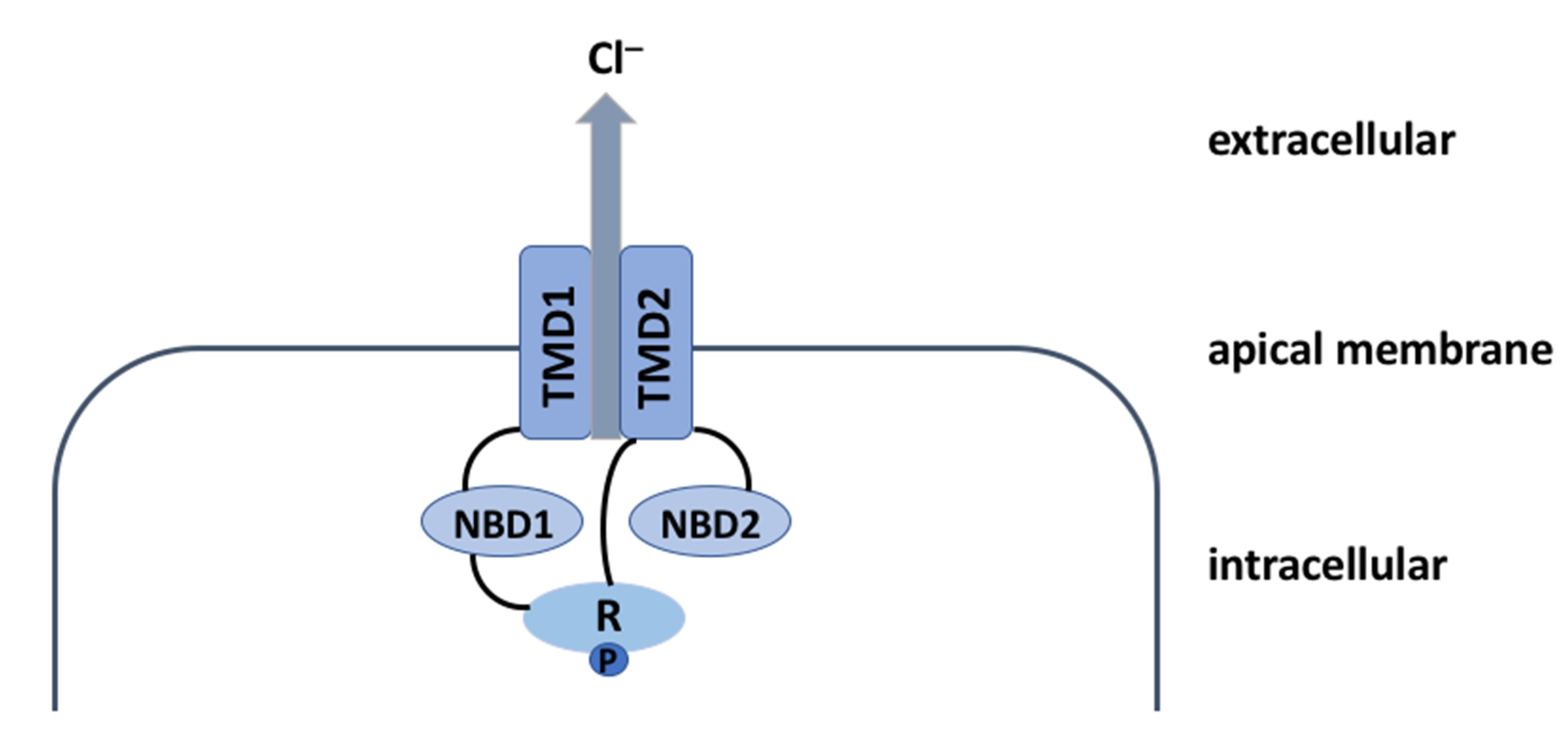
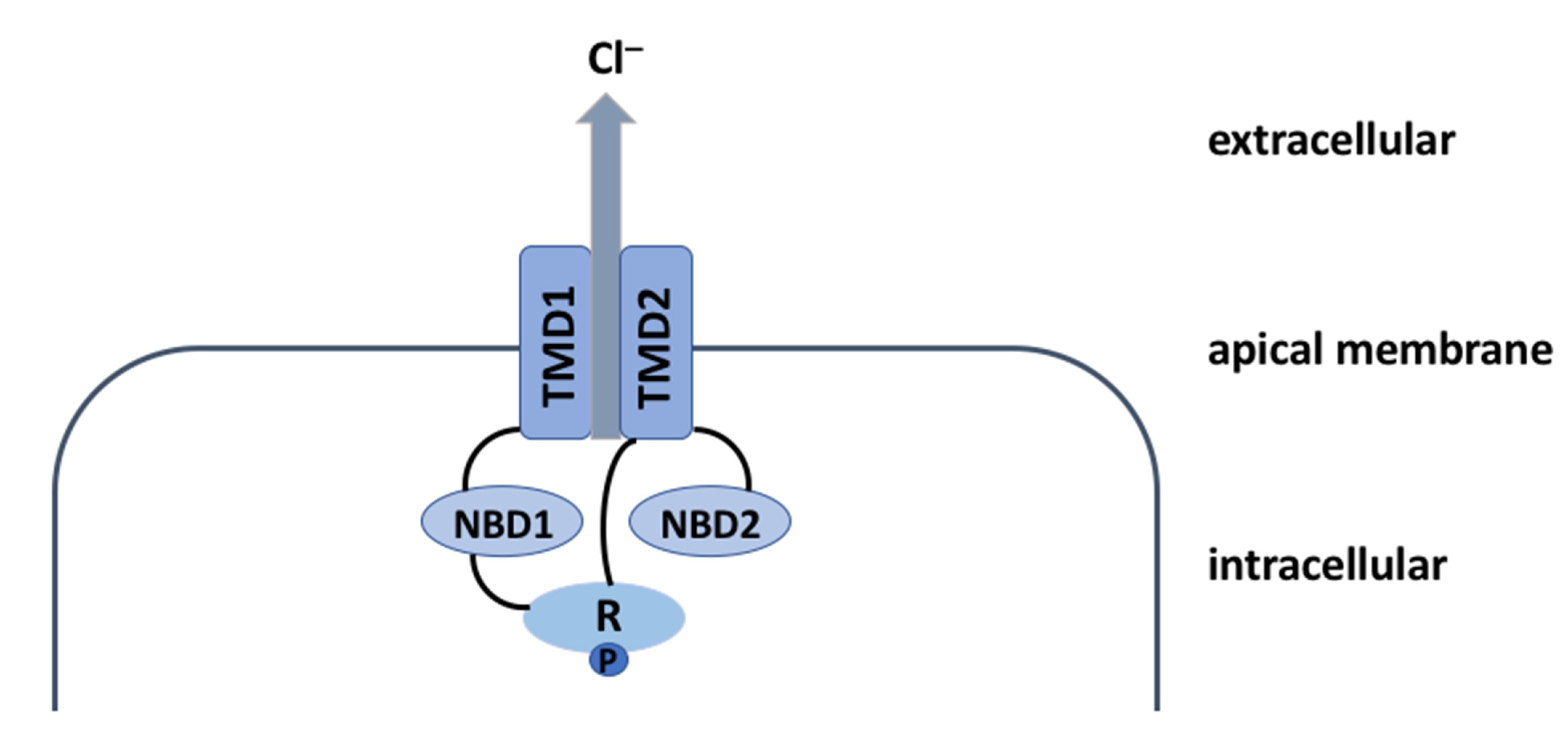
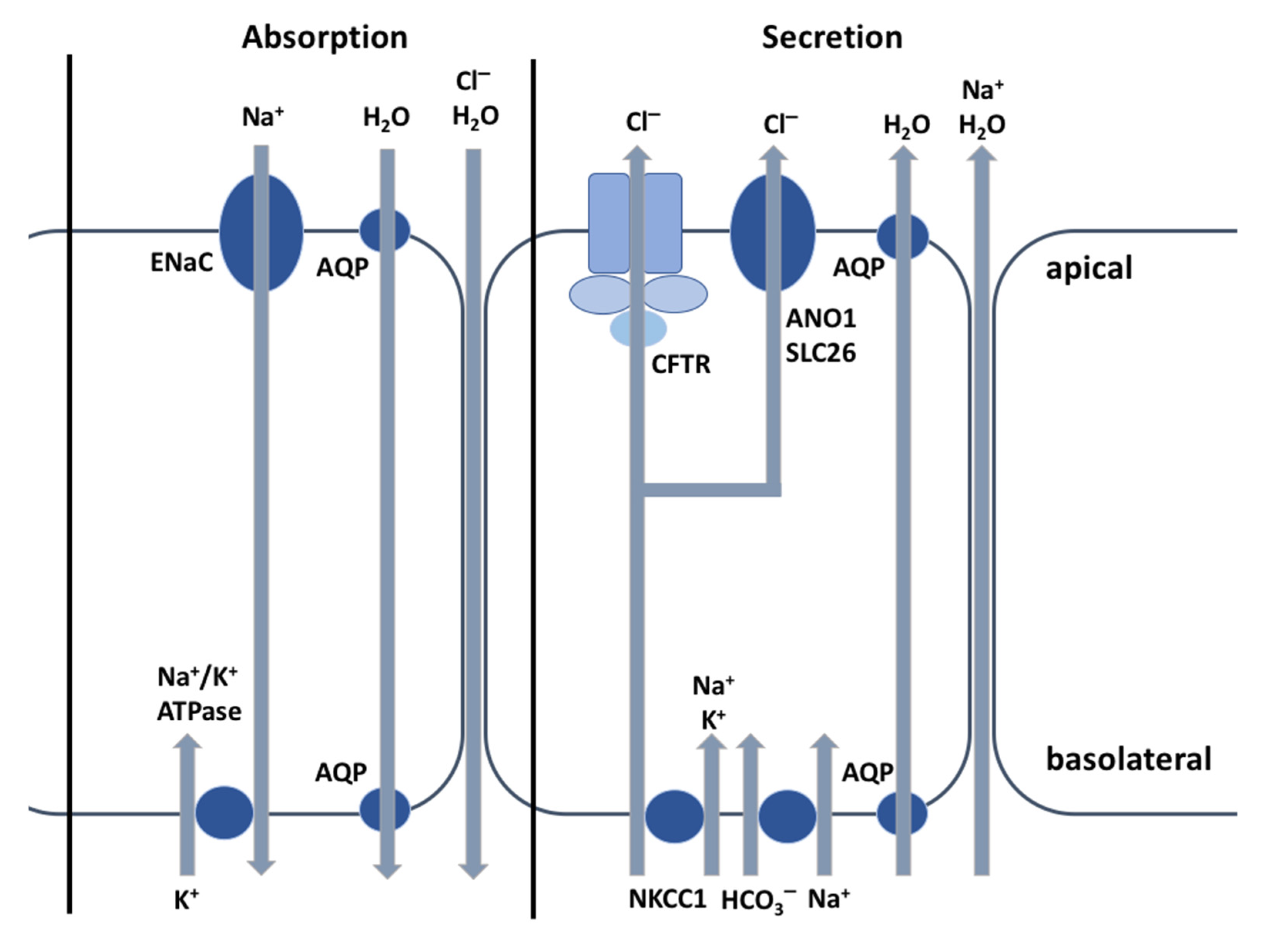
2. The CFTR Protein: A Chloride Channel
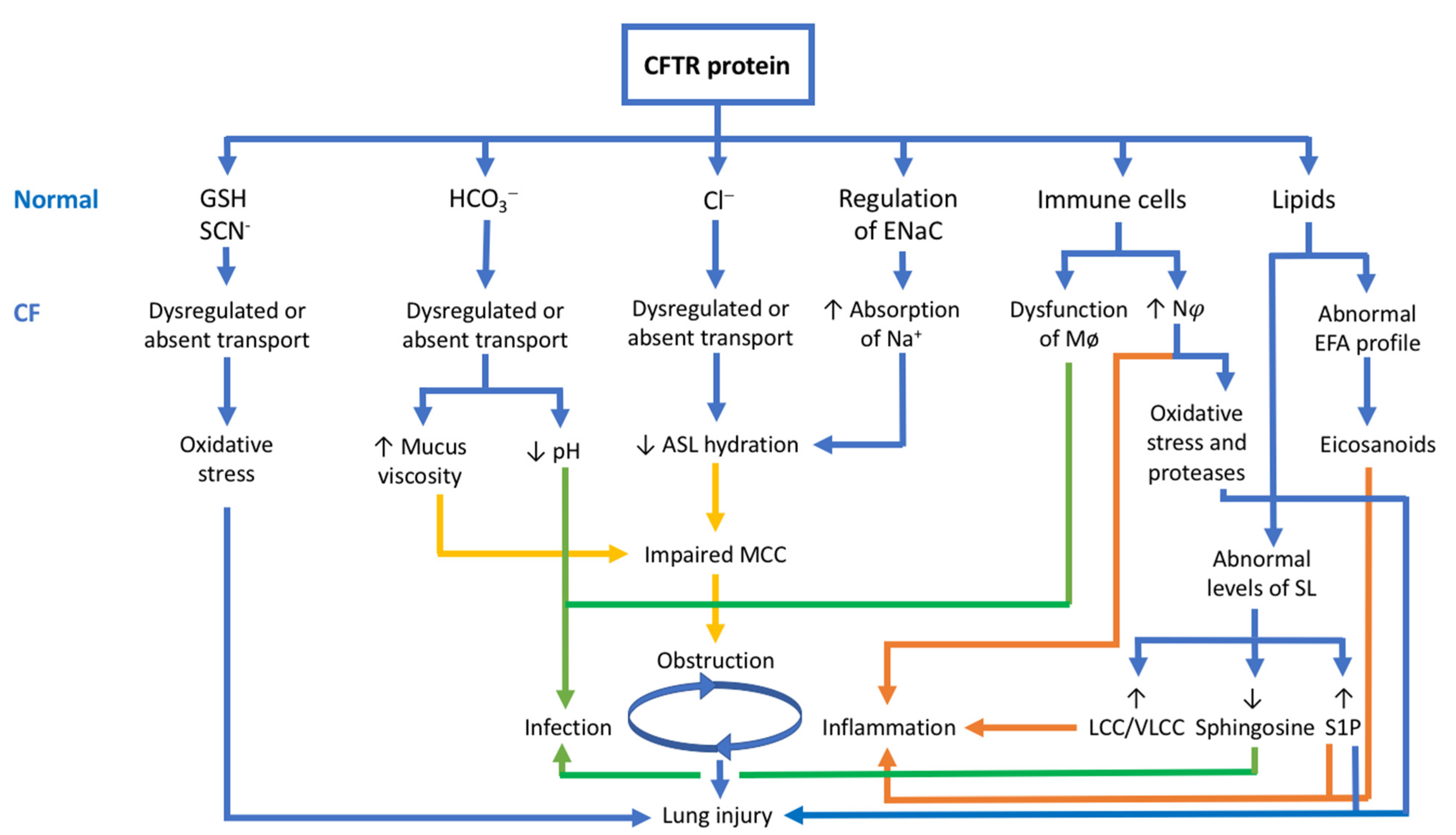
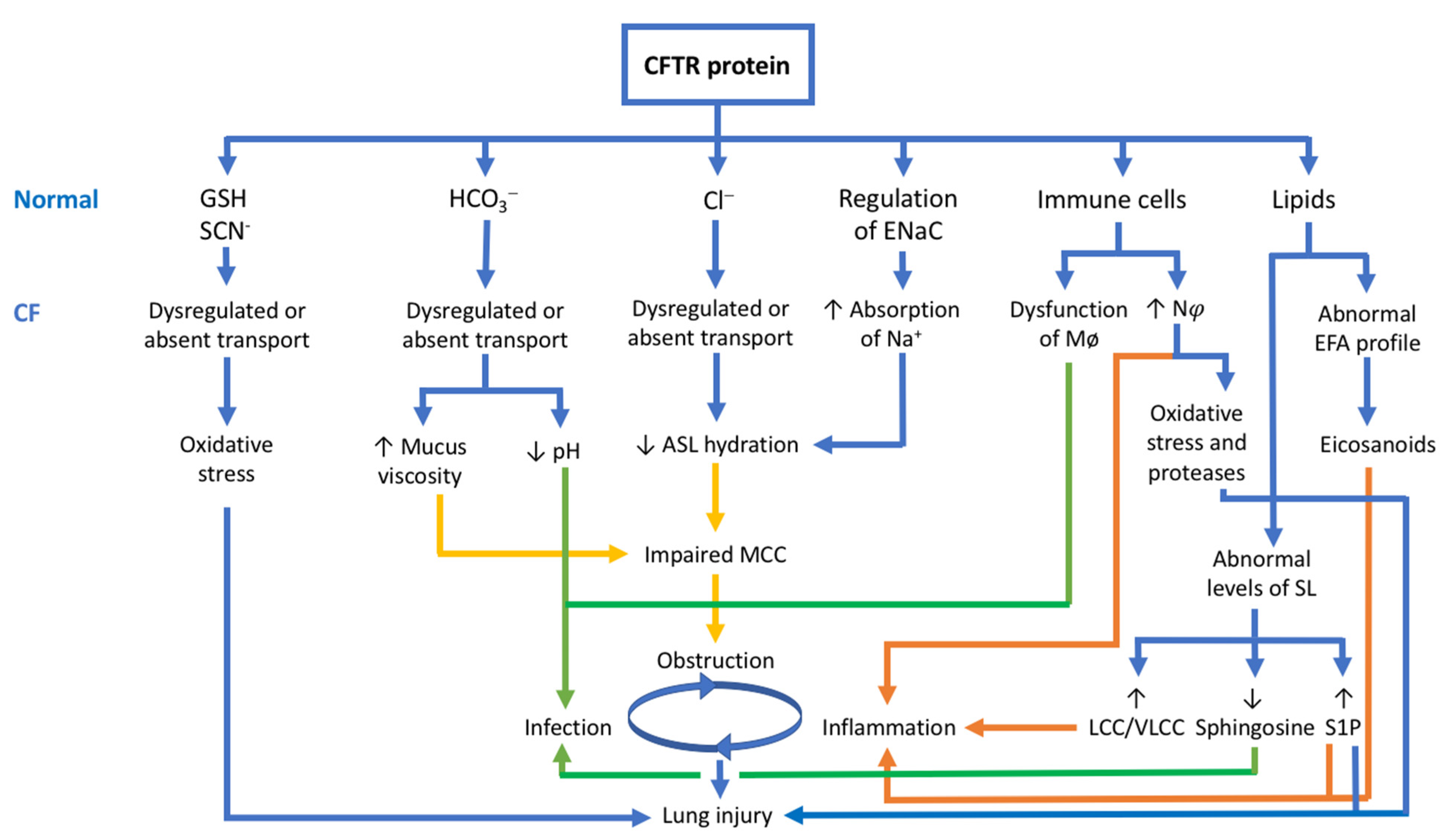
3. The CFTR Protein: Not Just a Chloride Channel
3.1. The CFTR Protein and Its Relationship with HCO3− Transport
3.2. The CFTR Protein and Its Relationship with GSH and SCN−
3.3. The CFTR Protein and Its Relationship with Immune Cells
3.4. The CFTR Protein and Its Relationship with Lipid Metabolism
3.5. The CFTR Protein and Its Relationship with the Pathophysiology of CF
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Naehrlich, L. The changing face of cystic fibrosis and its implications for screening. Int. J. Neonatal Screen 2020, 6, 54. [Google Scholar] [CrossRef]
- Fonseca, C.; Bicker, J.; Alves, G.; Falcão, A.; Fortuna, A. Cystic fibrosis: Physiopathology and the latest pharmacological treatments. Pharmacol. Res. 2020, 162, 105267. [Google Scholar] [CrossRef]
- Andersen, D.H. Cystic fibrosis of the pancreas and its relation to celiac diseasea clinical and pathologic study. Am. J. Dis. Child. 1938, 56, 344–399. [Google Scholar] [CrossRef]
- Fanconi, G.; Uehlinger, E.; Knauer, C. Das Coeliakie-syndrom bei angeborener zystischer Pankreasfibromatose und Bronchiektasien. Wien. Med. Wchnschr. 1936, 86, 753–756. [Google Scholar]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet Resp. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic Fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic Fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Quinton, P.M. The neglected ion: HCO3−. Nat. Med. 2001, 7, 292–293. [Google Scholar] [CrossRef] [PubMed]
- Di Sant’Agnese, P.A.; Darling, R.C.; Perera, G.A.; Shea, E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 1953, 12, 549–563. [Google Scholar]
- Gibson, L.E.; Cooke, R.E. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959, 23, 545–549. [Google Scholar]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell. Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinton, P.M. Cystic fibrosis: Lessons from the sweat gland. Physiology 2007, 22, 212–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.B.; Wolfe, A.S. Physiological mechanisms determining eccrine sweat composition. Eur. J. Appl. Physiol. 2020, 120, 719–752. [Google Scholar] [CrossRef] [Green Version]
- Reihill, J.A.; Douglas, L.E.J.; Martin, S.L. Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis. Genes 2021, 12, 453. [Google Scholar] [CrossRef]
- Ertongur-Fauth, T.; Hochheimer, A.; Buescher, J.M.; Rapprich, S.; Krohn, M. A novel TMEM16A splice variant lacking the dimerization domain contributes to calcium-activated chloride secretion in human sweat gland epithelial cells. Exp. Dermatol. 2014, 23, 825–831. [Google Scholar] [CrossRef]
- Reddy, M.M.; Quinton, P.M. Functional interaction of CFTR and ENaC in sweat glands. Pflugers Arch. 2003, 445, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.; Dreano, E.; Edwards, A.; Planelles, G.; Sermet-Gaudelus, I. Airway Surface Liquid pH Regulation in Airway Epithelium Current Understandings and Gaps in Knowledge. Int. J. Mol. Sci. 2021, 22, 3384. [Google Scholar] [CrossRef]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway surface liquid homeostasis in cystic fibrosis: Pathophysiology and therapeutic targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Scudieri, P.; Musante, I.; Venturini, A.; Guidone, D.; Genovese, M.; Cresta, F.; Caci, E.; Palleschi, A.; Poeta, M.; Santamaria, F.; et al. Ionocytes and CFTR Chloride Channel Expression in Normal and Cystic Fibrosis Nasal and Bronchial Epithelial Cells. Cells 2020, 9, 2090. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Blanchard, M.G.; Alijevic, O.; Da Tan, C.; Kellenberger, S.; Bencharit, S.; Cao, R.; Kesimer, M.; Walton, W.G.; Henderson, A.G.; et al. Identification of the SPLUNC1 ENaC-inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airway epithelial cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L990–L1001. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Caballero, A.; Rasmussen, J.E.; Gaillard, E.; Watson, M.J.; Olsen, J.C.; Donaldson, S.H.; Stutts, M.J.; Tarran, R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. USA 2009, 106, 11412–11417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Gilmore, R.C.; Alexis, N.E.; Tarran, R. SPLUNC1 Loses Its Antimicrobial Activity in Acidic Cystic Fibrosis Airway Secretions. Am. J. Respire Crit. Care. Med. 2019, 200, 633–636. [Google Scholar] [CrossRef]
- Jiang, D.; Wenzel, S.E.; Wu, Q.; Bowler, R.P.; Schnell, C.; Chu, H.W. Human neutrophil elastase degrades SPLUNC1 and impairs airway epithelial defense against bacteria. PLoS ONE 2013, 8, e64689. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O’Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Doušová, T.; Bähr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Front. Pharmacol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lérias, J.; Pinto, M.; Benedetto, R.; Schreiber, R.; Amaral, M.; Aureli, M.; Kunzelmann, K. Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell. Signal 2018, 44, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Bakouh, N.; Bienvenu, T.; Thomas, A.; Ehrenfeld, J.; Liote, H.; Roussel, D.; Duquesnoy, P.; Farman, N.; Viel, M.; Cherif-Zahar, B.; et al. Characterization of SLC26A9 in patients with CF-like lung disease. Hum. Mutat. 2013, 34, 1404–1414. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of Cystic Fibrosis Lung Disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Elkins, M.R.; Robinson, M.; Rose, B.R.; Harbour, C.; Moriarty, C.P.; Marks, G.B.; Belousova, E.G.; Xuan, W.; Bye, P.T.P.; National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N. Engl. J. Med. 2006, 354, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, M.L.; Bellon, G.; De Boeck, K.; Flume, P.A.; Fox, H.G.; Geller, D.E.; Haarman, E.G.; Hebestreit, H.U.; Lapey, A.; Schou, I.M.; et al. Long-term inhaled dry powder mannitol in cystic fibrosis: An international randomized study. Am. J. Respire Crit. Care Med. 2012, 185, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, gating, and regulation of the CFTR anion channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Stoltz, D.A.; Karp, P.H.; Ernst, S.E.; Pezzulo, A.A.; Moninger, T.O.; Rector, M.V.; Reznikov, L.R.; Launspach, J.L.; Chaloner, K.; et al. Loss of anion transport without increased sodium absorption characterizes newborn porcine cystic fibrosis airway epithelia. Cell 2010, 143, 911–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, J.T.; Tyler, S.R.; Zhang, Y.; Lee, B.J.; Liu, X.; Sun, X.; Sui, H.; Liang, B.; Luo, M.; Xie, W.; et al. Bioelectric characterization of epithelia from neonatal CFTR knockout ferrets. Am. J. Respire Cell. Mol. Biol. 2013, 49, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itani, O.A.; Chen, J.H.; Karp, P.H.; Ernst, S.; Keshavjee, S.; Parekh, K.; Klesney-Tait, J.; Zabner, J.; Welsh, M.J. Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. Proc. Natl. Acad. Sci. USA 2011, 108, 10260–10265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.Y.; Muallem, D.; Kiselyov, K.; Lee, M.G.; Thomas, P.J.; Muallem, S. Aberrant CFTR-dependent HCO3- transport in mutations associated with cystic fibrosis. Nature 2001, 410, 94–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K.; Schreiber, R.; Hadorn, H.B. Very early findings pointed to a defect in bicarbonate transport in CF, but it took many more years to identify the underlying molecular mechanism for bicarbonate secretion. J. Cyst. Fibros. 2017, 16, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.S.; Ernst, S.; Tang, X.X.; Karp, P.H.; Parker, C.P.; Ostedgaard, L.S.; Welsh, M.J. Relationships among CFTR expression, HCO3- secretion, and host defense may inform gene and cell-based cystic fibrosis therapies. Proc. Natl. Acad. Sci. USA 2016, 113, 5382–5387. [Google Scholar] [CrossRef] [Green Version]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Abou Alaiwa, M.H.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Dobay, O.; Laub, K.; Stercz, B.; Kéri, A.; Balázs, B.; Tóthpál, A.; Kardos, S.; Jaikumpun, P.; Ruksakiet, K.; Quinton, P.M.; et al. Bicarbonate inhibits bacterial growth and biofilm formation of prevalent cystic fibrosis pathogens. Front. Microbiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tang, X.X.; Vargas Buonfiglio, L.G.; Comellas, A.P.; Thornell, I.M.; Ramachandran, S.; Karp, P.H.; Taft, P.J.; Sheets, K.; Abou Alaiwa, M.H.; et al. Electrolyte transport properties in distal small airways from cystic fibrosis pigs with implications for host defense. Am. J. Phys. Lung Cell. Mol. Phys. 2016, 310, L670–L679. [Google Scholar]
- Simonin, J.; Bille, E.; Crambert, G.; Noel, S.; Dreano, E.; Edwards, A.; Hatton, A.; Prank, I.; Villeret, B.; Cottart, C.H.; et al. Airway surface liquid acidification initiates host defense abnormalities in Cystic Fibrosis. Sci. Rep. 2019, 9, 6516. [Google Scholar] [CrossRef] [PubMed]
- Le Simple, P.; Goepp, J.; Palmer, M.L.; Fahrenkrug, S.C.; O’Grady, S.M.; Ferraro, P.; Robert, R.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator is expressed in mucin granules from Calu-3 and primary human airway epithelial cells. Am. J. Respire Cell. Mol. Biol. 2013, 49, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Shamsuddin, A.K.; Quinton, P.M. Native small airways secrete bicarbonate. Am. J. Respire Cell. Mol. Biol. 2014, 50, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Trout, L.; Gatzy, J.T.; Ballard, S.T. Acetylcholine-induced liquid secretion by bronchial epithelium: Role of Cl− and HCO3− transport. Am. J. Physiol. 1998, 275, L1095–L1099. [Google Scholar] [CrossRef]
- Birket, S.E.; Chu, K.K.; Liu, L.; Houser, G.H.; Diephuis, B.J.; Wilsterman, E.J.; Dierksen, G.; Mazur, M.; Shastry, S.; Li, Y.; et al. A functional anatomic defect of the cystic fibrosis airway. Am. J. Respire Crit. Care Med. 2014, 190, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respire J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [Green Version]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, Mucins and Cystic Fibrosis. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S84–S96. [Google Scholar] [CrossRef] [Green Version]
- Quinton, P.M. Cystic fibrosis: Impaired bicarbonate secretion and mucoviscidosis. Lancet 2008, 372, 415–417. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.Y.T.; Yang, N.; Quinton, P.M.; Chin, W.C. A new role for bicarbonate in mucus formation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L542–L549. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Duerr, J.; Johannesson, B.; Schubert, S.C.; Treis, D.; Harm, M.; Graeber, S.Y.; Dalpke, A.; Schultz, C.; Mall, M.A.; et al. The bENaC-overexpressing mouse as a model of cystic fibrosis lung disease. J. Cyst. Fibros. 2011, 10 (Suppl. 2), S172–S182. [Google Scholar] [CrossRef] [Green Version]
- Birket, S.E.; Davis, J.M.; Fernandez, C.M.; Tuggle, K.L.; Oden, A.M.; Chu, K.K.; Tearney, G.J.; Fanucchi, M.V.; Sorscher, E.J.; Rowe, S.M. Development of an airway mucus defect in the cystic fibrosis rat. JCI Insight 2018, 3, e97199. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, L.H.; Evans, J.R.; Wang, T.T.; Ford, A.A.; Makhov, A.M.; Nguyen, K.; Coakley, R.D.; Griffith, J.D.; Davis, C.W.; Ballard, S.T. Defective postsecretory maturation of MUC5B mucin in cystic fibrosis airways. JCI Insight 2017, 2, e89752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.; Sutto, Z.; Schmid, N.; Novak, L.; Ivonnet, P.; Horvath, G.; Conner, G.; Fregien, N.; Salathe, M. Decreased soluble adenylyl cyclase activity in cystic fibrosis is related to defective apical bicarbonate exchange and affects ciliary beat frequency regulation. J. Biol. Chem. 2010, 285, 29998–30007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsdell, P.; Hanrahan, J.W. Glutathione permeability of CFTR. Am. J. Physiol. 1998, 275, C323–C326. [Google Scholar] [CrossRef] [PubMed]
- Conner, G.E.; Wijkstrom-Frei, C.; Randell, S.H.; Fernandez, V.E.; Salathe, M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007, 581, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef]
- Xu, Y.; Szép, S.; Lu, Z. The antioxidant role of thiocyanate in the pathogenesis of cystic fibrosis and other inflammation-related diseases. Proc. Natl. Acad. Sci. USA 2009, 106, 20515–20519. [Google Scholar] [CrossRef] [Green Version]
- Roum, J.H.; Buhl, R.; McElvaney, N.G.; Borok, Z.; Crystal, R.G. Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol. 1993, 75, 2419–2424. [Google Scholar] [CrossRef]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef]
- Bruscia, E.M.; Bonfield, T.L. Cystic Fibrosis Lung Immunity: The Role of the Macrophage. J. Innate Immun. 2016, 8, 550–563. [Google Scholar] [CrossRef]
- Lévêque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The impact of impaired macrophage functions in cystic fibrosis disease progression. J. Cyst. Fibros. 2017, 16, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Shrestha, C.L.; Wisniewski, B.L.; Pham, H.; Hou, X.; Li, W.; Dong, Y.; Kopp, B.T. Consequences of CRISPR-Cas9-Mediated CFTR Knockout in Human Macrophages. Front. Immunol. 2020, 11, 1871. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, M.; Worlitzsch, D.; Viglio, S.; Siegmann, N.; Iadarola, P.; Shute, J.K.; Geiser, M.; Pier, G.B.; Friedel, G.; Barr, M.L.; et al. Alveolar inflammation in cystic fibrosis. J. Cyst. Fibros. 2010, 9, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Krause, A.; Hamai, H.; Harvey, B.-G.; Worgall, T.S.; Worgall, S. Proinflammatory phenotype and increased caveolin-1 in alveolar macrophages with silenced CFTR mRNA. PLoS ONE 2010, 5, e11004. [Google Scholar] [CrossRef] [Green Version]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell. Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Law, S.M.; Stanfield, S.J.; Hardisty, G.R.; Dransfield, I.; Campbell, C.J.; Gray, R.D. Human cystic fibrosis monocyte derived macrophages display no defect in acidification of phagolysosomes when measured by optical nanosensors. J. Cyst. Fibros. 2020, 19, 203–210. [Google Scholar] [CrossRef]
- Freedman, S.D.; Blanco, P.G.; Zaman, M.M.; Shea, J.C.; Ollero, M.; Hopper, I.K.; Weed, D.A.; Gelrud, A.; Regan, M.M.; Laposata, M.; et al. Association of cystic fibrosis with abnormalities in fatty acid metabolism. N. Engl. J. Med. 2004, 350, 560–569. [Google Scholar] [CrossRef]
- Strandvik, B. Fatty acid metabolism in cystic fibrosis. N. Engl. J. Med. 2004, 350, 605–607. [Google Scholar] [CrossRef]
- Roulet, M.; Frascarolo, P.; Rappaz, I.; Pilet, M. Essential fatty acid deficiency in well nourished young cystic fibrosis patients. Eur. J. Pediatr. 1997, 156, 952–956. [Google Scholar] [CrossRef]
- Peretti, N.; Marcil, V.; Drouin, E.; Levy, E. Mechanisms of lipid malabsorption in cystic fibrosis: The impact of essential fatty acids deficiency. Nut. Metab. 2005, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibault, C.; Wojewodka, G.; Saeed, Z.; Hajduch, M.; Matouk, E.; De Sanctis, J.B.; Radzioch, D. Cystic fibrosis fatty acid imbalance is linked to ceramide deficiency and corrected by fenretinide. Am. J. Respire Cell. Mol. Biol. 2008, 41, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Strandvik, B.; Gronowitz, E.; Enlund, F.; Martinsson, T.; Wahlström, J. Essential fatty acid deficiency in relation to genotype in patients with cystic fibrosis. J. Pediatr. 2001, 139, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Seegmiller, A.C. Abnormal unsaturated fatty acid metabolism in cystic fibrosis: Biochemical mechanisms and clinical implications. Int. J. Mol. Sci. 2014, 15, 16083–16099. [Google Scholar] [CrossRef] [Green Version]
- Umunakwe, O.C.; Seegmiller, A.C. Abnormal n-6 fatty acid metabolism in cystic fibrosis is caused by activation of AMP-activated protein kinase. J. Lipid Res. 2014, 55, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.A.; Austen, K.F.; Soberman, R.J. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N. Engl. J. Med. 1990, 325, 645–655. [Google Scholar]
- Cui, G.; Cottrill, K.A.; Strickland, K.M.; Mashburn, S.A.; Koval, M.; McCarty, N.A. Alteration of Membrane Cholesterol Content Plays a Key Role in Regulation of Cystic Fibrosis Transmembrane Conductance Regulator Channel Activity. Front. Physiol. 2021, 12, 652513. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Y.; Gulbins, E.; Grassmé, H. The Anti-Infectious Role of Sphingosine in Microbial Diseases. Cells 2021, 10, 1105. [Google Scholar] [CrossRef]
- Cottrill, K.A.; Farinha, C.M.; McCarty, N.A. The bidirectional relationship between CFTR and lipids. Commun. Biol. 2020, 3, 179. [Google Scholar] [CrossRef]
- Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Veltman, M.; De Sanctis, J.B.; Stolarczyk, M.; Klymiuk, N.; Bähr, A.; Brouwer, R.W.; Oole, E.; et al. CFTR Correctors and Antioxidants Partially Normalize Lipid Imbalance but not Abnormal Basal Inflammatory Cytokine Profile in CF Bronchial Epithelial Cells. Front. Physiol. 2021, 12, 619442. [Google Scholar] [CrossRef]
- Horati, H.; Janssens, H.M.; Margaroli, C.; Veltman, M.; Stolarczyk, M.; Kilgore, M.B.; Chou, J.; Peng, L.; Tiddens, H.A.M.W.; Chandler, J.D.; et al. Airway profile of bioactive lipids predicts early progression of lung disease in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Harikumar, K.B. Sphingosine 1-Phosphate: A Novel Target for Lung Disorders. Front. Immunol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respire J. 2016, 49, 1600903. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W. Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respire Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar]
- Rosen, B.H.; Evans, T.I.A.; Moll, S.R.; Gray, J.S.; Liang, B.; Sun, X.; Zhang, Y.; Jensen-Cody, C.W.; Swatek, A.M.; Zhou, W.; et al. Infection is not required for mucoinflammatory lung disease in CFTR-knockout ferrets. Am. J. Respire Crit. Care Med. 2018, 197, 1308–1318. [Google Scholar] [CrossRef]
- Balázs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54 (Suppl. 3), S5–S12. [Google Scholar] [CrossRef] [Green Version]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53 (Suppl. 3), S30–S50. [Google Scholar] [CrossRef] [Green Version]
- Ideozu, J.E.; Zhang, X.; McColley, S.; Levy, H. Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights. Genes 2019, 10, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerholz, D.K.; Stoltz, D.A.; Gansemer, N.D.; Ernst, S.E.; Cook, D.P.; Strub, M.D.; LeClair, E.N.; Barker, C.K.; Adam, R.J.; Leidinger, M.R.; et al. Lack of cystic fibrosis transmembrane conductance regulator disrupts fetal airway development in pigs. Lab. Investig. 2018, 98, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Meyerholz, D.K.; Stoltz, D.A.; Namati, E.; Ramachandran, S.; Pezzulo, A.A.; Smith, A.R.; Rector, M.V.; Suter, M.J.; Kao, S.; McLennan, G.; et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am. J. Respire Crit. Care Med. 2010, 182, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.J.; Michalski, A.S.; Bauer, C.; Abou Alaiwa, M.H.; Gross, T.J.; Awadalla, M.S.; Bouzek, D.C.; Gansemer, N.D.; Taft, P.J.; Hoegger, M.J.; et al. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am. J. Respire Crit. Care Med. 2013, 188, 1434–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Affected Organs | Manifestations |
|---|---|
| Reproductive tract | Absence of vas deferens, male (female) infertility |
| Sweat glands | Elevated sweat chloride |
| Lungs | Airway obstruction, chronic bacterial infection, bronchiectasis, pneumothorax, hemoptysis |
| Sinuses | Sinusitis, polyps |
| Pancreas | Exocrine insufficiency, cystic fibrosis-related diabetes |
| Liver | Obstructive biliary tract disease |
| Intestine | Meconium ileus, distal intestinal obstruction syndrome, rectal prolapse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. https://doi.org/10.3390/cells10112844
Hanssens LS, Duchateau J, Casimir GJ. CFTR Protein: Not Just a Chloride Channel? Cells. 2021; 10(11):2844. https://doi.org/10.3390/cells10112844
Chicago/Turabian StyleHanssens, Laurence S., Jean Duchateau, and Georges J. Casimir. 2021. "CFTR Protein: Not Just a Chloride Channel?" Cells 10, no. 11: 2844. https://doi.org/10.3390/cells10112844
APA StyleHanssens, L. S., Duchateau, J., & Casimir, G. J. (2021). CFTR Protein: Not Just a Chloride Channel? Cells, 10(11), 2844. https://doi.org/10.3390/cells10112844