
Novel Balance Mechanism Participates in Stem Cell Therapy to Alleviate Neuropathology and Cognitive Impairment in Animal Models with Alzheimer’s Disease

Abstract
:1. Alzheimer’s Disease and Stem Cell Therapy
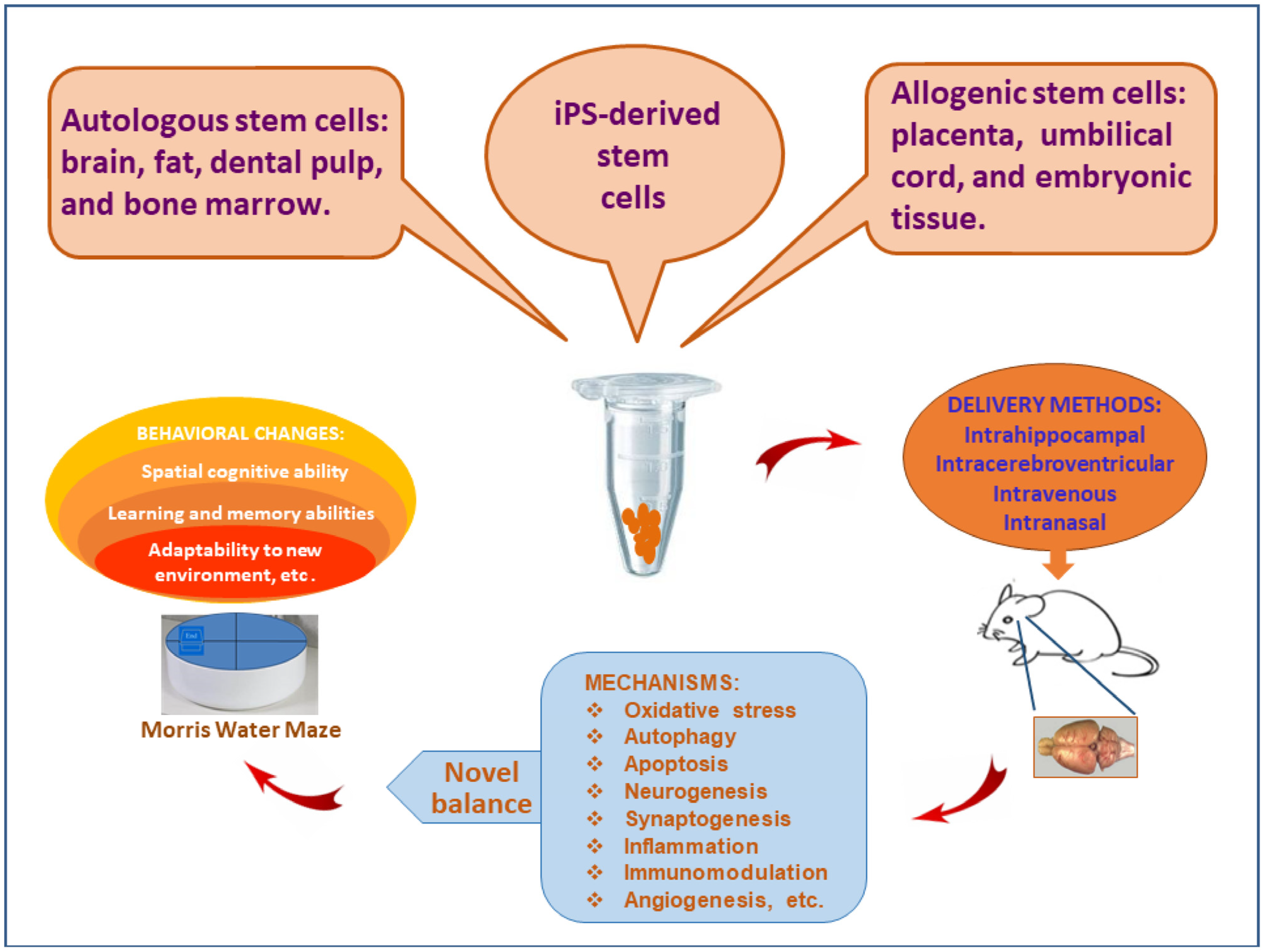
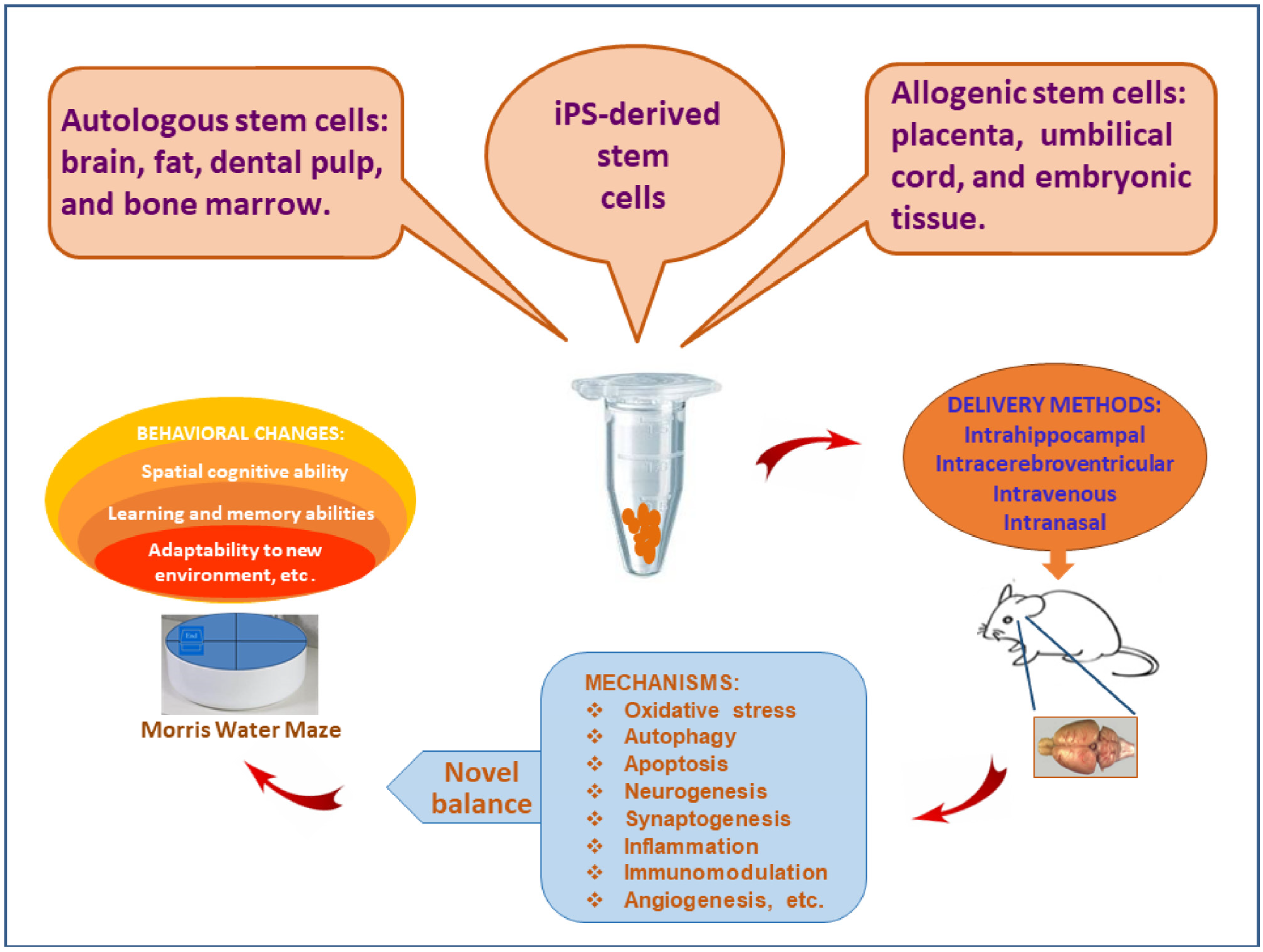
2. Participant Cell Types of New Balance Mechanism
- (1).
- Physical pressure. The local pressure of cerebral tissue can be increased after the stem cells are delivered through the intrahippocampal injection, but the same phenomenon is not found via the peripheral delivery. Nonetheless, the local physical pressure caused by mechanical force is almost negligible, since a similar therapeutic effects can be obtained through tail vein delivery as well [9,30,31].
- (2).
- Signaling molecules. The transportation of stem cells alters the microenvironment of cerebral tissue and stimulates the secretion of autocrine and paracrine cytokines, such as chemokines, leucocyte chemoattractant factors, transcription factors, inflammatory cytokines, fibrogenic cytokines, and growth factors (Table 1). Some factors are general products that can be secreted by all types of stem cells, whereas other cytokines are only produced by specific stem cells [32]. Those pragmatic cytokines participate in the establishment of new balance mechanisms. The secretion of autocrine and paracrine cytokines plays important roles in neurogenesis and synaptogenesis.
- (3).
- (a)
- Functional neurons play a central role in the brain. There are roughly 20 billion neurons in the human cortex. Each neuron has an average 7000 synaptic connections [33]. The number of synapses is relatively stabilized in adulthood. Neuronal synapses may decrease with aging, but they can also increase due to brain plasticity. The transplanted stem cells can stimulate neurogenesis and synapse formation. Newborn neurons may be from (i) the transdifferentiation of stem cells; and (ii) the activation of specialized multipotent stem cells in the brain. At present, stem cell therapy has overcome the concerns of uncertainty and safety, and its effectiveness has been validated as well.
- (b)
- Oligodendrocyte is a specific subtype of neuroglia. In the central nervous system, their branch structures wrap around the neuronal axons to form an insulating myelin sheath. The physiological function of oligodendrocytes is to maintain neuronal insulation during the excitement of nerve signals. The complete structure of the myelin sheath provides a safety measure for signal transmission among neuronal synapses. Stem cell therapy restores neuronal networks by way of synaptogenesis that is protected by the myelin sheaths from oligodendrocytes [34,35].
- (c)
- Astrocytes, also called astroglia, have projections covering local neurons. Astrocytes are the support system in the cerebral tissue to hold neurons in the position. Additionally, they can produce cytokines and interact with other cell types. For example, astrocytes participate in microglia-mediated inflammatory and immune processes [36]. Astrocytes are responsible for substance exchange. In the CNS, astrocytes contact both capillaries and neurons to transport nutrients. Moreover, the phagocytosis of astrocytes is implicated in the amyloid load of Alzheimer disease [37]. In the process of stem cell therapy, the precise roles of astrocytes are still unclear. After exposure to MSC-conditioned medium, the expression of pro-inflammatory factors such as IL-1β, TNF-α and IL-6 was attenuated in cultured astrocytes [38]. The transplanted stem cells acted on astrocytes to modify neuroimmune and relieve neuroinflammation in vivo [39].
- (d)
- Microglia are resident immune cells in the brain, equivalent to macrophages. Functional microglia take part in the neuroinflammation, immunomodulation, the elimination of Aβ proteins, and tau pathology. As the first line of the neuroimmune system, microglia remove cerebral debris and protect neurons from harmful invasion. In contrast, the inflammatory factors released by microglia can cause receptor-induced neuronal apoptosis [40]. Fortunately, microglial activity can be modulated by the transplanted stem cells. So, stem cell therapy suppresses neuroinflammation and controls neuroimmune overreaction. Furthermore, microglia can detect neuronal injury and play a critical role in the maintenance of neuronal health. As immune cells, microglia have duality in the pathogenesis of AD. They can not only protect neurons by engulfing detrimental Aβ proteins, but also damage neurons by secreting inflammatory cytokines [41,42,43]. The consequence may be beneficial or pernicious, which is determined by the comprehensive effect of multi-level signaling crosstalk.
3. Representative Signaling Pathways of New Balance Mechanism
3.1. The Transplantation of Stem Cells Mediates Cell Growth and Death
3.2. The Transplanted Stem Cells Regulate the Production and Removal of Aberrant Proteins
3.3. The Transplanted Stem Cells Can Produce Pro- and Anti-Inflammatory Cytokines
3.4. Immunoregulation Is Modulated by the Transplanted Stem Cells
3.5. The Transplanted Stem Cells Participate in Synaptic Plasticity
4. Perspective
4.1. Therapeutic Efficiency and Synergistic Effect
4.2. Stem Cell Viability
4.3. The Improvement of Delivery Methods
4.4. Exosomes
5. Challenges
- (1).
- The selection of surveillance biomarkers. Currently, monitoring markers (i.e., Aβ42, T-tau and P-tau, or exosomes in cerebrospinal fluid and/or peripheral bloodstream) need to be optimized for the evaluation of therapeutic effects.
- (2).
- The timeline of the new balance mechanism. Following the transplantation of stem cells, the pathological state is altered and then a new balance is developed. However, it is unsure how long the dynamic reconstruction can be maintained. Perhaps, it is necessary to repeatedly transplant stem cells to obtain reliable therapeutic effects. At this time, it is important to optimize the relevant parameters of stem cell transplantation, including cell concentration, time interval, inoculation position, and delivery method.
- (3).
- Uncertainty and perplexity. The therapeutic effect of transplanted stem cells involves multiple mechanisms, such as immunomodulation, inflammation, apoptosis, neurogenesis, autophagy, and angiogenesis. The integration of various mechanisms establishes a new balance and brings about beneficial improvements. Nowadays, most of the above-mentioned mechanisms have been investigated and their roles have been elucidated. Nevertheless, the details of relevant mechanisms still need to be explored, such as autophagy and immunomodulation, the interaction between astrocytes and microglia, microglial activation and synaptic remodeling, etc.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Boche, D.; Nicoll, J.A.R. Invited Review—Understanding cause and effect in Alzheimer’s pathophysiology: Implications for clinical trials. Neuropathol. Appl. Neurobiol. 2020, 46, 623–640. [Google Scholar] [CrossRef]
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Tanzi, R.E.; St George-Hyslop, P.H.; Gusella, J.F. Molecular genetic approaches to Alzheimer’s disease. Trends Neurosci. 1989, 12, 152–158. [Google Scholar] [CrossRef]
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651. [Google Scholar] [CrossRef] [Green Version]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Emilien, G.; Maloteaux, J.M.; Beyreuther, K.; Masters, C.L. Alzheimer disease: Mouse models pave the way for therapeutic opportunities. Arch. Neurol. 2000, 57, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Sandbrink, R.; Hartmann, T.; Masters, C.L.; Beyreuther, K. Genes contributing to Alzheimer’s disease. Mol. Psychiatry 1996, 1, 27–40. [Google Scholar]
- Qin, C.; Lu, Y.; Wang, K.; Bai, L.; Shi, G.; Huang, Y.; Li, Y. Transplantation of bone marrow mesenchymal stem cells improves cognitive deficits and alleviates neuropathology in animal models of Alzheimer’s disease: A meta-analytic review on potential mechanisms. Transl. Neurodegener. 2020, 9, 20. [Google Scholar] [CrossRef]
- Perdigao, C.; Barata, M.A.; Araujo, M.N.; Mirfakhar, F.S.; Castanheira, J.; Guimas Almeida, C. Intracellular Trafficking Mechanisms of Synaptic Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2020, 14, 72. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Larson, E.B.; Sonnen, J.A.; Shofer, J.B.; Petrie, E.C.; Schantz, A.; Peskind, E.R.; Raskind, M.A.; Breitner, J.C.; Montine, T.J. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology 2007, 69, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Storga, D.; Vrecko, K.; Birkmayer, J.G.; Reibnegger, G. Monoaminergic neurotransmitters, their precursors and metabolites in brains of Alzheimer patients. Neurosci. Lett. 1996, 203, 29–32. [Google Scholar] [CrossRef]
- Yew, D.T.; Li, W.P.; Webb, S.E.; Lai, H.W.; Zhang, L. Neurotransmitters, peptides, and neural cell adhesion molecules in the cortices of normal elderly humans and Alzheimer patients: A comparison. Exp. Gerontol. 1999, 34, 117–133. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Zhu, X.; Tabaton, M.; Liu, G.; McKeel, D.W., Jr.; Cohen, M.L.; Wang, X.; Siedlak, S.L.; Dwyer, B.E.; Hayashi, T.; et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J. Alzheimers Dis. 2010, 19, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghai, R.; Nagarajan, K.; Arora, M.; Grover, P.; Ali, N.; Kapoor, G. Current Strategies and Novel Drug Approaches for Alzheimer Disease. CNS Neurol. Disord. Drug Targets 2020, 19, 676–690. [Google Scholar] [CrossRef]
- Alexander, G.C.; Emerson, S.; Kesselheim, A.S. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA 2021, 325, 1717–1718. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Park, H.J.; Kim, H.N.; Oh, S.H.; Bae, J.S.; Ha, H.J.; Lee, P.H. Mesenchymal stem cells enhance autophagy and increase beta-amyloid clearance in Alzheimer disease models. Autophagy 2014, 10, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Kubota, K.; Kobayashi, E.; Chikenji, T.S.; Saito, Y.; Konari, N.; Fujimiya, M. Bone marrow-derived mesenchymal stem cells improve cognitive impairment in an Alzheimer’s disease model by increasing the expression of microRNA-146a in hippocampus. Sci. Rep. 2020, 10, 10772. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, S.; Yang, B.; Huang, T.; Meng, N.; Xu, L.; Xing, Q.; Zhang, Y.; Zhang, K.; Li, Q.; et al. Resveratrol promotes hUC-MSCs engraftment and neural repair in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2018, 339, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, N.; Shimizu, J.; Takai, K.; Arimitsu, N.; Ueda, Y.; Wakisaka, S.; Suzuki, T.; Suzuki, N. Cellular and molecular mechanisms of the restoration of human APP transgenic mouse cognitive dysfunction after transplant of human iPS cell-derived neural cells. Exp. Neurol. 2015, 271, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gu, G.J.; Zhang, Q.; Liu, J.H.; Zhang, B.; Guo, Y.; Wang, M.Y.; Gong, Q.Y.; Xu, J.R. NSCs promote hippocampal neurogenesis, metabolic changes and synaptogenesis in APP/PS1 transgenic mice. Hippocampus 2017, 27, 1250–1263. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Wang, X.; Xu, S. Therapeutic Effects of Transplantation of As-MiR-937-Expressing Mesenchymal Stem Cells in Murine Model of Alzheimer’s Disease. Cell. Physiol. Biochem. 2015, 37, 321–330. [Google Scholar] [CrossRef]
- Nasiri, E.; Alizadeh, A.; Roushandeh, A.M.; Gazor, R.; Hashemi-Firouzi, N.; Golipoor, Z. Melatonin-pretreated adipose-derived mesenchymal stem cells efficeintly improved learning, memory, and cognition in an animal model of Alzheimer’s disease. Metab. Brain Dis. 2019, 34, 1131–1143. [Google Scholar] [CrossRef]
- Van den Hurk, M.; Erwin, J.A.; Yeo, G.W.; Gage, F.H.; Bardy, C. Patch-Seq Protocol to Analyze the Electrophysiology, Morphology and Transcriptome of Whole Single Neurons Derived From Human Pluripotent Stem Cells. Front. Mol. Neurosci. 2018, 11, 261. [Google Scholar] [CrossRef]
- Bae, J.S.; Jin, H.K.; Lee, J.K.; Richardson, J.C.; Carter, J.E. Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-beta deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 524–531. [Google Scholar] [CrossRef]
- Lampron, A.; Pimentel-Coelho, P.M.; Rivest, S. Migration of bone marrow-derived cells into the central nervous system in models of neurodegeneration. J. Comp. Neurol. 2013, 521, 3863–3876. [Google Scholar] [CrossRef]
- Hwang, J.H.; Shim, S.S.; Seok, O.S.; Lee, H.Y.; Woo, S.K.; Kim, B.H.; Song, H.R.; Lee, J.K.; Park, Y.K. Comparison of cytokine expression in mesenchymal stem cells from human placenta, cord blood, and bone marrow. J. Korean Med. Sci. 2009, 24, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Drachman, D.A. Do we have brain to spare? Neurology 2005, 64, 2004–2005. [Google Scholar] [CrossRef]
- Jaldeep, L.; Lipi, B.; Prakash, P. Potential role of NGF, BDNF and their receptors in Oligodendrocytes differentiation from neural stem cell—An in-vitro study. Cell Biol. Int. 2020, 45, 432–446. [Google Scholar] [CrossRef]
- Klein, R.; Mahlberg, N.; Ohren, M.; Ladwig, A.; Neumaier, B.; Graf, R.; Hoehn, M.; Albrechtsen, M.; Rees, S.; Fink, G.R.; et al. The Neural Cell Adhesion Molecule-Derived (NCAM)-Peptide FG Loop (FGL) Mobilizes Endogenous Neural Stem Cells and Promotes Endogenous Regenerative Capacity after Stroke. J. Neuroimmune Pharmacol. 2016, 11, 708–720. [Google Scholar] [CrossRef]
- Lian, H.; Litvinchuk, A.; Chiang, A.C.; Aithmitti, N.; Jankowsky, J.L.; Zheng, H. Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J. Neurosci. 2016, 36, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflamm. 2016, 13, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, S.; Calas, A.G.; Vergouts, M.; Hermans, E. Immunomodulatory influence of bone marrow-derived mesenchymal stem cells on neuroinflammation in astrocyte cultures. J. Neuroimmunol. 2012, 249, 40–48. [Google Scholar] [CrossRef]
- Konttinen, H.; Gureviciene, I.; Oksanen, M.; Grubman, A.; Loppi, S.; Huuskonen, M.T.; Korhonen, P.; Lampinen, R.; Keuters, M.; Belaya, I.; et al. PPARbeta/delta-agonist GW0742 ameliorates dysfunction in fatty acid oxidation in PSEN1DeltaE9 astrocytes. Glia 2019, 67, 146–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, P.; Wang, X.; Wang, S.; Wei, Y.; Feng, J.; Zhu, M. Maresin 1 Improves Cognitive Decline and Ameliorates Inflammation in a Mouse Model of Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 466. [Google Scholar] [CrossRef] [Green Version]
- Sierksma, A.; Lu, A.; Mancuso, R.; Fattorelli, N.; Thrupp, N.; Salta, E.; Zoco, J.; Blum, D.; Buee, L.; De Strooper, B.; et al. Novel Alzheimer risk genes determine the microglia response to amyloid-beta but not to TAU pathology. EMBO Mol. Med. 2020, 12, e10606. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell. Neurosci. 2018, 12, 531. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Ying, M.; Gu, S.; Yin, W.; Li, Y.; Yuan, H.; Fang, S.; Li, M. Cysteinyl Leukotriene Receptor 2 is Involved in Inflammation and Neuronal Damage by Mediating Microglia M1/M2 Polarization through NF-kappaB Pathway. Neuroscience 2019, 422, 99–118. [Google Scholar] [CrossRef]
- Kan, I.; Barhum, Y.; Melamed, E.; Offen, D. Mesenchymal stem cells stimulate endogenous neurogenesis in the subventricular zone of adult mice. Stem Cell Rev. Rep. 2011, 7, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Bavamian, S.; Mellios, N.; Lalonde, J.; Fass, D.M.; Wang, J.; Sheridan, S.D.; Madison, J.M.; Zhou, F.; Rueckert, E.H.; Barker, D.; et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol. Psychiatry 2015, 20, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cao, J.X.; Sun, B.; Li, H.L.; Xia, Y.; Wu, Z.; Tang, C.L.; Hu, J. Mesenchymal stem cells inhibition of chronic ethanol-induced oxidative damage via upregulation of phosphatidylinositol-3-kinase/Akt and modulation of extracellular signal-regulated kinase 1/2 activation in PC12 cells and neurons. Neuroscience 2010, 167, 1115–1124. [Google Scholar] [CrossRef]
- Singh, P.; Fukuda, S.; Liu, L.; Chitteti, B.R.; Pelus, L.M. Survivin Is Required for Mouse and Human Bone Marrow Mesenchymal Stromal Cell Function. Stem Cells 2018, 36, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Xue, F.; Shi, C.; Chen, Q.; Hang, W.; Xia, L.; Wu, Y.; Tao, S.Z.; Zhou, J.; Shi, A.; Chen, J. Melatonin Mediates Protective Effects against Kainic Acid-Induced Neuronal Death through Safeguarding ER Stress and Mitochondrial Disturbance. Front. Mol. Neurosci. 2017, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Polis, B.; Samson, A.O. Neurogenesis versus neurodegeneration: The broken balance in Alzheimer’s disease. Neural Regen. Res. 2021, 16, 496–497. [Google Scholar] [CrossRef]
- Yu, S.; Hei, Y.; Liu, W. Upregulation of seladin-1 and nestin expression in bone marrow mesenchymal stem cell transplantation via the ERK1/2 and PI3K/Akt signaling pathways in an Alzheimer’s disease model. Oncol. Lett. 2018, 15, 7443–7449. [Google Scholar] [CrossRef]
- Gomez Del Pulgar, T.; De Ceballos, M.L.; Guzman, M.; Velasco, G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [Green Version]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef]
- Erlich, S.; Mizrachy, L.; Segev, O.; Lindenboim, L.; Zmira, O.; Adi-Harel, S.; Hirsch, J.A.; Stein, R.; Pinkas-Kramarski, R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007, 3, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ma, K.; Qi, T.; Wei, X.; Zhang, Q.; Li, G.; Chiu, J.F. P62 regulates resveratrol-mediated Fas/Cav-1 complex formation and transition from autophagy to apoptosis. Oncotarget 2015, 6, 789–801. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A.; Gao, F.B. Regulation of Abeta pathology by beclin 1: A protective role for autophagy? J. Clin. Investig. 2008, 118, 2015–2018. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, A.; Yamamoto, S.; Ting, T.; Fan, Y.; Sadleir, K.; Wang, Y.; Zhang, W.; Huang, S.; Levine, B.; Vassar, R.; et al. A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease. PLoS Genet. 2017, 13, e1006962. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Liang, Y.; Liang, F.; Shen, N.; Shinozuka, K.; Yu, J.T.; Ran, C.; Quan, Q.; Tanzi, R.E.; Zhang, C. A Curcumin Analog Reduces Levels of the Alzheimer’s Disease-Associated Amyloid-beta Protein by Modulating AbetaPP Processing and Autophagy. J. Alzheimer’s Dis. 2019, 72, 761–771. [Google Scholar] [CrossRef]
- Esler, W.P.; Wolfe, M.S. A portrait of Alzheimer secretases—New features and familiar faces. Science 2001, 293, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Wu, P.Y.; Chen, Y.W.; Chang, Y.T.; Bhore, N.; Wu, P.F.; Liao, Y.F. Quantitative Measurement of gamma-Secretase-mediated Amyloid Precursor Protein and Notch Cleavage in Cell-based Luciferase Reporter Assay Platforms. J. Vis. Exp. 2018, 56795. [Google Scholar] [CrossRef]
- Morroni, F.; Sita, G.; Tarozzi, A.; Rimondini, R.; Hrelia, P. Early effects of Abeta1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav. Brain Res. 2016, 314, 106–115. [Google Scholar] [CrossRef]
- Yamamoto, N.; Tanida, M.; Kasahara, R.; Sobue, K.; Suzuki, K. Leptin inhibits amyloid beta-protein fibrillogenesis by decreasing GM1 gangliosides on the neuronal cell surface through PI3K/Akt/mTOR pathway. J. Neurochem. 2014, 131, 323–332. [Google Scholar] [CrossRef]
- Esmaeilzade, B.; Artimani, T.; Amiri, I.; Najafi, R.; Shahidi, S.; Sabec, M.; Farzadinia, P.; Zare, M.; Zahiri, M.; Soleimani Asl, S. Dimethyloxalylglycine preconditioning enhances protective effects of bone marrow-derived mesenchymal stem cells in Abeta- induced Alzheimer disease. Physiol. Behav. 2019, 199, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Verret, L.; Jankowsky, J.L.; Xu, G.M.; Borchelt, D.R.; Rampon, C. Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci. 2007, 27, 6771–6780. [Google Scholar] [CrossRef]
- Verret, L.; Trouche, S.; Zerwas, M.; Rampon, C. Hippocampal neurogenesis during normal and pathological aging. Psychoneuroendocrinology 2007, 32 (Suppl. 1), S26–S30. [Google Scholar] [CrossRef]
- Gustavsson, T.; Syvanen, S.; O’Callaghan, P.; Sehlin, D. SPECT imaging of distribution and retention of a brain-penetrating bispecific amyloid-beta antibody in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 37. [Google Scholar] [CrossRef]
- Honjo, Y.; Horibe, T.; Torisawa, A.; Ito, H.; Nakanishi, A.; Mori, H.; Komiya, T.; Takahashi, R.; Kawakami, K. Protein disulfide isomerase P5-immunopositive inclusions in patients with Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 38, 601–609. [Google Scholar] [CrossRef]
- Kuang, H.; Tan, C.Y.; Tian, H.Z.; Liu, L.H.; Yang, M.W.; Hong, F.F.; Yang, S.L. Exploring the bi-directional relationship between autophagy and Alzheimer’s disease. CNS Neurosci. Ther. 2020, 26, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierzynowska, K.; Podlacha, M.; Gaffke, L.; Majkutewicz, I.; Mantej, J.; Wegrzyn, A.; Osiadly, M.; Myslinska, D.; Wegrzyn, G. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic Alzheimer’s disease. Neuropharmacology 2019, 148, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Wang, Z.H.; Tian, Q. Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourmaud, S.; Shou, H.; Irwin, D.J.; Sansalone, K.; Jacobs, L.M.; Lucas, T.H.; Marsh, E.D.; Davis, K.A.; Jensen, F.E.; Talos, D.M. Alzheimer-Like amyloid and tau alterations associated with cognitive deficit in temporal lobe epilepsy. Brain 2020, 143, 191–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Orejana, L.; Barros-Minones, L.; Aguirre, N.; Puerta, E. Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp. Gerontol. 2013, 48, 565–571. [Google Scholar] [CrossRef]
- Wu, C.; Gong, W.G.; Wang, Y.J.; Sun, J.J.; Zhou, H.; Zhang, Z.J.; Ren, Q.G. Escitalopram alleviates stress-induced Alzheimer’s disease-like tau pathologies and cognitive deficits by reducing hypothalamic-pituitary-adrenal axis reactivity and insulin/GSK-3beta signal pathway activity. Neurobiol. Aging 2018, 67, 137–147. [Google Scholar] [CrossRef]
- Ye, T.; Li, X.; Zhou, P.; Ye, S.; Gao, H.; Hua, R.; Ma, J.; Wang, Y.; Cai, B. Chrysophanol improves memory ability of d-galactose and Abeta25-35 treated rat correlating with inhibiting tau hyperphosphorylation and the CaM-CaMKIV signal pathway in hippocampus. 3 Biotech 2020, 10, 111. [Google Scholar] [CrossRef]
- Lee, I.S.; Jung, K.; Kim, I.S.; Lee, H.; Kim, M.; Yun, S.; Hwang, K.; Shin, J.E.; Park, K.I. Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol. Neurodegener. 2015, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Jin, H.K.; Endo, S.; Schuchman, E.H.; Carter, J.E.; Bae, J.S. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells 2010, 28, 329–343. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Demirci, S.; Aynali, A.; Demirci, K.; Demirci, S.; Aridogan, B.C. The Serum Levels of Resistin and Its Relationship with Other Proinflammatory Cytokines in Patients with Alzheimer’s Disease. Clin. Psychopharmacol. Neurosci. 2017, 15, 59–63. [Google Scholar] [CrossRef]
- Ng, A.; Tam, W.W.; Zhang, M.W.; Ho, C.S.; Husain, S.F.; McIntyre, R.S.; Ho, R.C. IL-1beta, IL-6, TNF- alpha and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Sci. Rep. 2018, 8, 12050. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Interleukin-10: New perspectives on an old cytokine. Immunol. Rev. 2008, 226, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [Green Version]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma + TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa beta as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Z.; Hong, Y.; Zhang, K.; Wang, J.; Zheng, L.; Zhang, Z.; Hu, Z.; Han, X.; Han, Y.; Chen, T.; et al. BAG-1M co-activates BACE1 transcription through NF-kappaB and accelerates Abeta production and memory deficit in Alzheimer’s disease mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2398–2407. [Google Scholar] [CrossRef]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Barton Whittle, T.; Nicolas Crain, C.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2017, 55, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Nogueras-Ortiz, C.J.; Mahairaki, V.; Delgado-Peraza, F.; Das, D.; Avgerinos, K.; Eren, E.; Hentschel, M.; Goetzl, E.J.; Mattson, M.P.; Kapogiannis, D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells 2020, 9, 1618. [Google Scholar] [CrossRef]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2020, 34, 2436–2450. [Google Scholar] [CrossRef] [Green Version]
- Qiao, P.; Ma, J.; Wang, Y.; Huang, Z.; Zou, Q.; Cai, Z.; Tang, Y. Curcumin Prevents Neuroinflammation by Inducing Microglia to Transform into the M2-phenotype via CaMKKbeta-dependent Activation of the AMP-Activated Protein Kinase Signal Pathway. Curr. Alzheimer Res. 2020, 17, 735–752. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Zeng, H.; Liu, N.; Yang, Y.Y.; Xing, H.Y.; Liu, X.X.; Li, F.; La, G.Y.; Huang, M.J.; Zhou, M.W. Lentivirus-mediated downregulation of alpha-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J. Neuroinflamm. 2019, 16, 283. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Xue, T.F.; Guo, X.D.; Yang, J.; Guo, R.B.; Wang, J.; Huang, J.Y.; Zhao, X.J.; Sun, X.L. Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 2018, 17, e12774. [Google Scholar] [CrossRef]
- Zhang, Y.; He, M.L. Deferoxamine enhances alternative activation of microglia and inhibits amyloid beta deposits in APP/PS1 mice. Brain Res. 2017, 1677, 86–92. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Mee, A.P.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Miklossy, J.; Khalili, K.; Gern, L.; Ericson, R.L.; Darekar, P.; Bolle, L.; Hurlimann, J.; Paster, B.J. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J. Alzheimer’s Dis. 2004, 6, 639–649; discussion 673–681. [Google Scholar] [CrossRef] [Green Version]
- Pisa, D.; Alonso, R.; Rabano, A.; Rodal, I.; Carrasco, L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Sci. Rep. 2015, 5, 15015. [Google Scholar] [CrossRef] [Green Version]
- Linard, M.; Letenneur, L.; Garrigue, I.; Doize, A.; Dartigues, J.F.; Helmer, C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 200–208. [Google Scholar] [CrossRef]
- Carter, C.J. Genetic, Transcriptome, Proteomic, and Epidemiological Evidence for Blood-Brain Barrier Disruption and Polymicrobial Brain Invasion as Determinant Factors in Alzheimer’s Disease. J. Alzheimer’s Rep. 2017, 1, 125–157. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Bushman, M.; Kishore, N.; Niehus, R.; de Salazar, P.M.; Cowling, B.J.; Lipsitch, M.; Leung, G.M. Estimating clinical severity of COVID-19 from the transmission dynamics in Wuhan, China. Nat. Med. 2020, 26, 506–510. [Google Scholar] [CrossRef] [Green Version]
- Caratozzolo, S.; Zucchelli, A.; Turla, M.; Cotelli, M.S.; Fascendini, S.; Zanni, M.; Bianchetti, A.; Psy, M.P.; Rozzini, R.; Boffelli, S.; et al. The impact of COVID-19 on health status of home-dwelling elderly patients with dementia in East Lombardy, Italy: Results from COVIDEM network. Aging Clin. Exp. Res. 2020, 32, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Bergman, M.; Salman, H.; Beloosesky, Y.; Djaldetti, M.; Bessler, H. Are peripheral blood cells from patients with Alzheimer disease more sensitive to apoptotic stimuli? Alzheimer Assoc. Disord. 2002, 16, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; White, C.C.; Winn, P.A.; Cimpean, M.; Replogle, J.M.; Glick, L.R.; Cuerdon, N.E.; Ryan, K.J.; Johnson, K.A.; Schneider, J.A.; et al. CD33 modulates TREM2: Convergence of Alzheimer loci. Nat. Neurosci. 2015, 18, 1556–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsonego, A.; Zota, V.; Karni, A.; Krieger, J.I.; Bar-Or, A.; Bitan, G.; Budson, A.E.; Sperling, R.; Selkoe, D.J.; Weiner, H.L. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J. Clin. Investig. 2003, 112, 415–422. [Google Scholar] [CrossRef] [Green Version]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 5370. [Google Scholar] [CrossRef]
- Chen, H.; Lin, W.; Zhang, Y.; Lin, L.; Chen, J.; Zeng, Y.; Zheng, M.; Zhuang, Z.; Du, H.; Chen, R.; et al. IL-10 Promotes Neurite Outgrowth and Synapse Formation in Cultured Cortical Neurons after the Oxygen-Glucose Deprivation via JAK1/STAT3 Pathway. Sci. Rep. 2016, 6, 30459. [Google Scholar] [CrossRef] [Green Version]
- Naaldijk, Y.; Jager, C.; Fabian, C.; Leovsky, C.; Bluher, A.; Rudolph, L.; Hinze, A.; Stolzing, A. Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS1 Alzheimer mice. Neuropathol. Appl. Neurobiol. 2017, 43, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Rong, Y.; Wang, J.; Zhou, Z.; Ge, X.; Ji, C.; Jiang, D.; Gong, F.; Li, L.; Chen, J.; et al. Exosome-shuttled miR-216a-5p from hypoxic preconditioned mesenchymal stem cells repair traumatic spinal cord injury by shifting microglial M1/M2 polarization. J. Neuroinflamm. 2020, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Baufeld, C.; O’Loughlin, E.; Calcagno, N.; Madore, C.; Butovsky, O. Differential contribution of microglia and monocytes in neurodegenerative diseases. J. Neural Transm. Vienna 2018, 125, 809–826. [Google Scholar] [CrossRef]
- Zhong, Z.; Chen, A.; Fa, Z.; Ding, Z.; Xie, J.; Sun, Y.; Zhang, R.; Wang, Q. Adipose-Derived Stem Cells Modulate BV2 Microglial M1/M2 Polarization by Producing GDNF. Stem Cells Dev. 2020, 29, 714–727. [Google Scholar] [CrossRef]
- Petukhova, E.O.; Mukhamedshina, Y.O.; Salafutdinov, I.; Garanina, E.E.; Kaligin, M.S.; Leushina, A.V.; Rizvanov, A.A.; Reis, H.J.; Palotas, A.; Zefirov, A.L.; et al. Effects of Transplanted Umbilical Cord Blood Mononuclear Cells Overexpressing GDNF on Spatial Memory and Hippocampal Synaptic Proteins in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 443–453. [Google Scholar] [CrossRef]
- Ren, C.; Gu, X.; Li, H.; Lei, S.; Wang, Z.; Wang, J.; Yin, P.; Zhang, C.; Wang, F.; Liu, C. The role of DKK1 in Alzheimer’s disease: A potential intervention point of brain damage prevention? Pharmacol. Res. 2019, 144, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Zhang, J.; Li, H.; Sun, X. Silencing of aquaporin1 activates the Wnt signaling pathway to improve cognitive function in a mouse model of Alzheimer’s disease. Gene 2020, 755, 144904. [Google Scholar] [CrossRef]
- Dobrowolski, R.; Vick, P.; Ploper, D.; Gumper, I.; Snitkin, H.; Sabatini, D.D.; De Robertis, E.M. Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2012, 2, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnes, M.; Casas Tinto, S. Aberrant Wnt signaling: A special focus in CNS diseases. J. Neurogenet. 2017, 31, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liang, G.; Liang, S.; Mund, R.; Shi, Y.; Wei, H. Dantrolene Ameliorates Impaired Neurogenesis and Synaptogenesis in Induced Pluripotent Stem Cell Lines Derived from Patients with Alzheimer’s Disease. Anesthesiology 2020, 132, 1062–1079. [Google Scholar] [CrossRef]
- Hayashi, Y.; Lin, H.T.; Lee, C.C.; Tsai, K.J. Effects of neural stem cell transplantation in Alzheimer’s disease models. J. Biomed. Sci. 2020, 27, 29. [Google Scholar] [CrossRef] [Green Version]
- Corbett, N.J.; Gabbott, P.L.; Klementiev, B.; Davies, H.A.; Colyer, F.M.; Novikova, T.; Stewart, M.G. Amyloid-beta induced CA1 pyramidal cell loss in young adult rats is alleviated by systemic treatment with FGL, a neural cell adhesion molecule-derived mimetic peptide. PLoS ONE 2013, 8, e71479. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Blaschke, S.; Neumaier, B.; Endepols, H.; Graf, R.; Keuters, M.; Hucklenbroich, J.; Albrechtsen, M.; Rees, S.; Fink, G.R.; et al. The synthetic NCAM mimetic peptide FGL mobilizes neural stem cells in vitro and in vivo. Stem Cell Rev. Rep. 2014, 10, 539–547. [Google Scholar] [CrossRef]
- Jiang, Y.; Gao, H.; Yuan, H.; Xu, H.; Tian, M.; Du, G.; Xie, W. Amelioration of postoperative cognitive dysfunction in mice by mesenchymal stem cell-conditioned medium treatments is associated with reduced inflammation, oxidative stress and increased BDNF expression in brain tissues. Neurosci. Lett. 2019, 709, 134372. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.; Smith, T.N.; Wang, N.; Chua, J.Y.; Westbroek, E.; Wang, K.; Guzman, R. BDNF Pretreatment of Human Embryonic-Derived Neural Stem Cells Improves Cell Survival and Functional Recovery After Transplantation in Hypoxic-Ischemic Stroke. Cell Transplant. 2015, 24, 2449–2461. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Pan, C.; Xuan, A.; Xu, L.; Bao, G.; Liu, F.; Fang, J.; Long, D. Treatment Efficacy of NGF Nanoparticles Combining Neural Stem Cell Transplantation on Alzheimer’s Disease Model Rats. Med. Sci. Monit. 2015, 21, 3608–3615. [Google Scholar] [CrossRef] [Green Version]
- Danielyan, L.; Beer-Hammer, S.; Stolzing, A.; Schafer, R.; Siegel, G.; Fabian, C.; Kahle, P.; Biedermann, T.; Lourhmati, A.; Buadze, M.; et al. Intranasal delivery of bone marrow-derived mesenchymal stem cells, macrophages, and microglia to the brain in mouse models of Alzheimer’s and Parkinson’s disease. Cell Transplant. 2014, 23 (Suppl. 1), S123–S139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.; Ban, J.J.; Yang, S.; Im, W.; Kim, M. The exosome of adipose-derived stem cells reduces beta-amyloid pathology and apoptosis of neuronal cells derived from the transgenic mouse model of Alzheimer’s disease. Brain Res. 2018, 1691, 87–93. [Google Scholar] [CrossRef]
- Wang, X.; Yang, G. Bone marrow mesenchymal stem cells-derived exosomes reduce Abeta deposition and improve cognitive function recovery in mice with Alzheimer’s disease by activating sphingosine kinase/sphingosine-1-phosphate signaling pathway. Cell Biol. Int. 2020, 45, 775–784. [Google Scholar] [CrossRef]
- Losurdo, M.; Pedrazzoli, M.; D’Agostino, C.; Elia, C.A.; Massenzio, F.; Lonati, E.; Mauri, M.; Rizzi, L.; Molteni, L.; Bresciani, E.; et al. Intranasal delivery of mesenchymal stem cell-derived extracellular vesicles exerts immunomodulatory and neuroprotective effects in a 3xTg model of Alzheimer’s disease. Stem Cells Transl. Med. 2020, 9, 1068–1084. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Examples | Function | References |
|---|---|---|---|
| Inflammatory cytokines | TNFα, IL-1, IL-2, IL-6, IL-10 | To regulate inflammatory and immune responses, to participate in the regulation of cell growth and apoptosis, etc. | J. Clin. Endocrinol. Metab. 1998 Jun;83(6):2043–51; Immunotherapy. 2018 Sep;10(12):1053–1064; |
| Fibrogenic cytokines | FGF, TIMP-1 | Proliferation of fibroblasts, collagen synthesis and extracellular fibrosis, immune mediators. | PLoS ONE. 2019 Apr 22;14(4):e0215678; Brain Res. 2004 Apr 16;1005(1–2):21–8. |
| Chemokines | CCL5, CXCL-10, CXCL-12, | Chemo-attractants, to guide the migration of cells, to regulate immunity, inflammation, angiogenesis, etc. | Stem Cells. 2012 Jul;30(7):1544–55; Cancer Res. 2011 Jun 1;71(11):3831–40; J. Cell Physiol. 2019 Aug;234(10):18707–18719 |
| Leucocyte chemoattractant factors | CINC-1, G-CSF, SCF, GM-CSF | To participate in immune/inflammatory cascade. | PLoS ONE. 2019 Apr 22;14(4):e0215678; Blood. 2000 Nov 15;96(10):3422–30. |
| Transcription factors | GATA-4, Nkx2.5, MEF2C | Response to intercellular and extracellular signals, transcriptional regulation in development, cell cycle, and pathogenesis. | Mol. Med. Rep. 2015 Aug;12(2):2607–21; Tissue Eng. Part A. 2011 Jan;17(1–2):45–58. |
| Growth factors | HGF, IGF-1 | Signaling molecules promote cell differentiation and maturation. | Stem Cells Dev. 2010 Jul;19(7):1035–42; Int. J. Stem Cells. 2009 May;2(1):59–68. |
| Vascular endothelial growth factor | VEGF | To stimulate the formation of blood vessels. | Int. J. Stem Cells. 2009 May;2(1):59–68; Brain Res. 2004 Apr 16;1005(1–2):21–8. |
| Other | MCP-1, OPG | Selectively recruiting monocytes, to regulate bone metabolism. | Int. J. Stem Cells. 2009 May;2(1):59–68; J. Interferon Cytokine Res. 2009 Jun;29(6):313–26; Cell. 1997 Apr 18;89(2):309–19. |
| Mechanisms | Cell Types | Signaling Pathways | References |
|---|---|---|---|
| Immunoregulation | Neurons, Microglia, Astrocytes, Oligodendrocytes | To facilitate microglial M1/M2 polarization; to regulate the crosstalk between T cells and microglia; to mediate synaptic plasticity. | Neuroscience. 2019 Dec 1;422:99–118; Proc. Natl. Acad. Sci. USA. 2006 Mar 28;103(13): 5048–5053; Front. Synaptic Neurosci. 2018 Jun 13;10:14. |
| Inflammation | Neurons, Microglia, Astrocytes, Oligodendrocytes | To decrease the level of NF-κB in astrocytes; to reduce the levels of TNF-α, IL-6, and MCP-1; to regulate cell growth and apoptosis. | Neuropathol. Appl. Neurobiol. 2017 Jun;43(4):299–314; Sci. Rep. 2020 Jul 1;10(1):10772; DOI:10.1186/s13024-015-0035-6. |
| Neurogenesis | Neurons, Microglia, Astrocytes, Oligodendrocytes | To increase IGF-1 expression in the hippocampus; to increase N-acetylaspartate and Glutamate; to induce the expression of synaptophysin. | Exp. Ther. Med. 2017 Nov; 14(5): 4312–4320; Transl. Neurodegener. 2020 May 27;9(1):20; Hippocampus. 2017 Dec;27(12):1250–1263 |
| Autophagy | Neurons, Microglia, Astrocytes, Oligodendrocytes | To increase cellular viability and LC3-II expression; to upregulate BECN1/Beclin 1 expression; to enhance mitophagy. | Autophagy. 2014 Jan;10(1):32–44; Mol. Neurobiol. 2019 Dec;56(12):8220–8236; Autophagy. 2021 Jan 19;1–20. |
| Apoptosis | Neurons, Microglia, Astrocytes, Oligodendrocytes | To regulate expression of hippocampal SIRT1, PCNA, p53, ac-p53, p21, and p16; to target caspase pathway; Ca2+ signaling. | Behav. Brain Res. 2018 Feb 26;339:297–304; Front. Neurosci. 2018 May 22;12:333; Curr. Alzheimer Res. 2010 Sep;7(6):540–8; Sci. Rep. 2016 Aug 12;6:31450. |
| Angiogenesis | Neurons, Microglia, Astrocytes, Oligodendrocytes | BMSCs secrete VEGF, BDNF, NT-3, IGF-1, bFGF, GDNF and TGF. VEGF is the most important mitogen in the process of angiogenesis. | Brain Res. 2011 Jan 7; 1367:103–113; Int. J. Mol. Med. 2013 May;31(5):1087–96; Neuroreport. 2015 May 6;26(7):399–404. |
| Synaptogenesis | Neurons, Microglia, Astrocytes, Oligodendrocytes | To stimulate the production of BDNF and NGF for remyelination; peptide FG loop (FGL) amplifies remyelination and modulates neuroinflammation. | Cell Biol. Int. 2021 Feb;45(2):432–446; J. Neuroimmune. Pharmacol. 2016 Dec;11(4):708–720; Front. Cell Dev. Biol. 2021 Jul 2;9:680301. |
| Names | Function | References |
|---|---|---|
| TREM2 | Transmembrane glycoprotein. To mediate immune and inflammatory responses as microglial receptor. | Neurobiol. Dis. 2020 Nov;145:105072; Neurobiol. Dis. 2019 Jul;127:432–448. |
| CR1 | To regulate complement cascade and mediate immune adherence as well as phagocytosis. | Stem Cell Res. 2016 Nov;17(3):560–563. |
| HLA-DRB5 | To encode major histocompatibility complex class II protein involved in immune responses. | Neurol. Genet. 2018 Jan 18;4(1):e211; JAMA Neurol. 2015 Jan;72(1):15–24. |
| CD33 | Microglial receptor converged on immune-inflammatory pathways. | Neurobiol. Dis. 2019 Jul;127:432–448; Gerontology. 2019;65(4):323–331 |
| MS4A | Belonging to a class of four-transmembrane spanning proteins. | Aging Cell. 2019 Aug;18(4):e12964. |
| INPP5D | At the plasma membrane, the protein hydrolyzes the 5′ phosphate and regulates multiple signaling pathways. | EMBO Mol. Med. 2020 Mar 6;12(3):e10606. |
| EPHA1 | To regulate the developmental of nervous system. | Int. J. Comput. Biol. Drug Des. 2020;13(1):58–70; J. Immunol. 2020 Sep 1;205(5):1318–1322. |
| CLU | Diverse functions such as protein chaperoning, apoptosis, complement activation, etc. | Mol Neurodegener. 2015 Jul 16;10:30; Turk J Med Sci. 2015;45(5):1082–6. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, C.; Li, Y.; Wang, K. Novel Balance Mechanism Participates in Stem Cell Therapy to Alleviate Neuropathology and Cognitive Impairment in Animal Models with Alzheimer’s Disease. Cells 2021, 10, 2757. https://doi.org/10.3390/cells10102757
Qin C, Li Y, Wang K. Novel Balance Mechanism Participates in Stem Cell Therapy to Alleviate Neuropathology and Cognitive Impairment in Animal Models with Alzheimer’s Disease. Cells. 2021; 10(10):2757. https://doi.org/10.3390/cells10102757
Chicago/Turabian StyleQin, Chuan, Yongning Li, and Kewei Wang. 2021. "Novel Balance Mechanism Participates in Stem Cell Therapy to Alleviate Neuropathology and Cognitive Impairment in Animal Models with Alzheimer’s Disease" Cells 10, no. 10: 2757. https://doi.org/10.3390/cells10102757