
Plectin in Skin Fragility Disorders

Abstract
:1. Introduction
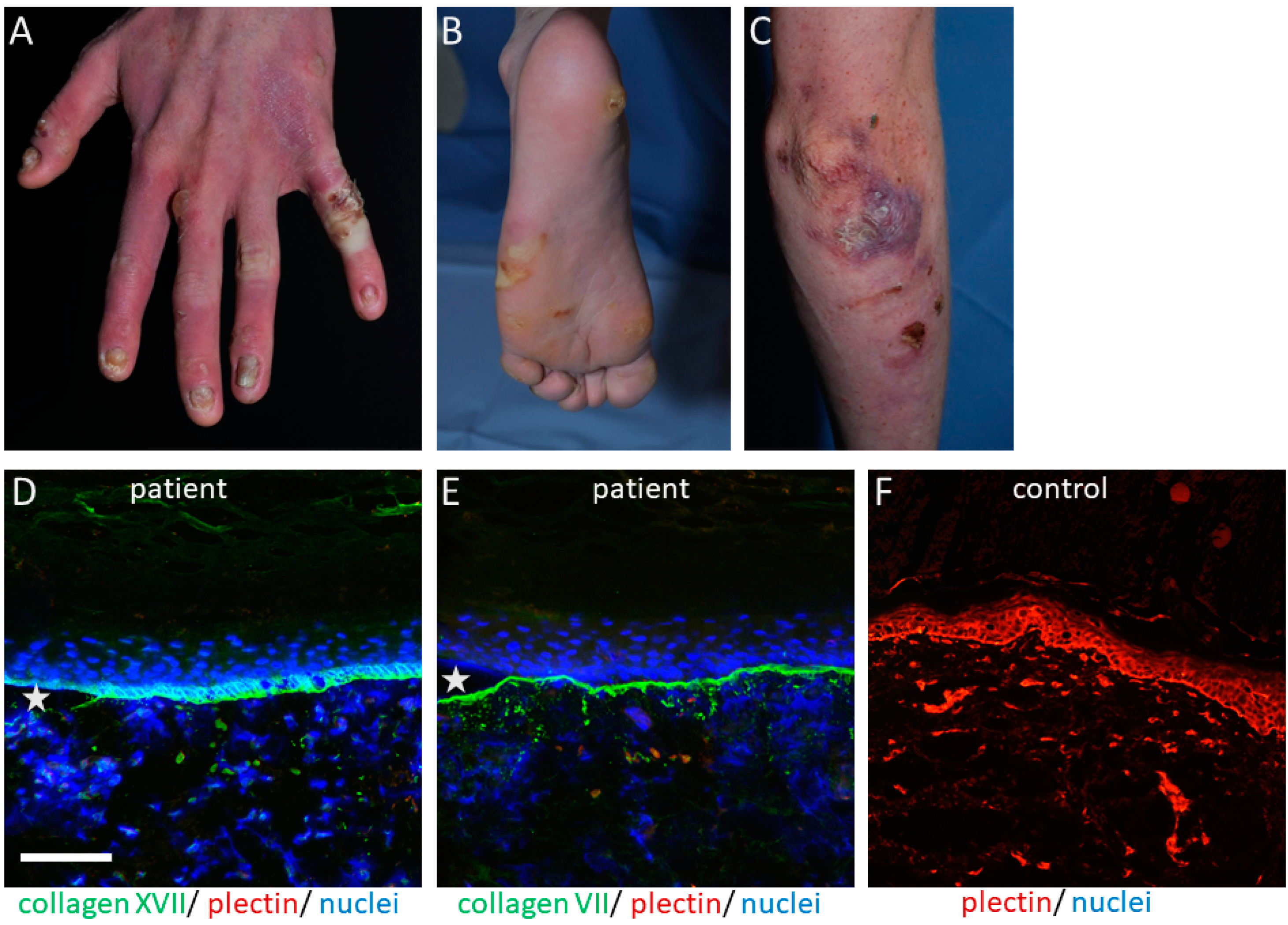
2. Plectin in Hereditary Skin Fragility Disorders

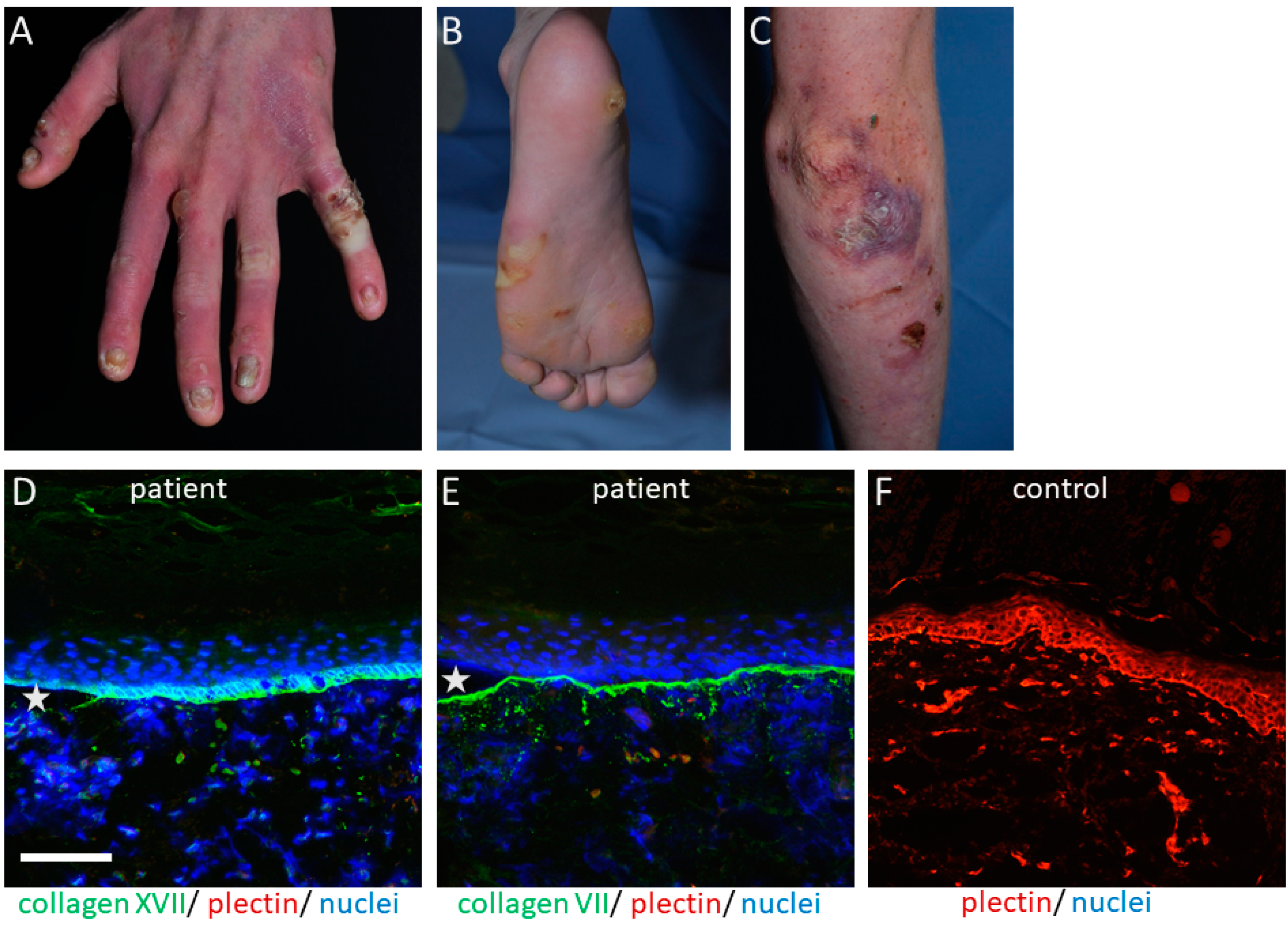{kind=link}
| Subtype | Plectin Isotype Affected | Mode of Inheritance | Organs Affected | Mouse Model |
|---|---|---|---|---|
| EBS-PA | 1 | AR | Skin, mucosa, pylorus, urinary tract (muscle) | [45], die 2–3 days after birth |
| EBS-MD | 1 | AR | Skin, mucosa, muscle, enamel, laryngotracheal and gastrointestinal | [45], died 2–3 days after birth |
| Limb girdle muscular dystrophy type 2Q | 1f | AR | Muscle | [46] |
| EBS-MD with myasthenic symptoms | 1 | AR | Muscle | NA |
| EBS Ogna | rod domain | AD | Skin | [45] |
| EBS-plectin 1a | 1a | AR | Skin | [17] |
3. Plectin in Autoimmune Skin Fragility Disorders
4. The Role of Plectin in Fibroblast Organization
5. Treatment Option for Patients with EBS-MD
6. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Castañón, M.J.; Walko, G.; Winter, L.; Wiche, G. Plectin-Intermediate Filament Partnership in Skin, Skeletal Muscle, and Peripheral Nerve. Histochem. Cell Biol. 2013, 140, 33–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiche, G.; Becker, B.; Luber, K.; Weitzer, G.; Castañon, M.J.; Hauptmann, R.; Stratowa, C.; Stewart, M. Cloning and Sequencing of Rat Plectin Indicates a 466-KD Polypeptide Chain with a Three-Domain Structure Based on a Central Alpha-Helical Coiled Coil. J. Cell Biol. 1991, 114, 83–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsuga, K. Plectin-Related Skin Diseases. J. Dermatol Sci 2015, 77, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Winter, L. Plectin Isoforms as Organizers of Intermediate Filament Cytoarchitecture. Bioarchitecture 2011, 1, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Geerts, D.; Fontao, L.; Nievers, M.G.; Schaapveld, R.Q.; Purkis, P.E.; Wheeler, G.N.; Lane, E.B.; Leigh, I.M.; Sonnenberg, A. Binding of Integrin Alpha6beta4 to Plectin Prevents Plectin Association with F-Actin but Does Not Interfere with Intermediate Filament Binding. J. Cell Biol. 1999, 147, 417–434. [Google Scholar] [CrossRef] [Green Version]
- Koster, J.; van Wilpe, S.; Kuikman, I.; Litjens, S.H.M.; Sonnenberg, A. Role of Binding of Plectin to the Integrin Beta4 Subunit in the Assembly of Hemidesmosomes. Mol. Biol. Cell 2004, 15, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Geerts, D.; Favre, B.; Borradori, L.; Sonnenberg, A. Analysis of the Interactions between BP180, BP230, Plectin and the Integrin Alpha6beta4 Important for Hemidesmosome Assembly. J. Cell Sci. 2003, 116, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouameur, J.-E.; Favre, B.; Fontao, L.; Lingasamy, P.; Begré, N.; Borradori, L. Interaction of Plectin with Keratins 5 and 14: Dependence on Several Plectin Domains and Keratin Quaternary Structure. J. Investig. Dermatol. 2014, 134, 2776–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijikata, T.; Murakami, T.; Ishikawa, H.; Yorifuji, H. Plectin Tethers Desmin Intermediate Filaments onto Subsarcolemmal Dense Plaques Containing Dystrophin and Vinculin. Histochem. Cell Biol. 2003, 119, 109–123. [Google Scholar] [CrossRef]
- Te Molder, L.; Hoekman, L.; Kreft, M.; Bleijerveld, O.; Sonnenberg, A. Comparative Interactomics Analysis Reveals Potential Regulators of A6β4 Distribution in Keratinocytes. Biol. Open 2020, 9. [Google Scholar] [CrossRef]
- Winter, L.; Wiche, G. The Many Faces of Plectin and Plectinopathies: Pathology and Mechanisms. Acta Neuropathol. 2013, 125, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Borradori, L.; Sonnenberg, A. Structure and Function of Hemidesmosomes: More than Simple Adhesion Complexes. J. Investig. Dermatol. 1999, 112, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin Isoform 1b Mediates Mitochondrion-Intermediate Filament Network Linkage and Controls Organelle Shape. J. Cell Biol. 2008, 181, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, R.; Kunz, W.S.; Rouan, F.; Pfendner, E.; Tolksdorf, K.; Kappes-Horn, K.; Altenschmidt-Mehring, M.; Knoblich, R.; van der Ven, P.F.M.; Reimann, J.; et al. Disorganization of the Desmin Cytoskeleton and Mitochondrial Dysfunction in Plectin-Related Epidermolysis Bullosa Simplex with Muscular Dystrophy. J. Neuropathol. Exp. Neurol. 2002, 61, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolling, M.C.; Jongbloed, J.D.H.; Boven, L.G.; Diercks, G.F.H.; Smith, F.J.D.; Irwin McLean, W.H.; Jonkman, M.F. Plectin Mutations Underlie Epidermolysis Bullosa Simplex in 8% of Patients. J. Investig. Dermatol. 2014, 134, 273–276. [Google Scholar] [CrossRef] [Green Version]
- Koss-Harnes, D.; Hoyheim, B.; Anton-Lamprecht, I.; Gjesti, A.; Jorgensen, R.S.; Jahnsen, F.L.; Olaisen, B.; Wiche, G.; Gedde-Dahl, T., Jr. A Site-Specific Plectin Mutation Causes Dominant Epidermolysis Bullosa Simplex Ogna: Two Identical de Novo Mutations. J. Investig. Dermatol. 2002, 118, 87–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gostyńska, K.B.; Nijenhuis, M.; Lemmink, H.; Pas, H.H.; Pasmooij, A.M.G.; Lang, K.K.; Castañón, M.J.; Wiche, G.; Jonkman, M.F. Mutation in Exon 1a of PLEC, Leading to Disruption of Plectin Isoform 1a, Causes Autosomal-Recessive Skin-Only Epidermolysis Bullosa Simplex. Hum. Mol. Genet. 2015, 24, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- McMillan, J.R.; Akiyama, M.; Rouan, F.; Mellerio, J.E.; Lane, E.B.; Leigh, I.M.; Owaribe, K.; Wiche, G.; Fujii, N.; Uitto, J.; et al. Plectin Defects in Epidermolysis Bullosa Simplex with Muscular Dystrophy. Muscle Nerve 2007, 35, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, P.; et al. Consensus Re-Classification of Inherited Epidermolysis Bullosa and Other Disorders with Skin Fragility. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamura, D.; Goto, M.; Sakai, K.; Nakamura, H.; McMillan, J.R.; Akiyama, M.; Shirado, O.; Oyama, N.; Satoh, M.; Kaneko, F.; et al. Possible Involvement of Exon 31 Alternative Splicing in Phenotype and Severity of Epidermolysis Bullosa Caused by Mutations in PLEC1. J. Investig. Dermatol. 2007, 127, 1537–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.J.; Uitto, J. Epidermolysis Bullosa with Pyloric Atresia. Dermatol. Clin. 2010, 28, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, A.; Chiaverini, C.; Chevrant-Breton, J.; DelRio, M.; Diociaiuti, A.; Dupuis, R.P.; El Hachem, M.; Le Fiblec, B.; Sankari-Ho, A.M.; Valhquist, A.; et al. Epidermolysis Bullosa Simplex with PLEC Mutations: New Phenotypes and New Mutations. Br. J. Dermatol. 2013, 168, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Ketema, M.; Secades, P.; Kreft, M.; Nahidiazar, L.; Janssen, H.; Jalink, K.; de Pereda, J.M.; Sonnenberg, A. The Rod Domain Is Not Essential for the Function of Plectin in Maintaining Tissue Integrity. Mol. Biol. Cell 2015, 26, 2402–2417. [Google Scholar] [CrossRef] [Green Version]
- Lateo, S.; Pace, J.; Aquilina, J.; Debono, A.G.; Bologna, F.A. Plectin Deficiency Disease. A Case Report. Adv. Exp. Med. Biol. 1999, 455, 551–555. [Google Scholar] [PubMed]
- Pfendner, E.; Rouan, F.; Uitto, J. Progress in Epidermolysis Bullosa: The Phenotypic Spectrum of Plectin Mutations. Exp. Dermatol. 2005, 14, 241–249. [Google Scholar] [CrossRef]
- Kyrova, J.; Kopeckova, L.; Buckova, H.; Mrazova, L.; Vesely, K.; Hermanova, M.; Oslejskova, H.; Fajkusova, L. Epidermolysis Bullosa Simplex with Muscular Dystrophy. Review of the Literature and a Case Report. J. Dermatol. Case Rep. 2016, 10, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Argyropoulou, Z.; Liu, L.; Ozoemena, L.; Branco, C.C.; Senra, R.; Reis-Rego, Â.; Mota-Vieira, L. A Novel PLEC Nonsense Homozygous Mutation (c.7159G > T; p.Glu2387*) Causes Epidermolysis Bullosa Simplex with Muscular Dystrophy and Diffuse Alopecia: A Case Report. BMC Dermatol. 2018, 18, 1. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Ren, Y.; Lin, Z.; Wang, H.; Zhou, Y.; Yang, Y. Compound Heterozygous PLEC Mutations in a Patient of Consanguineous Parentage with Epidermolysis Bullosa Simplex with Muscular Dystrophy and Diffuse Alopecia. Int. J. Dermatol. 2015, 54, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Yiu, E.M.; Klausegger, A.; Waddell, L.B.; Grasern, N.; Lloyd, L.; Tran, K.; North, K.N.; Bauer, J.W.; McKelvie, P.; Chow, C.W.; et al. Epidermolysis Bullosa with Late-Onset Muscular Dystrophy and Plectin Deficiency. Muscle Nerve 2011, 44, 135–141. [Google Scholar] [CrossRef]
- Gundesli, H.; Talim, B.; Korkusuz, P.; Balci-Hayta, B.; Cirak, S.; Akarsu, N.A.; Topaloglu, H.; Dincer, P. Mutation in Exon 1f of PLEC, Leading to Disruption of Plectin Isoform 1f, Causes Autosomal-Recessive Limb-Girdle Muscular Dystrophy. Am. J. Hum. Genet. 2010, 87, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Türk, M.; Harter, P.N.; Mittelbronn, M.; Kornblum, C.; Norwood, F.; Jungbluth, H.; Thiel, C.T.; Schlötzer-Schrehardt, U.; Schröder, R. Downstream Effects of Plectin Mutations in Epidermolysis Bullosa Simplex with Muscular Dystrophy. Acta Neuropathol. Commun. 2016, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Staszewska, I.; Mihailovska, E.; Fischer, I.; Goldmann, W.H.; Schroder, R.; Wiche, G. Chemical Chaperone Ameliorates Pathological Protein Aggregation in Plectin-Deficient Muscle. J. Clin. Investig. 2014, 124, 1144–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolling, M.C.; Pas, H.H.; de Visser, M.; Aronica, E.; Pfendner, E.G.; van den Berg, M.P.; Diercks, G.F.; Suurmeijer, A.J.; Jonkman, M.F. PLEC1 Mutations Underlie Adult-Onset Dilated Cardiomyopathy in Epidermolysis Bullosa Simplex with Muscular Dystrophy. J. Investig. Dermatol. 2010, 130, 1178–1181. [Google Scholar] [CrossRef] [Green Version]
- Villa, C.R.; Ryan, T.D.; Collins, J.J.; Taylor, M.D.; Lucky, A.W.; Jefferies, J.L. Left Ventricular Non-Compaction Cardiomyopathy Associated with Epidermolysis Bullosa Simplex with Muscular Dystrophy and PLEC1 Mutation. Neuromuscul. Disord. 2015, 25, 165–168. [Google Scholar] [CrossRef]
- Thorolfsdottir, R.B.; Sveinbjornsson, G.; Sulem, P.; Helgadottir, A.; Gretarsdottir, S.; Benonisdottir, S.; Magnusdottir, A.; Davidsson, O.B.; Rajamani, S.; Roden, D.M.; et al. A Missense Variant in PLEC Increases Risk of Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Hoorntje, E.T.; Posafalvi, A.; Syrris, P.; van der Velde, K.J.; Bolling, M.C.; Protonotarios, A.; Boven, L.G.; Amat-Codina, N.; Groeneweg, J.A.; Wilde, A.A.; et al. No Major Role for Rare Plectin Variants in Arrhythmogenic Right Ventricular Cardiomyopathy. PLoS ONE 2018, 13, e0203078. [Google Scholar] [CrossRef]
- Banwell, B.L.; Russel, J.; Fukudome, T.; Shen, X.M.; Stilling, G.; Engel, A.G. Myopathy, Myasthenic Syndrome, and Epidermolysis Bullosa Simplex Due to Plectin Deficiency. J. Neuropathol. Exp. Neurol. 1999, 58, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Garcia, A.; Tutmaher, M.S.; Upadhyayula, S.R.; Sanchez Russo, R.; Verma, S. Novel PLEC Gene Variants Causing Congenital Myasthenic Syndrome. Muscle Nerve 2019, 60, E40–E43. [Google Scholar] [CrossRef]
- Selcen, D.; Juel, V.C.; Hobson-Webb, L.D.; Smith, E.C.; Stickler, D.E.; Bite, A.V.; Ohno, K.; Engel, A.G. Myasthenic Syndrome Caused by Plectinopathy. Neurology 2011, 76, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrest, K.; Mellerio, J.E.; Robb, S.; Dopping-Hepenstal, P.J.; McGrath, J.A.; Liu, L.; Buk, S.J.; Al-Sarraj, S.; Wraige, E.; Jungbluth, H. Congenital Muscular Dystrophy, Myasthenic Symptoms and Epidermolysis Bullosa Simplex (EBS) Associated with Mutations in the PLEC1 Gene Encoding Plectin. Neuromuscul. Disord. 2010, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Vukasinovic, N.; Gross, K.; Fischer, I.; Sibitz, S.; Fuchs, P.; Reipert, S.; Jungwirth, U.; Berger, W.; Salzer, U.; et al. Targeted Proteolysis of Plectin Isoform 1a Accounts for Hemidesmosome Dysfunction in Mice Mimicking the Dominant Skin Blistering Disease EBS-Ogna. PLoS Genet. 2011, 7, e1002396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koss-Harnes, D.; Jahnsen, F.L.; Wiche, G.; Søyland, E.; Brandtzaeg, P.; Gedde-Dahl, T. Plectin Abnormality in Epidermolysis Bullosa Simplex Ogna: Non-Responsiveness of Basal Keratinocytes to Some Anti-Rat Plectin Antibodies. Exp. Dermatol. 1997, 6, 41–48. [Google Scholar] [CrossRef]
- Kiritsi, D.; Pigors, M.; Tantcheva-Poor, I.; Wessel, C.; Arin, M.J.; Kohlhase, J.; Bruckner-Tuderman, L.; Has, C. Epidermolysis Bullosa Simplex Ogna Revisited. J. Investig. Dermatol. 2013, 133, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamsberg, C.; Fuchs, P.; Osmanagic-Myers, S.; Fischer, I.; Propst, F.; Elbe-Bürger, A.; Wiche, G. Targeted Ablation of Plectin Isoform 1 Uncovers Role of Cytolinker Proteins in Leukocyte Recruitment. Proc. Natl. Acad. Sci. USA 2005, 102, 18449–18454. [Google Scholar] [CrossRef] [Green Version]
- Andrä, K.; Lassmann, H.; Bittner, R.; Shorny, S.; Fässler, R.; Propst, F.; Wiche, G. Targeted Inactivation of Plectin Reveals Essential Function in Maintaining the Integrity of Skin, Muscle, and Heart Cytoarchitecture. Genes Dev. 1997, 11, 3143–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konieczny, P.; Fuchs, P.; Reipert, S.; Kunz, W.S.; Zeold, A.; Fischer, I.; Paulin, D.; Schroder, R.; Wiche, G. Myofiber Integrity Depends on Desmin Network Targeting to Z-Disks and Costameres via Distinct Plectin Isoforms. J. Cell Bio. 2008, 181, 667–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goletz, S.; Zillikens, D.; Schmidt, E. Structural Proteins of the Dermal-Epidermal Junction Targeted by Autoantibodies in Pemphigoid Diseases. Exp. Dermatol. 2017, 26, 1154–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijsrogge, J.J.A.; de Jong, M.C.J.M.; Kloosterhuis, G.J.; Vermeer, M.H.; Koster, J.; Sonnenberg, A.; Jonkman, M.F.; Pas, H.H. Antiplectin Autoantibodies in Subepidermal Blistering Diseases. Br. J. Dermatol. 2009, 161, 762–771. [Google Scholar] [CrossRef]
- Schmidt, E.; Zillikens, D. Pemphigoid Diseases. Lancet 2013, 381, 320–332. [Google Scholar] [CrossRef]
- Anhalt, G.J.; Kim, S.C.; Stanley, J.R.; Korman, N.J.; Jabs, D.A.; Kory, M.; Izumi, H.; Ratrie, H.; Mutasim, D.; Ariss-Abdo, L. Paraneoplastic Pemphigus. An Autoimmune Mucocutaneous Disease Associated with Neoplasia. N. Engl. J. Med. 1990, 323, 1729–1735. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Ndoye, A.; Bassler, K.D.; Shultz, L.D.; Shields, M.C.; Ruben, B.S.; Webber, R.J.; Pittelkow, M.R.; Lynch, P.J.; Grando, S.A. Classification, Clinical Manifestations, and Immunopathological Mechanisms of the Epithelial Variant of Paraneoplastic Autoimmune Multiorgan Syndrome: A Reappraisal of Paraneoplastic Pemphigus. Arch. Dermatol. 2001, 137, 193–206. [Google Scholar]
- Paolino, G.; Didona, D.; Magliulo, G.; Iannella, G.; Didona, B.; Mercuri, S.R.; Moliterni, E.; Donati, M.; Ciofalo, A.; Granata, G.; et al. Paraneoplastic Pemphigus: Insight into the Autoimmune Pathogenesis, Clinical Features and Therapy. Int. J. Mol. Sci. 2017, 18, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didona, D.; DI Zenzo, G.; Joly, P. Paraneoplastic Autoimmune Multiorgan Syndrome. Ital. J. Dermatol. Venerol. 2021, 156, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, C.; Ruhrberg, C.; Nie, Z.; Karashima, T.; Mori, O.; Nishikawa, T.; Green, K.J.; Anhalt, G.J.; DiColandrea, T.; Watt, F.M.; et al. Envoplakin and Periplakin Are Components of the Paraneoplastic Pemphigus Antigen Complex. J. Invest. Dermatol. 1998, 111, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Joly, P.; Richard, C.; Gilbert, D.; Courville, P.; Chosidow, O.; Roujeau, J.C.; Beylot-Barry, M.; D’incan, M.; Martel, P.; Lauret, P.; et al. Sensitivity and Specificity of Clinical, Histologic, and Immunologic Features in the Diagnosis of Paraneoplastic Pemphigus. J. Am. Acad. Dermatol. 2000, 43, 619–626. [Google Scholar] [CrossRef]
- Numata, S.; Teye, K.; Tsuruta, D.; Sogame, R.; Ishii, N.; Koga, H.; Natsuaki, Y.; Tsuchisaka, A.; Hamada, T.; Karashima, T.; et al. Anti-α-2-Macroglobulin-like-1 Autoantibodies Are Detected Frequently and May Be Pathogenic in Paraneoplastic Pemphigus. J. Investig. Dermatol. 2013, 133, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohzono, A.; Sogame, R.; Li, X.; Teye, K.; Tsuchisaka, A.; Numata, S.; Koga, H.; Kawakami, T.; Tsuruta, D.; Ishii, N.; et al. Clinical and Immunological Findings in 104 Cases of Paraneoplastic Pemphigus. Br. J. Dermatol. 2015, 173, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the Vimentin Network under Control: Cell-Matrix Adhesion-Associated Plectin 1f Affects Cell Shape and Polarity of Fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, S.; Chowdhury, F.; Tay, B.; Ouyang, M.; Gregor, M.; Wang, Y.; Wiche, G.; Wang, N. Plectin Contributes to Mechanical Properties of Living Cells. Am. J. Physiol. Cell Physiol. 2009, 296, C868–C877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurny, R.; Abdoulrahman, K.; Janda, L.; Rünzler, D.; Köhler, G.; Castañón, M.J.; Wiche, G. Oxidation and Nitrosylation of Cysteines Proximal to the Intermediate Filament (IF)-Binding Site of Plectin: Effects on Structure and Vimentin Binding and Involvement in IF Collapse. J. Biol. Chem. 2007, 282, 8175–8187. [Google Scholar] [CrossRef] [Green Version]
- Spörrer, M.; Prochnicki, A.; Tölle, R.C.; Nyström, A.; Esser, P.R.; Homberg, M.; Athanasiou, I.; Zingkou, E.; Schilling, A.; Gerum, R.; et al. Treatment of Keratinocytes with 4-Phenylbutyrate in Epidermolysis Bullosa: Lessons for Therapies in Keratin Disorders. EBioMedicine 2019, 44, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wally, V.; Klausegger, A.; Koller, U.; Lochmüller, H.; Krause, S.; Wiche, G.; Mitchell, L.G.; Hintner, H.; Bauer, J.W. 5′ Trans-Splicing Repair of the PLEC1 Gene. J. Investig. Dermatol. 2008, 128, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiritsi, D.; Tsakiris, L.; Schauer, F. Plectin in Skin Fragility Disorders. Cells 2021, 10, 2738. https://doi.org/10.3390/cells10102738
Kiritsi D, Tsakiris L, Schauer F. Plectin in Skin Fragility Disorders. Cells. 2021; 10(10):2738. https://doi.org/10.3390/cells10102738
Chicago/Turabian StyleKiritsi, Dimitra, Leonidas Tsakiris, and Franziska Schauer. 2021. "Plectin in Skin Fragility Disorders" Cells 10, no. 10: 2738. https://doi.org/10.3390/cells10102738
APA StyleKiritsi, D., Tsakiris, L., & Schauer, F. (2021). Plectin in Skin Fragility Disorders. Cells, 10(10), 2738. https://doi.org/10.3390/cells10102738