
A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Materials
2.3. Antibodies
2.4. Mitochondrial Isolation
2.5. Mitochondrial Matrix Swelling by Calcium Overload
2.6. Release of Cytochrome c from Mitochondrial Suspensions
2.7. Mitochondrial Respiration Assay
2.8. SDS–PAGE and Western Blotting
2.9. Immunoelectron Microscopy (EM)
2.10. Statistical Analysis
3. Results
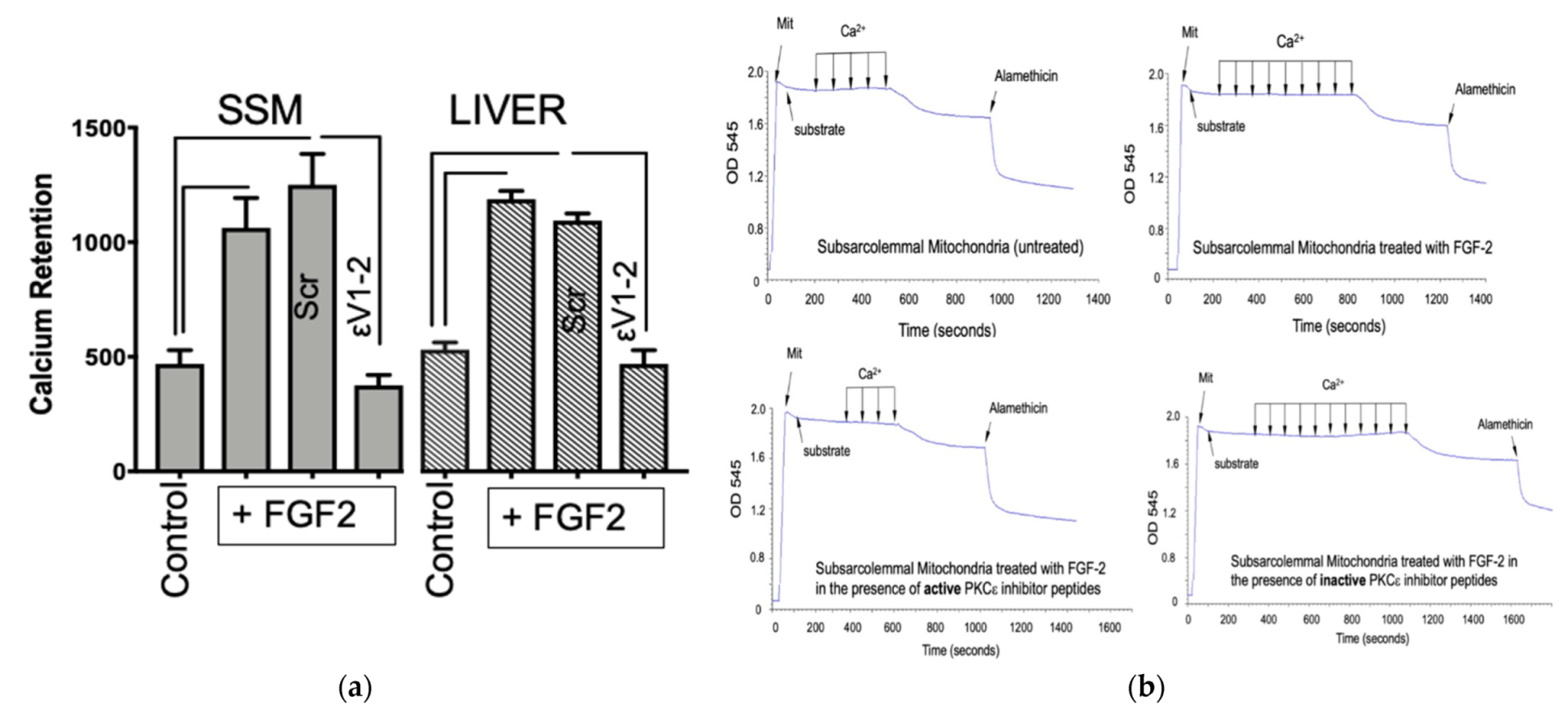
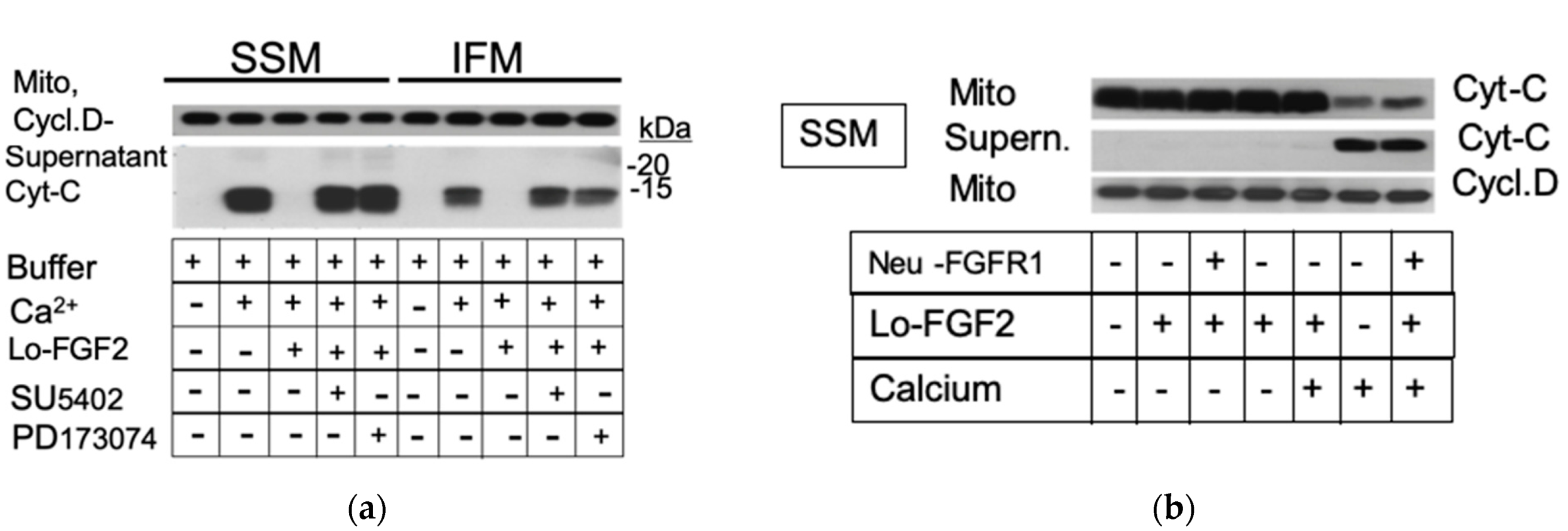
3.1. The Effect of Lo-FGF2 on Mitochondrial Resistance to Calcium-Overload-Induced Formation of mPTP—The Role of Mitochondrial Protein Kinase C (PKC) ε
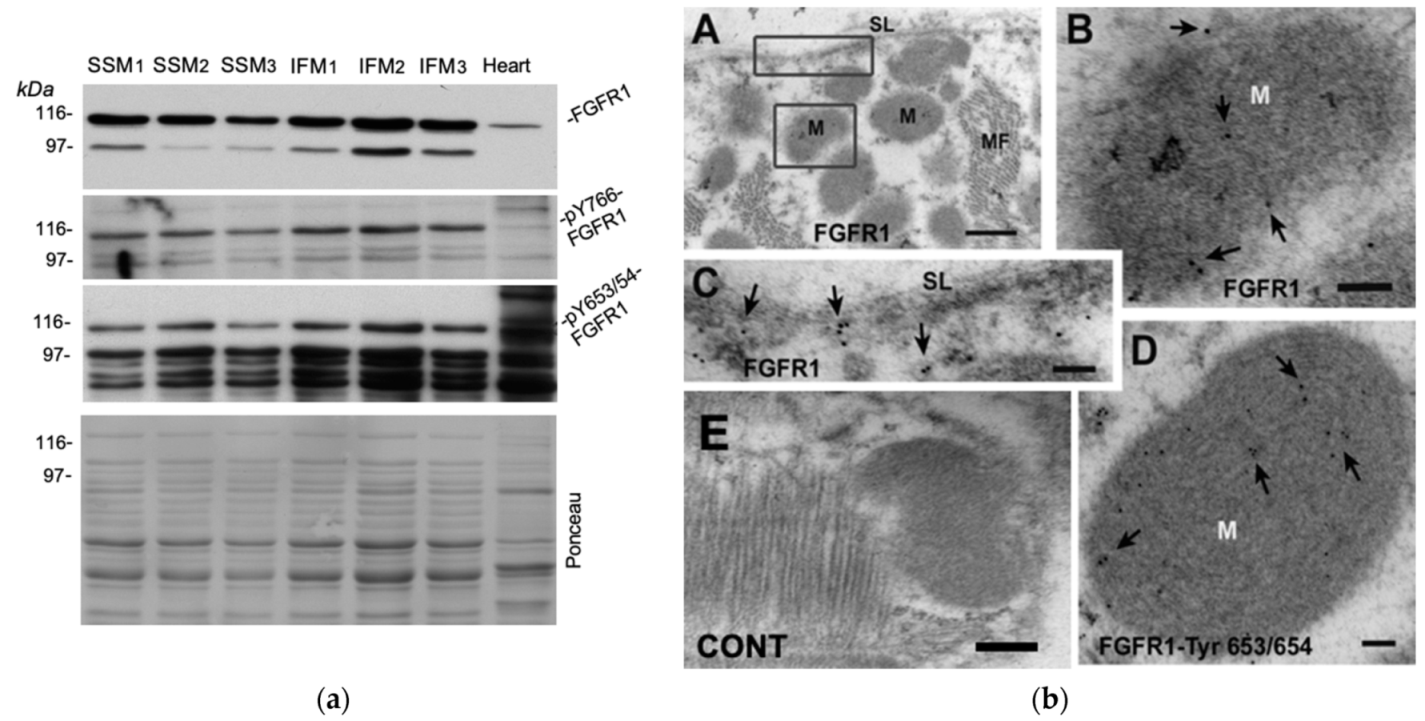
3.2. Probing for a Mitochondrial FGF2 Receptor, Mito-FGFR1
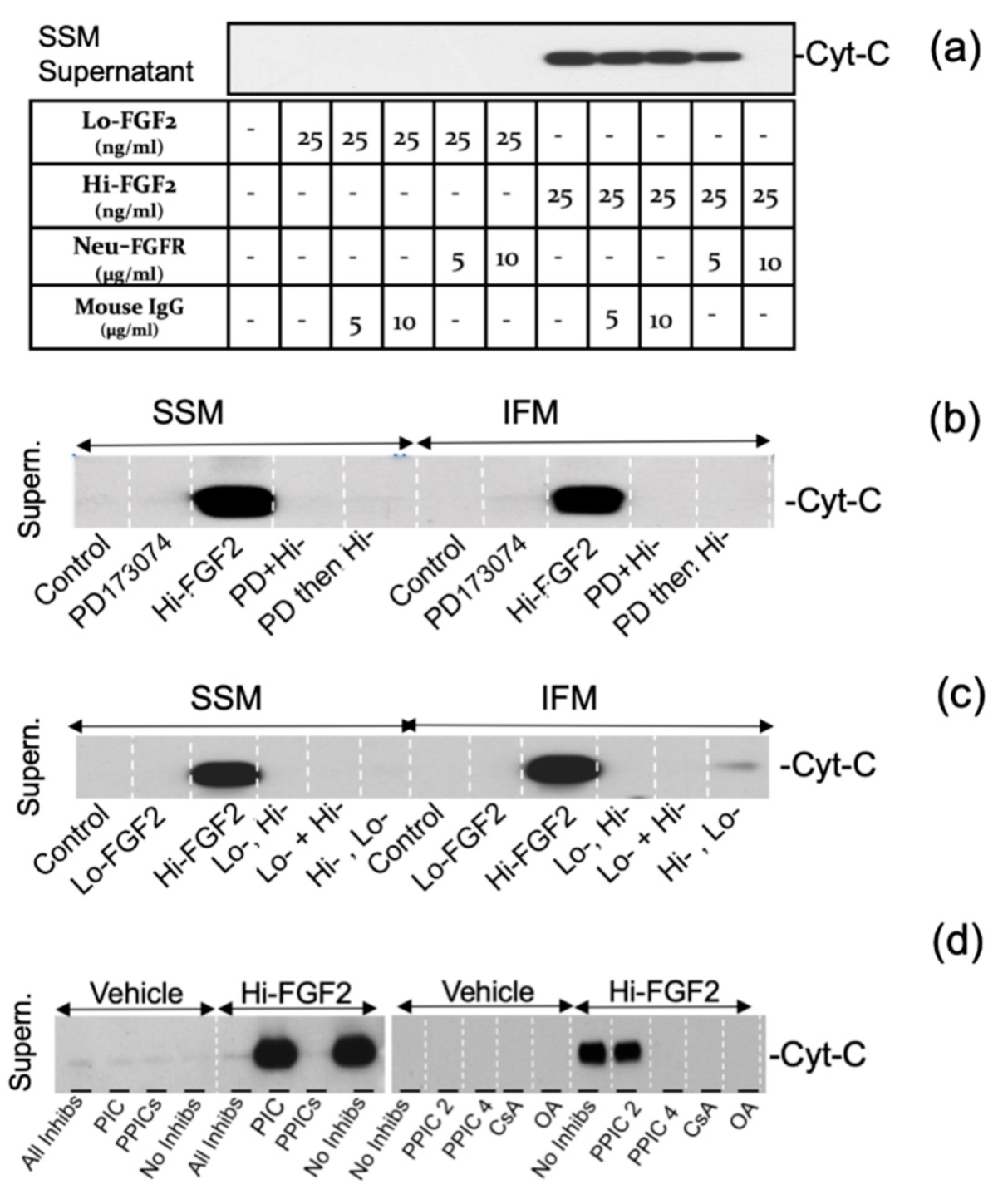
3.3. The Effects of Hi-FGF2 on Cardiac Mitochondria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef]
- Song, R.; Zhang, L. Cardiac ECM: Its Epigenetic Regulation and Role in Heart Development and Repair. Int. J. Mol. Sci. 2020, 21, 8610. [Google Scholar] [CrossRef]
- Kardami, E.; Detillieux, K.; Ma, X.; Jiang, Z.; Santiago, J.J.; Jimenez, S.K.; Cattini, P.A. Fibroblast growth factor-2 and cardioprotection. Heart Fail. Rev. 2007, 12, 267–277. [Google Scholar] [CrossRef]
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of Fgf signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.J.; Ma, X.; McNaughton, L.J.; Nickel, B.E.; Bestvater, B.P.; Yu, L.; Fandrich, R.R.; Netticadan, T.; Kardami, E. Preferential accumulation and export of high molecular weight FGF-2 by rat cardiac non-myocytes. Cardiovasc. Res. 2011, 89, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C.; et al. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Pasumarthi, K.B.; Bock, M.E.; Lytras, A.; Kardami, E.; Cattini, P.A. Cloning and expression of fibroblast growth factor receptor-1 isoforms in the mouse heart: Evidence for isoform switching during heart development. J. Mol. Cell. Cardiol. 1994, 26, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Pasumarthi, K.B.; Padua, R.R.; Massaeli, H.; Fandrich, R.R.; Pierce, G.N.; Cattini, P.A.; Kardami, E. Adult cardiomyocytes express functional high-affinity receptors for basic fibroblast growth factor. Am. J. Physiol. 1995, 268, H1927–H1938. [Google Scholar] [CrossRef] [PubMed]
- Szybowska, P.; Kostas, M.; Wesche, J.; Haugsten, E.M.; Wiedlocha, A. Negative Regulation of FGFR (Fibroblast Growth Factor Receptor) Signaling. Cells 2021, 10, 1342. [Google Scholar] [CrossRef]
- Csanaky, K.; Hess, M.W.; Klimaschewski, L. Membrane-Associated, Not Cytoplasmic or Nuclear, FGFR1 Induces Neuronal Differentiation. Cells 2019, 8, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kole, D.; Grella, A.; Dolivo, D.; Shumaker, L.; Hermans, W.; Dominko, T. High molecular weight FGF2 isoforms demonstrate canonical receptor-mediated activity and support human embryonic stem cell self-renewal. Stem Cell Res. 2017, 21, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.S.; Jeyaraman, M.; Wen, G.B.; Fandrich, R.R.; Dixon, I.M.; Cattini, P.A.; Kardami, E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J. Mol. Cell. Cardiol. 2007, 42, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Bodmer, J.; Pietras, D.; Azhar, M.; Doetschman, T.; Schultz Jel, J. Biological functions of the low and high molecular weight protein isoforms of fibroblast growth factor-2 in cardiovascular development and disease. Dev. Dyn. 2009, 238, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Porter, D.; Scott, A.; Newman, G.; Doetschman, T.; Schultz Jel, J. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: The role of JNK signaling. J. Mol. Cell. Cardiol. 2007, 42, 106–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srisakuldee, W.; Makazan, Z.; Nickel, B.E.; Zhang, F.; Thliveris, J.A.; Pasumarthi, K.B.; Kardami, E. The FGF-2-triggered protection of cardiac subsarcolemmal mitochondria from calcium overload is mitochondrial connexin 43-dependent. Cardiovasc. Res. 2014, 103, 72–80. [Google Scholar] [CrossRef] [Green Version]
- Koleini, N.; Nickel, B.E.; Wang, J.; Roveimiab, Z.; Fandrich, R.R.; Kirshenbaum, L.A.; Cattini, P.A.; Kardami, E. Fibroblast growth factor-2-mediated protection of cardiomyocytes from the toxic effects of doxorubicin requires the mTOR/Nrf-2/HO-1 pathway. Oncotarget 2017, 8, 87415–87430. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Bodmer, J.R.; Azhar, M.; Newman, G.; Coffin, J.D.; Doetschman, T.; Schultz Jel, J. The influence of FGF2 high molecular weight (HMW) isoforms in the development of cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2010, 48, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Nusayr, E.; Sadideen, D.T.; Doetschman, T. FGF2 modulates cardiac remodeling in an isoform- and sex-specific manner. Physiol. Rep. 2013, 1, 1–14. [Google Scholar] [CrossRef]
- Koleini, N.; Nickel, B.E.; Nagalingam, R.S.; Landry, N.M.; Fandrich, R.R.; Cheung, D.Y.C.; Dixon, I.M.; Czubryt, M.P.; Jassal, D.S.; Cattini, P.A.; et al. Elimination of endogenous high molecular weight FGF2 prevents pressure-overload-induced systolic dysfunction, linked to increased FGFR1 activity and NR1D1 expression. Cell Tissue Res. 2021, 1–16. [Google Scholar] [CrossRef]
- Koleini, N.; Santiago, J.J.; Nickel, B.E.; Sequiera, G.L.; Wang, J.; Fandrich, R.R.; Jassal, D.S.; Dhingra, S.; Kirshenbaum, L.A.; Cattini, P.A.; et al. Elimination or neutralization of endogenous high-molecular-weight FGF2 mitigates doxorubicin-induced cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H279–H288. [Google Scholar] [CrossRef]
- Pasumarthi, K.B.; Kardami, E.; Cattini, P.A. High and low molecular weight fibroblast growth factor-2 increase proliferation of neonatal rat cardiac myocytes but have differential effects on binucleation and nuclear morphology. Evidence for both paracrine and intracrine actions of fibroblast growth factor-2. Circ. Res. 1996, 78, 126–136. [Google Scholar] [CrossRef]
- Hirst, C.J.; Herlyn, M.; Cattini, P.A.; Kardami, E. High levels of CUG-initiated FGF-2 expression cause chromatin compaction, decreased cardiomyocyte mitosis, and cell death. Mol. Cell Biochem. 2003, 246, 111–116. [Google Scholar] [CrossRef]
- Ma, X.; Dang, X.; Claus, P.; Hirst, C.; Fandrich, R.R.; Jin, Y.; Grothe, C.; Kirshenbaum, L.A.; Cattini, P.A.; Kardami, E. Chromatin compaction and cell death by high molecular weight FGF-2 depend on its nuclear localization, intracrine ERK activation, and engagement of mitochondria. J. Cell Physiol. 2007, 213, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc. Natl Acad Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Pasumarthi, K.B. Ultrastructural and immunocharacterization of undifferentiated myocardial cells in the developing mouse heart. J. Cell Mol. Med. 2007, 11, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Zhang, J.; Wang, O.L.; Guo, Y.; Bolli, R.; Cardwell, E.M.; Ping, P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Lacerda, L.; McCarthy, J.; Mungly, S.F.; Lynn, E.G.; Sack, M.N.; Opie, L.H.; Lecour, S. TNFalpha protects cardiac mitochondria independently of its cell surface receptors. Basic Res. Cardiol. 2010, 105, 751–762. [Google Scholar] [CrossRef] [Green Version]
- Hitosugi, T.; Fan, J.; Chung, T.W.; Lythgoe, K.; Wang, X.; Xie, J.; Ge, Q.; Gu, T.L.; Polakiewicz, R.D.; Roesel, J.L.; et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 2011, 44, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Srisakuldee, W. Studies on the Role of Connexin43 Phosphorylation in the Injury-Resistant Heart. Ph.D. Thesis, University of Manitoba, Winnipeg, MB, Canada, 2014. [Google Scholar]
- Marek, L.; Ware, K.E.; Fritzsche, A.; Hercule, P.; Helton, W.R.; Smith, J.E.; McDermott, L.A.; Coldren, C.D.; Nemenoff, R.A.; Merrick, D.T.; et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol. Pharmacol. 2009, 75, 196–207. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamont, F.R.; Tomlinson, D.C.; Cooper, P.A.; Shnyder, S.D.; Chester, J.D.; Knowles, M.A. Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br. J. Cancer 2011, 104, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Kee, W.J.; Facci, L.; Macdonald, G.; Doherty, P.; Walsh, F.S. The FGFR1 inhibitor PD 173074 selectively and potently antagonizes FGF-2 neurotrophic and neurotropic effects. J. Neurochem. 2000, 75, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Schuller, A.C.; Ahmed, Z.; Levitt, J.A.; Suen, K.M.; Suhling, K.; Ladbury, J.E. Indirect recruitment of the signalling adaptor Shc to the fibroblast growth factor receptor 2 (FGFR2). Biochem. J. 2008, 416, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paneni, F.; Mocharla, P.; Akhmedov, A.; Costantino, S.; Osto, E.; Volpe, M.; Luscher, T.F.; Cosentino, F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ. Res. 2012, 111, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.D.; Garlid, K.D. Intramitochondrial signaling: Interactions among mitoKATP, PKCepsilon, ROS, and MPT. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H874–H882. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, F.R.; Siemen, D.; Mawrin, C.; Horn, T.F.; Dietzmann, K. The neurotrophin receptor TrkB is colocalized to mitochondrial membranes. Int. J. Biochem. Cell Biol. 2006, 38, 610–620. [Google Scholar] [CrossRef]
- Carito, V.; Pingitore, A.; Cione, E.; Perrotta, I.; Mancuso, D.; Russo, A.; Genchi, G.; Caroleo, M.C. Localization of nerve growth factor (NGF) receptors in the mitochondrial compartment: Characterization and putative role. Biochim. Biophys. Acta 2012, 1820, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell Biol. 2004, 24, 7059–7071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef]
- Dunham-Ems, S.M.; Lee, Y.W.; Stachowiak, E.K.; Pudavar, H.; Claus, P.; Prasad, P.N.; Stachowiak, M.K. Fibroblast growth factor receptor-1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Mol. Biol. Cell 2009, 20, 2401–2412. [Google Scholar] [CrossRef] [Green Version]
- Maher, P.A. Identification and characterization of a novel, intracellular isoform of fibroblast growth factor receptor-1(FGFR-1). J. Cell Physiol. 1996, 169, 380–390. [Google Scholar] [CrossRef]
- Laederich, M.B.; Degnin, C.R.; Lunstrum, G.P.; Holden, P.; Horton, W.A. Fibroblast growth factor receptor 3 (FGFR3) is a strong heat shock protein 90 (Hsp90) client: Implications for therapeutic manipulation. J. Biol. Chem. 2011, 286, 19597–19604. [Google Scholar] [CrossRef] [Green Version]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem. 2009, 284, 36592–36604. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Smith, K.R.; Lim, S.T.; Tian, R.; Lu, J.; Tan, M. Regulation of mitochondrial functions by protein phosphorylation and dephosphorylation. Cell BioSci. 2016, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugi, S.; Imamura, T.; Ricketts, W.; Olefsky, J.M. Protein phosphatase 2A forms a molecular complex with Shc and regulates Shc tyrosine phosphorylation and downstream mitogenic signaling. Mol. Cell Biol. 2002, 22, 2375–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srisakuldee, W.; Nickel, B.E.; Fandrich, R.R.; Zhang, F.; Pasumarthi, K.B.S.; Kardami, E. A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition. Cells 2021, 10, 2735. https://doi.org/10.3390/cells10102735
Srisakuldee W, Nickel BE, Fandrich RR, Zhang F, Pasumarthi KBS, Kardami E. A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition. Cells. 2021; 10(10):2735. https://doi.org/10.3390/cells10102735
Chicago/Turabian StyleSrisakuldee, Wattamon, Barbara E. Nickel, Robert R. Fandrich, Feixong Zhang, Kishore B. S. Pasumarthi, and Elissavet Kardami. 2021. "A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition" Cells 10, no. 10: 2735. https://doi.org/10.3390/cells10102735
APA StyleSrisakuldee, W., Nickel, B. E., Fandrich, R. R., Zhang, F., Pasumarthi, K. B. S., & Kardami, E. (2021). A Cardiac Mitochondrial FGFR1 Mediates the Antithetical Effects of FGF2 Isoforms on Permeability Transition. Cells, 10(10), 2735. https://doi.org/10.3390/cells10102735