
The SZT2 Interactome Unravels New Functions of the KICSTOR Complex

 , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines, Cell Culture, and Treatment of Cells
2.2. Generation of HEK293 Flp-In™ T-Rex™ Cell Line Inducibly Expressing SH.SZT2WT
2.3. Affinity Purification and Sample Preparation for Mass Spectrometry
2.4. LC-MS/MS Analysis
2.5. Data Analysis
2.6. Generation of KO Cell Lines
2.7. Total Cell Lysates and Immunoblotting
2.8. Immunofluorescence and Microscopy
2.9. Cell Diameter Analysis
2.10. Statistical Analysis
3. Results
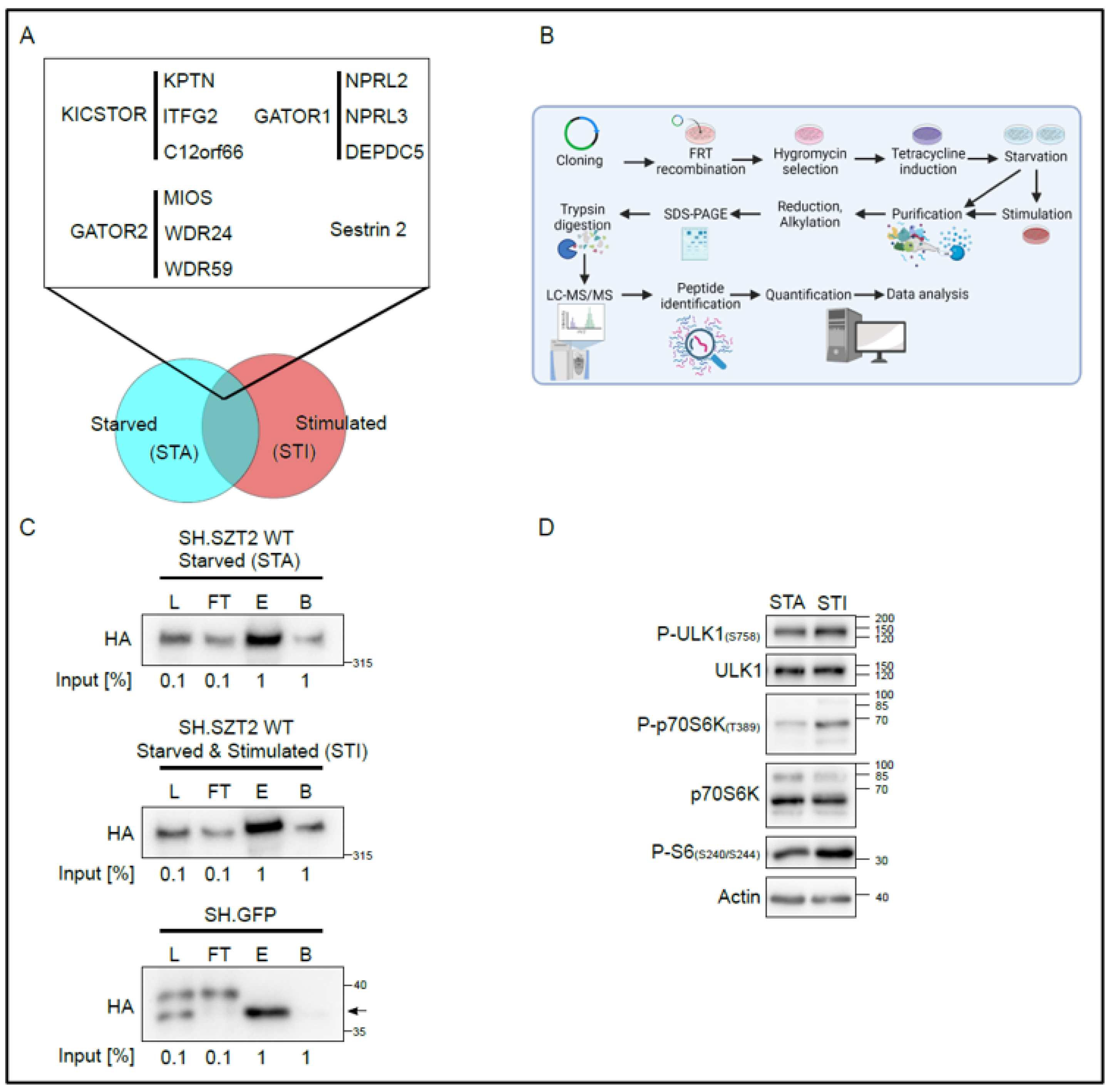
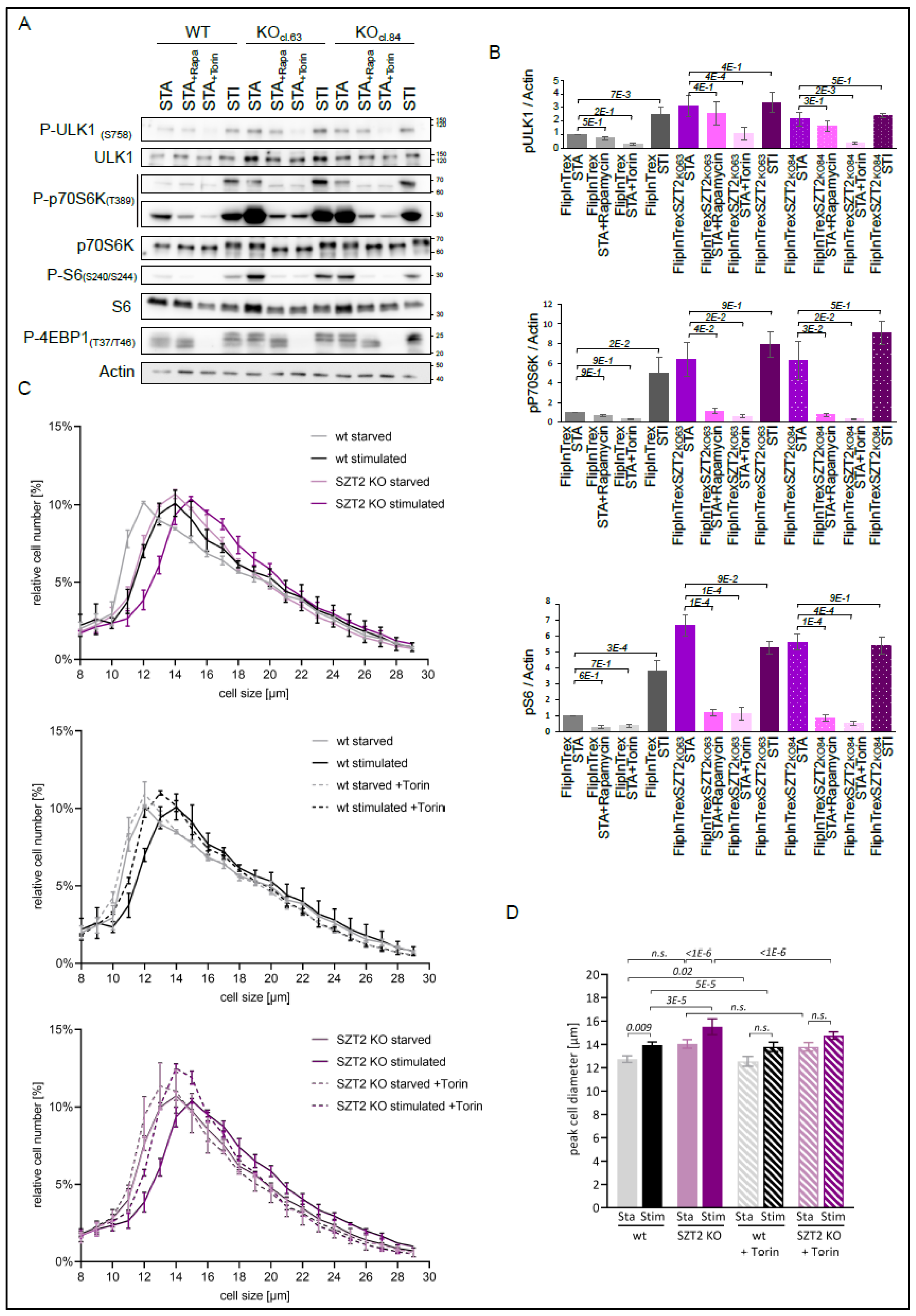
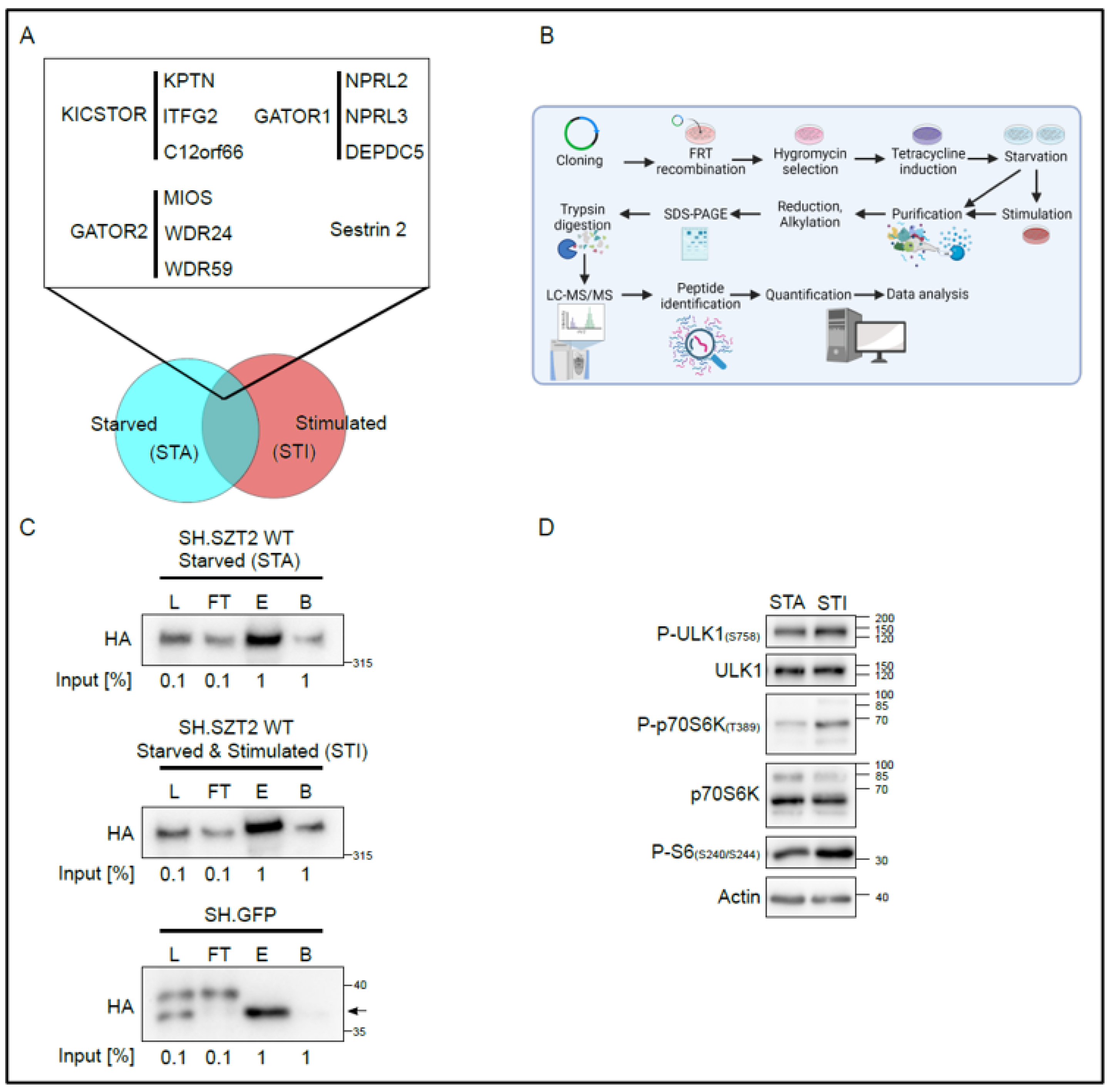
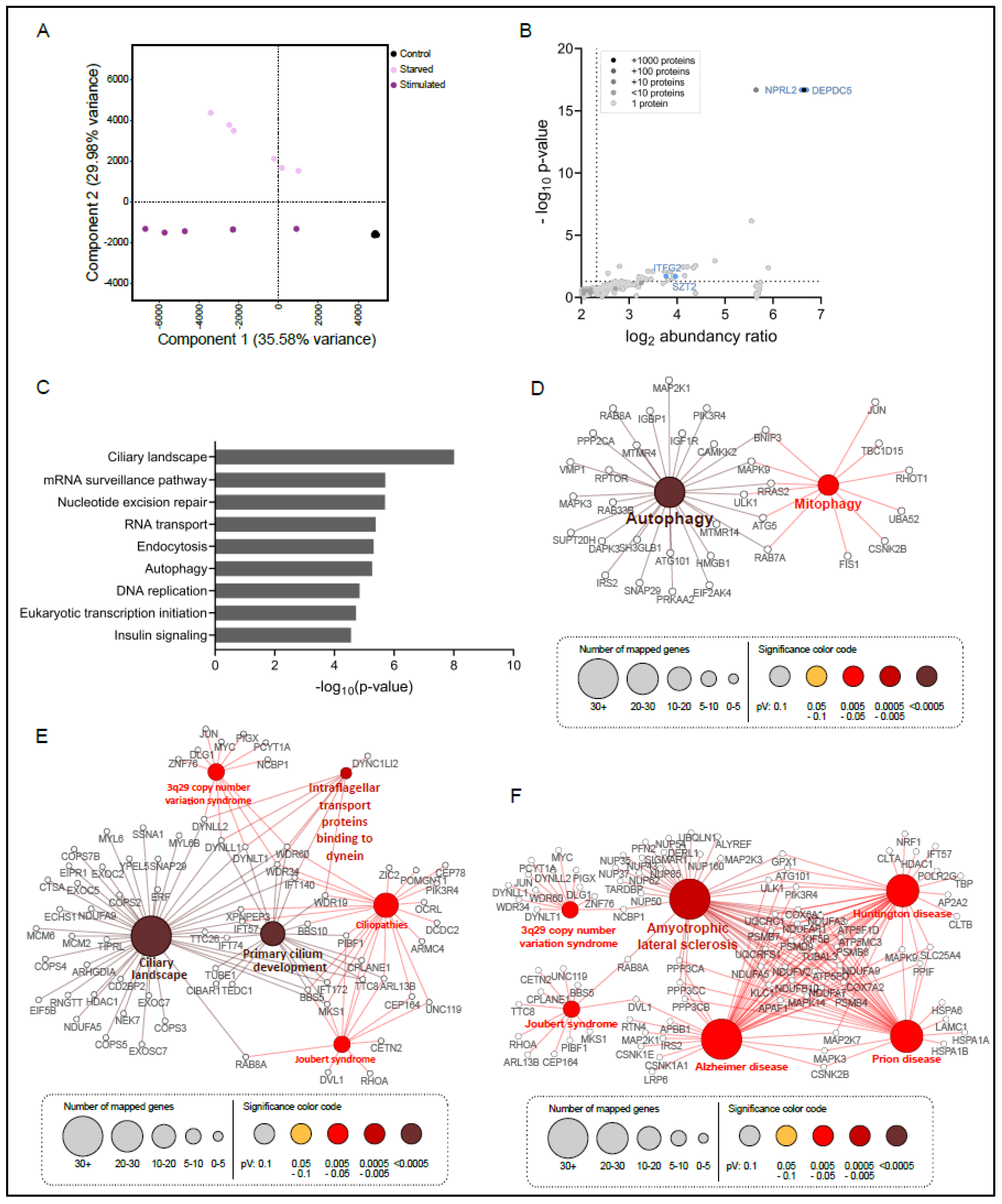
3.1. Establishment of the SZT2 Interactome
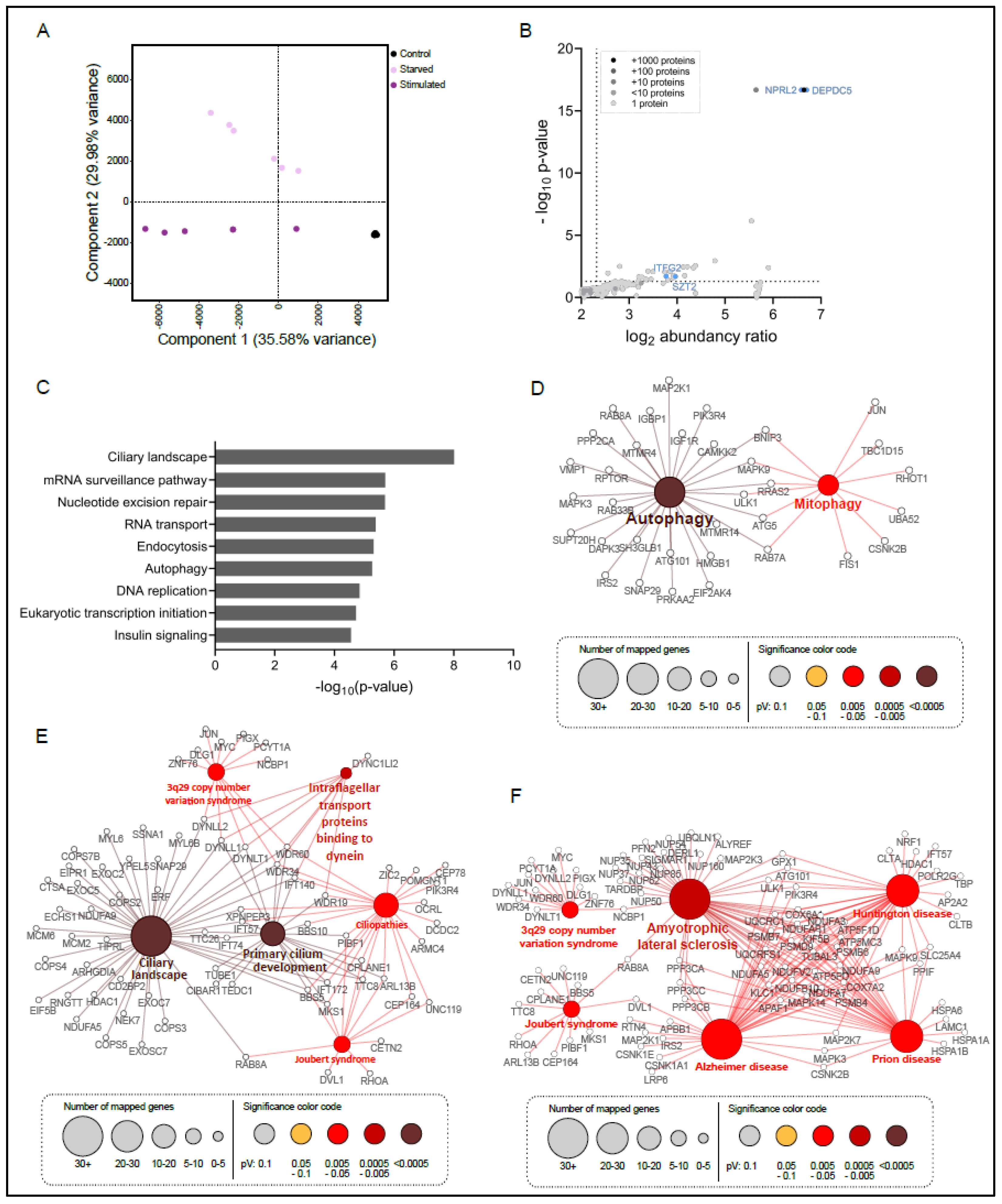
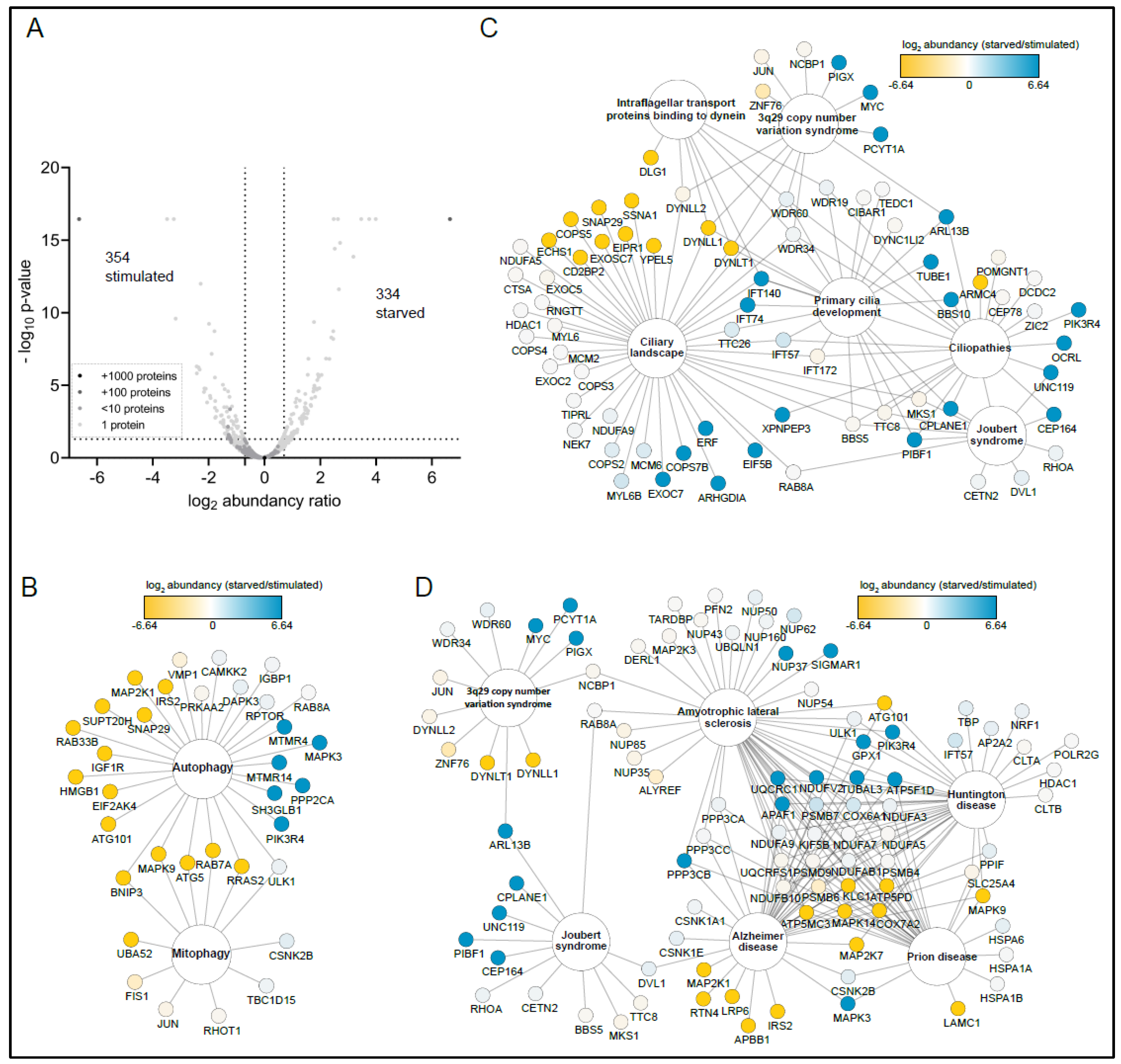
3.2. SZT2 Interactome Analysis
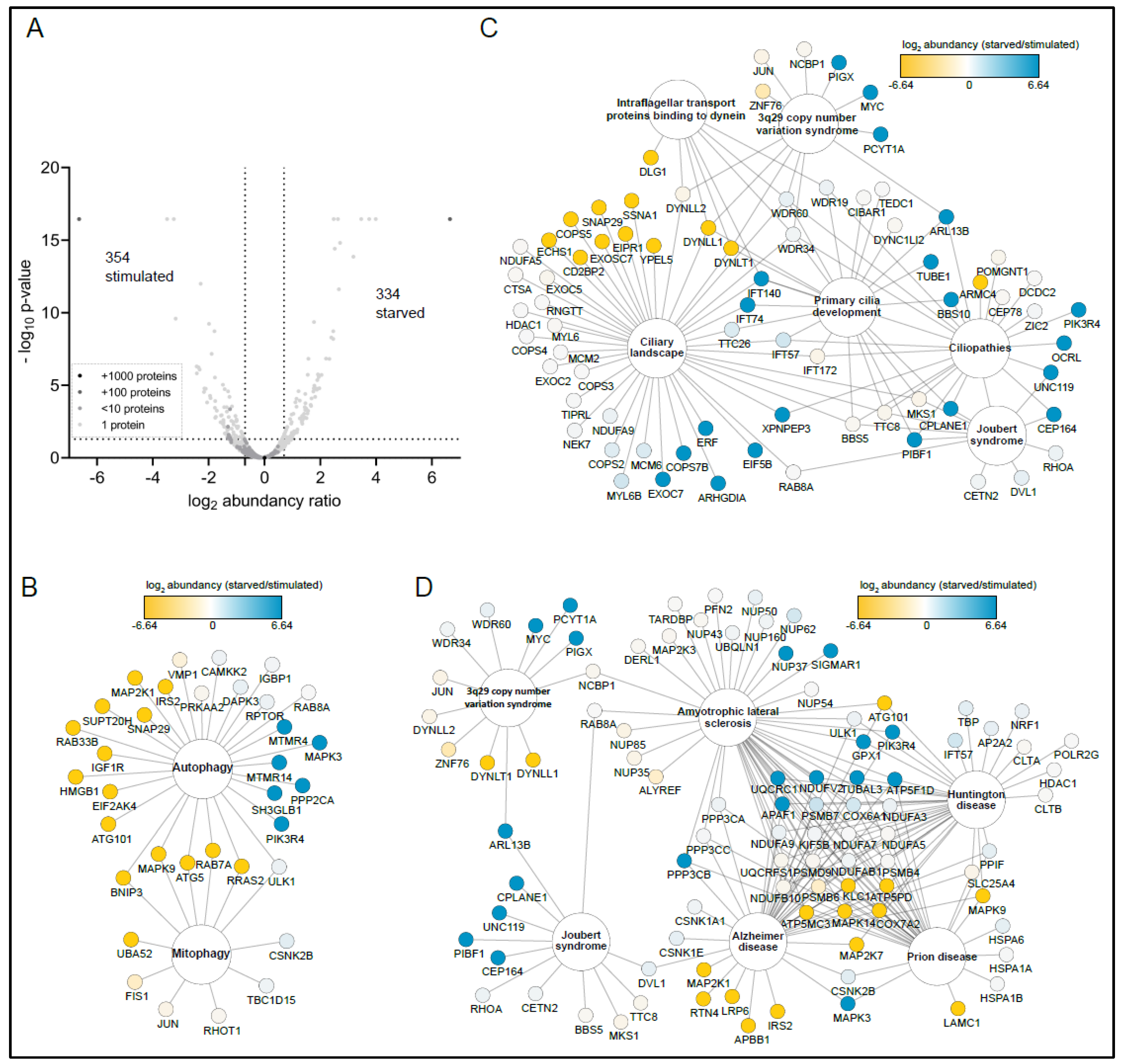
3.3. Metabolic State Alters the SZT2 Interactome
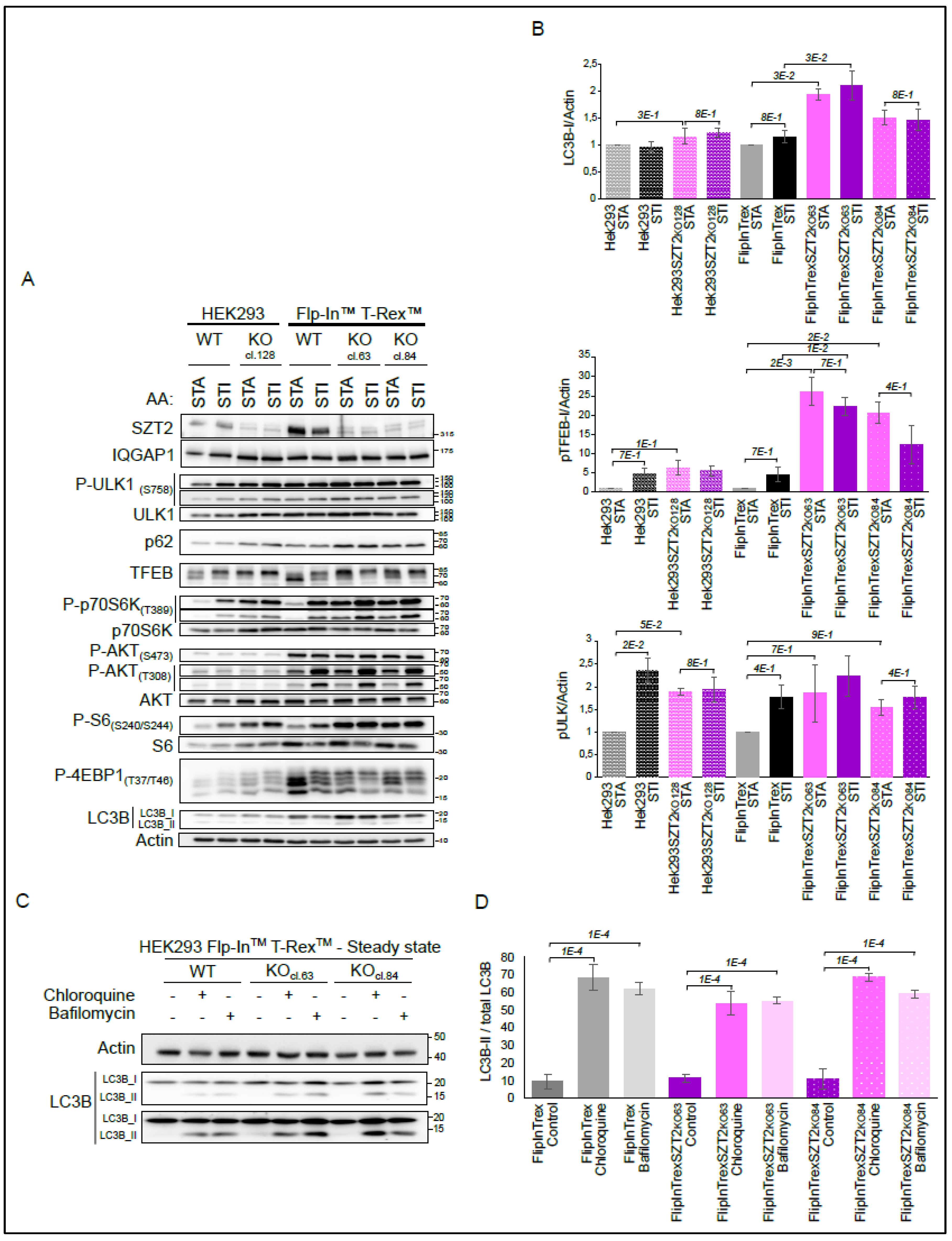
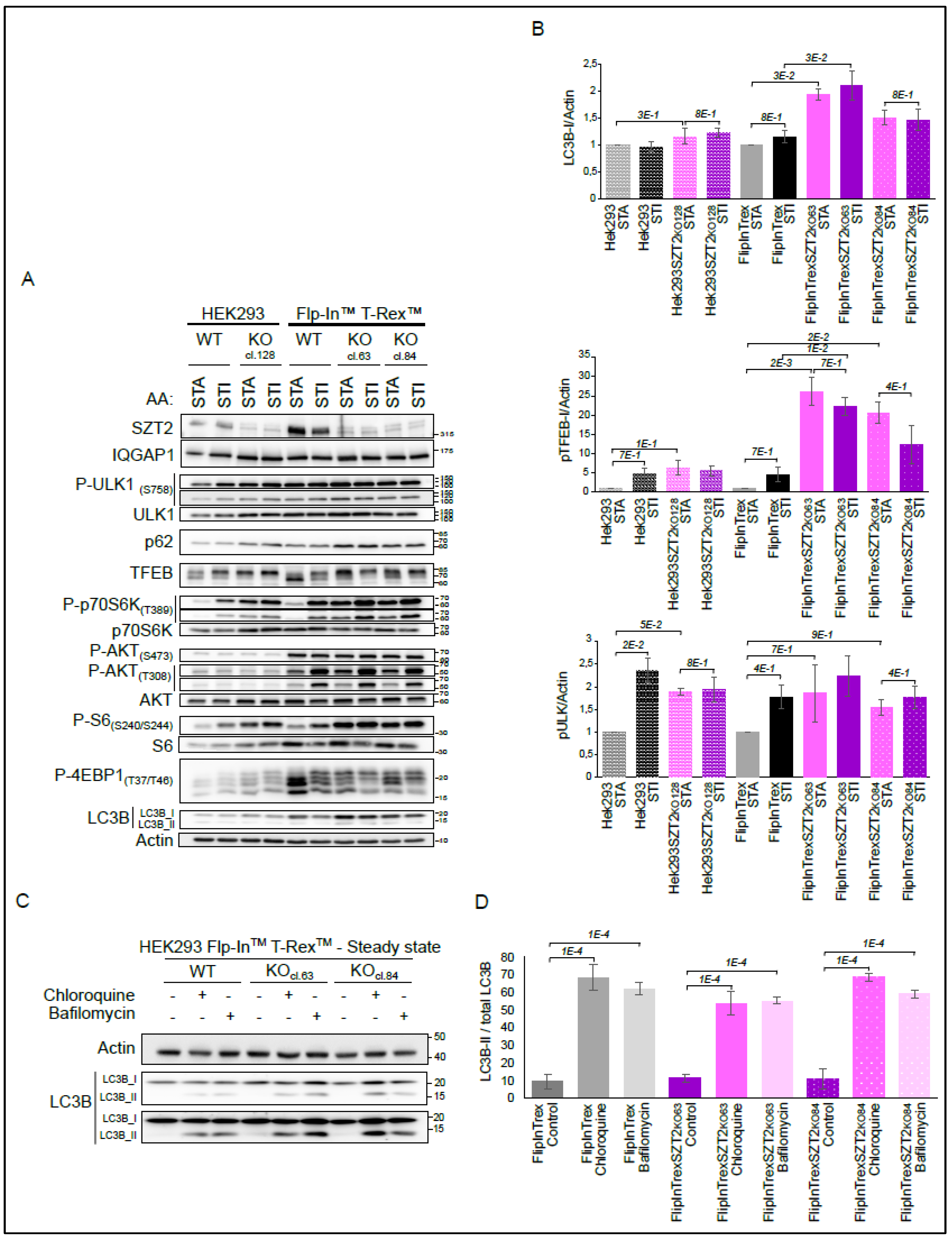
3.4. Characterization of SZT2 KO Cells
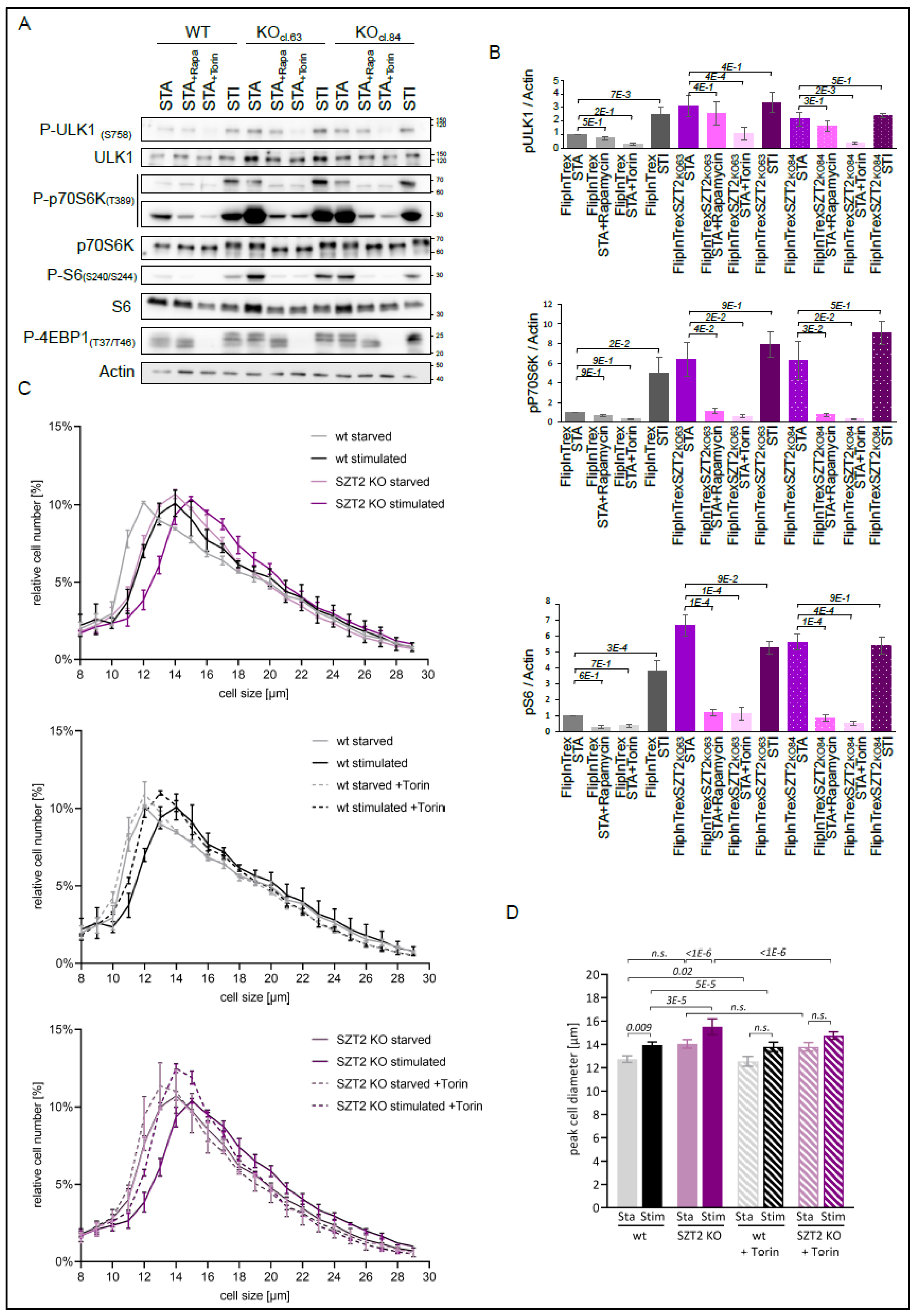
3.5. Effects of mTORC1 Inhibitors on SZT2 KO Cells
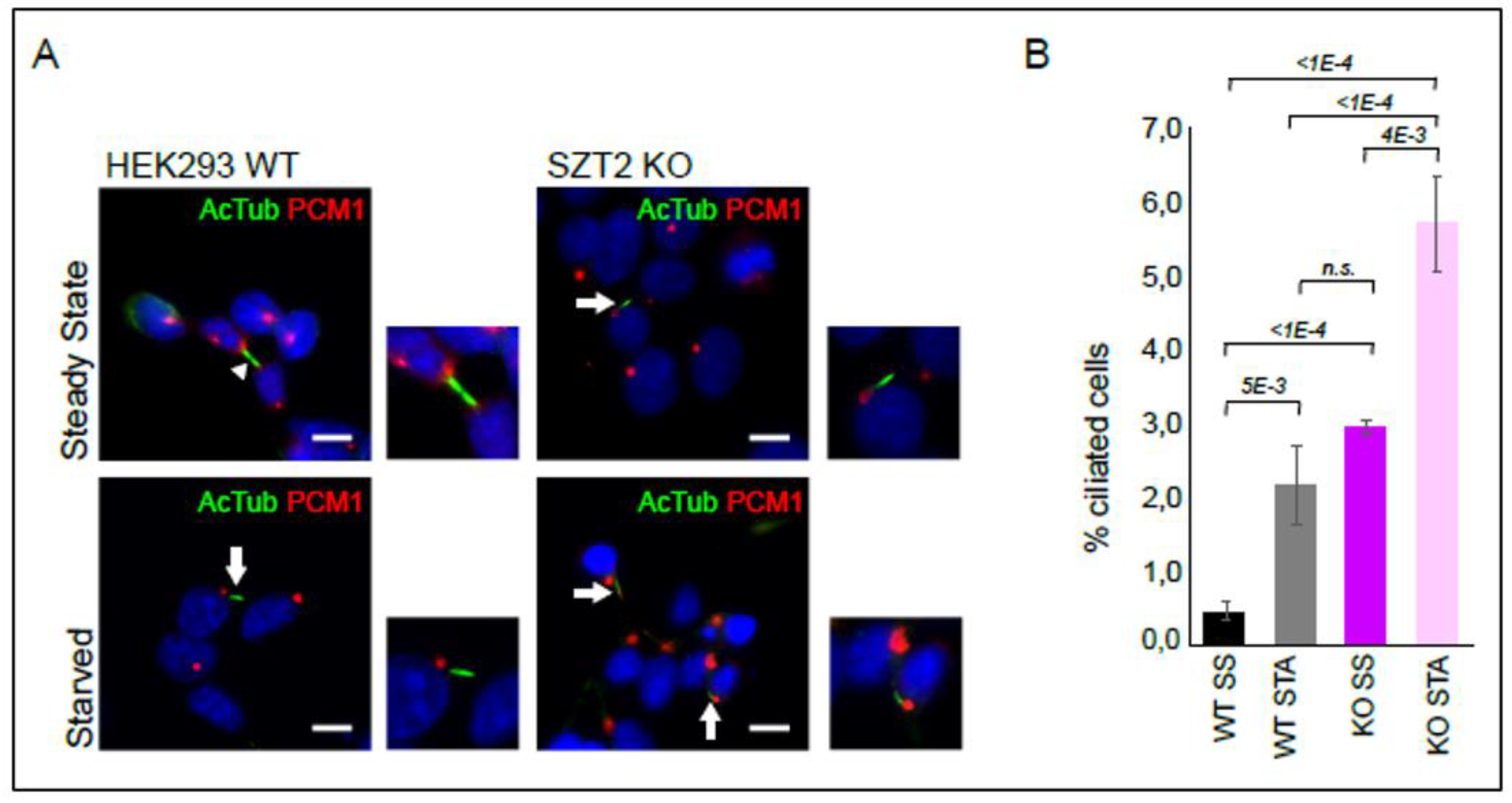
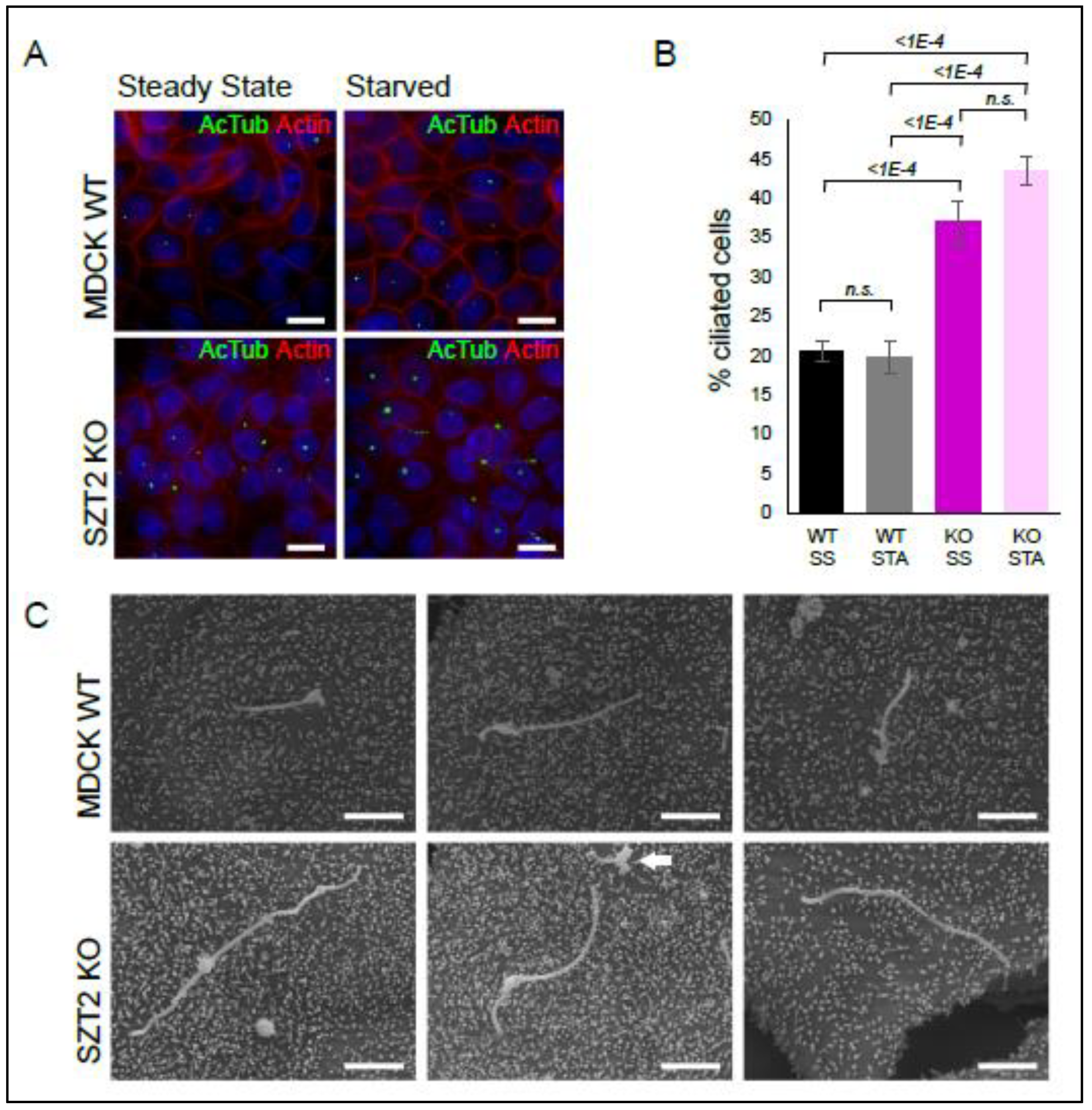
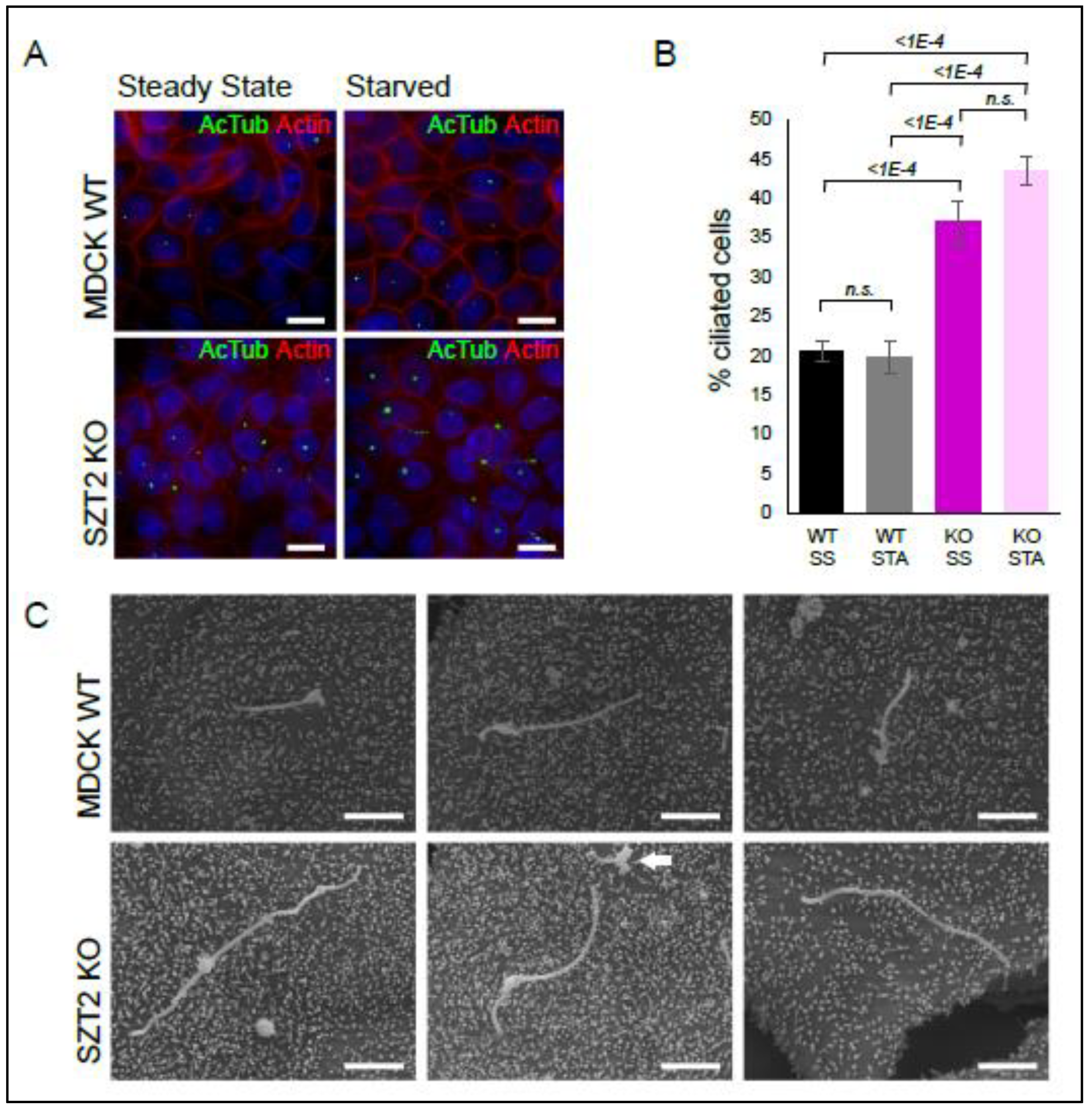
3.6. SZT2 in Ciliogenesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frankel, W.N.; Yang, Y.; Mahaffey, C.L.; Beyer, B.J.; O’Brien, T.P. Szt2, a novel gene for seizure threshold in mice. Genes Brain Behav. 2009, 8, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Toutzaris, D.; Lewerenz, J.; Albrecht, P.; Jensen, L.T.; Letz, J.; Geerts, A.; Golz, S.; Methner, A. A novel giant peroxisomal superoxide dismutase motif-containing protein. Free Radic. Biol. Med. 2010, 48, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kim, J.; Alexander, A.; Cai, S.; Tripathi, D.N.; Dere, R.; Tee, A.R.; Tait-Mulder, J.; Di Nardo, A.; Han, J.M.; et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nat. Cell Biol. 2013, 15, 1186–1196. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Guertin, D.A. mTORC1 gRABs the Golgi. Cancer Cell 2014, 26, 601–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.B.; Burgos, P.; Parker, A.W.; Iadevaia, V.; Proud, C.G.; Allen, R.A.; O’Connell, J.P.; Jeshtadi, A.; Stubbs, C.D.; Botchway, S.W. mTOR direct interactions with Rheb-GTPase and raptor: Sub-cellular localization using fluorescence lifetime imaging. BMC Cell Biol. 2013, 14, 3. [Google Scholar] [CrossRef]
- Rosner, M.; Hengstschlager, M. Detection of cytoplasmic and nuclear functions of mTOR by fractionation. Methods Mol. Biol. 2012, 821, 105–124. [Google Scholar]
- Tsang, C.K.; Liu, H.; Zheng, X.F. mTOR binds to the promoters of RNA polymerase I- and III-transcribed genes. Cell Cycle 2010, 9, 953–957. [Google Scholar] [CrossRef] [Green Version]
- Kantidakis, T.; Ramsbottom, B.A.; Birch, J.L.; Dowding, S.N.; White, R.J. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc. Natl. Acad. Sci. USA 2010, 107, 11823–11828. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Zoncu, R.; Chang, S.; Gumper, I.; Snitkin, H.; Wolfson, R.L.; Kirak, O.; Sabatini, D.D.; Sabatini, D.M. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 2013, 493, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Manifava, M.; Smith, M.; Rotondo, S.; Walker, S.; Niewczas, I.; Zoncu, R.; Clark, J.; Ktistakis, N.T. Dynamics of mTORC1 activation in response to amino acids. eLife 2016, 5, e19960. [Google Scholar] [CrossRef]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teis, D.; Wunderlich, W.; Huber, L.A. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 2002, 3, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Teis, D.; Taub, N.; Kurzbauer, R.; Hilber, D.; de Araujo, M.E.; Erlacher, M.; Offterdinger, M.; Villunger, A.; Geley, S.; Bohn, G.; et al. p14-MP1-MEK1 signaling regulates endosomal traffic and cellular proliferation during tissue homeostasis. J. Cell Biol. 2006, 175, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, W.; Fialka, I.; Teis, D.; Alpi, A.; Pfeifer, A.; Parton, R.G.; Lottspeich, F.; Huber, L.A. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J. Cell Biol. 2001, 152, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nada, S.; Hondo, A.; Kasai, A.; Koike, M.; Saito, K.; Uchiyama, Y.; Okada, M. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009, 28, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.L.; Tee, A.R.; Short, J.D.; Bergeron, J.M.; Kim, J.; Shen, J.; Guo, R.; Johnson, C.L.; Kiguchi, K.; Walker, C.L. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J. Cell Biol. 2006, 173, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Y.; Shin, H.R.; Berdan, C.A.; Ford, B.; Ward, C.C.; Olzmann, J.A.; Zoncu, R.; Nomura, D.K. Covalent targeting of the vacuolar H(+)-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 2019, 15, 776–785. [Google Scholar] [CrossRef]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Chantranupong, L.; Knockenhauer, K.E.; Schwartz, T.U.; Sabatini, D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature 2016, 536, 229–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; French, J.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; Perucca, E.; Tomson, T.; Wiebe, S.; Zhang, Y.H.; et al. Classification of the epilepsies: New concepts for discussion and debate-Special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia Open 2016, 1, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensadoun, A.; Weinstein, D. Assay of proteins in the presence of interfering materials. Anal. Biochem. 1976, 70, 241–250. [Google Scholar] [CrossRef]
- Hellman, U. Sample preparation by SDS/PAGE and in-gel digestion. EXS 2000, 88, 43–54. [Google Scholar]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Filipek, P.A.; de Araujo, M.E.G.; Vogel, G.F.; De Smet, C.H.; Eberharter, D.; Rebsamen, M.; Rudashevskaya, E.L.; Kremser, L.; Yordanov, T.; Tschaikner, P.; et al. LAMTOR/Ragulator is a negative regulator of Arl8b- and BORC-dependent late endosomal positioning. J. Cell Biol. 2017, 216, 4199–4215. [Google Scholar] [CrossRef]
- Haller, T.; Dietl, P.; Pfaller, K.; Frick, M.; Mair, N.; Paulmichl, M.; Hess, M.W.; Furst, J.; Maly, K. Fusion pore expansion is a slow, discontinuous, and Ca2+-dependent process regulating secretion from alveolar type II cells. J. Cell Biol. 2001, 155, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.; Morse, S.; Ararat, M.; Graham, F.L. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 2002, 16, 869–871. [Google Scholar] [CrossRef]
- Pitcher, J.A.; Hall, R.A.; Daaka, Y.; Zhang, J.; Ferguson, S.S.; Hester, S.; Miller, S.; Caron, M.G.; Lefkowitz, R.J.; Barak, L.S. The G protein-coupled receptor kinase 2 is a microtubule-associated protein kinase that phosphorylates tubulin. J. Biol. Chem. 1998, 273, 12316–12324. [Google Scholar] [CrossRef] [Green Version]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- van Koppen, C.; Meyer zu Heringdorf, M.; Laser, K.T.; Zhang, C.; Jakobs, K.H.; Bunemann, M.; Pott, L. Activation of a high affinity Gi protein-coupled plasma membrane receptor by sphingosine-1-phosphate. J. Biol. Chem. 1996, 271, 2082–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachter, J.B.; Sromek, S.M.; Nicholas, R.A.; Harden, T.K. HEK293 human embryonic kidney cells endogenously express the P2Y1 and P2Y2 receptors. Neuropharmacology 1997, 36, 1181–1187. [Google Scholar] [CrossRef]
- Anderson, L.; Alexander, C.L.; Faccenda, E.; Eidne, K.A. Rapid desensitization of the thyrotropin-releasing hormone receptor expressed in single human embryonal kidney 293 cells. Biochem. J. 1995, 311 Pt 2, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Pullen, N.; Dennis, P.B.; Andjelkovic, M.; Dufner, A.; Kozma, S.C.; Hemmings, B.A.; Thomas, G. Phosphorylation and activation of p70s6k by PDK1. Science 1998, 279, 707–710. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.A.; Quinsay, M.N.; Orogo, A.M.; Giang, K.; Rikka, S.; Gustafsson, A.B. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 2012, 287, 19094–19104. [Google Scholar] [CrossRef] [Green Version]
- Xian, H.; Yang, Q.; Xiao, L.; Shen, H.M.; Liou, Y.C. STX17 dynamically regulated by Fis1 induces mitophagy via hierarchical macroautophagic mechanism. Nat. Commun. 2019, 10, 2059. [Google Scholar] [CrossRef] [Green Version]
- Tsang, W.Y.; Bossard, C.; Khanna, H.; Peranen, J.; Swaroop, A.; Malhotra, V.; Dynlacht, B.D. CP110 suppresses primary cilia formation through its interaction with CEP290, a protein deficient in human ciliary disease. Dev. Cell 2008, 15, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; De Nicolo, A. The Centrosome and the Primary Cilium: The Yin and Yang of a Hybrid Organelle. Cells 2019, 8, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Frenkel, E.M.; Laqtom, N.N.; Dharamdasani, V.; Lewis, C.A.; Chan, S.H.; Heinze, I.; Ori, A.; Sabatini, D.M. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 2018, 360, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, J.; von Stockum, S.; Marchesan, E.; Caicci, F.; Ferrari, V.; Rakovic, A.; Klein, C.; Antonini, A.; Bubacco, L.; Ziviani, E. USP14 inhibition corrects an in vivo model of impaired mitophagy. EMBO Mol. Med. 2018, 10, e9014. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 2019, 74, 891–908.e810. [Google Scholar] [CrossRef] [PubMed]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e906. [Google Scholar] [CrossRef]
- Zutphen, T.; Veenhuis, M.; van der Klei, I.J. Pex14 is the sole component of the peroxisomal translocon that is required for pexophagy. Autophagy 2008, 4, 63–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellu, A.R.; Komori, M.; van der Klei, I.J.; Kiel, J.A.; Veenhuis, M. Peroxisome biogenesis and selective degradation converge at Pex14p. J. Biol. Chem. 2001, 276, 44570–44574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Wang, B.; Li, N.; Wu, Y.; Jia, J.; Suo, T.; Chen, Q.; Liu, Y.J.; Tang, J. RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1. PLoS ONE 2011, 6, e24367. [Google Scholar] [CrossRef]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Inoki, K.; Guan, K.L. mTOR pathway as a target in tissue hypertrophy. Annu. Rev. Pharm. Toxicol. 2007, 47, 443–467. [Google Scholar] [CrossRef]
- Wiegering, A.; Dildrop, R.; Kalfhues, L.; Spychala, A.; Kuschel, S.; Lier, J.M.; Zobel, T.; Dahmen, S.; Leu, T.; Struchtrup, A.; et al. Cell type-specific regulation of ciliary transition zone assembly in vertebrates. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Rodriguez-Boulan, E.; Kreitzer, G.; Musch, A. Organization of vesicular trafficking in epithelia. Nat. Rev. Mol. Cell Biol. 2005, 6, 233–247. [Google Scholar] [CrossRef]
- Gadadhar, S.; Dadi, H.; Bodakuntla, S.; Schnitzler, A.; Bieche, I.; Rusconi, F.; Janke, C. Tubulin glycylation controls primary cilia length. J. Cell Biol. 2017, 216, 2701–2713. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.; Lippincott-Schwartz, J. Visualization of live primary cilia dynamics using fluorescence microscopy. Curr. Protoc. Cell Biol. 2012, 57, 4–26. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zou, Y.; Mao, D.; Sun, D.; Gao, G.; Shi, J.; Liu, X.; Zhu, C.; Yang, M.; Ye, W.; et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J. Cell Biol. 2014, 206, 173–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiprilov, E.N.; Awan, A.; Desprat, R.; Velho, M.; Clement, C.A.; Byskov, A.G.; Andersen, C.Y.; Satir, P.; Bouhassira, E.E.; Christensen, S.T.; et al. Human embryonic stem cells in culture possess primary cilia with hedgehog signaling machinery. J. Cell Biol. 2008, 180, 897–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beese, C.J.; Brynjolfsdottir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2019, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Ocak, U.; Gao, L.; Tu, S.; Lenahan, C.J.; Zhang, J.; Shao, A. Selective autophagy as a therapeutic target for neurological diseases. Cell Mol. Life Sci. 2021, 78, 1369–1392. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Uittenbogaard, M.; Gropman, A.; Brantner, C.A.; Chiaramello, A. Novel metabolic signatures of compound heterozygous Szt2 variants in a case of early-onset of epileptic encephalopathy. Clin. Case Rep. 2018, 6, 2376–2384. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, A.; Wertz, M.H.; Kwiatkowski, E.; Tsai, P.T.; Leech, J.D.; Greene-Colozzi, E.; Goto, J.; Dilsiz, P.; Talos, D.M.; Clish, C.B.; et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 2014, 23, 3865–3874. [Google Scholar] [CrossRef] [Green Version]
- Alesi, N.; Akl, E.W.; Khabibullin, D.; Liu, H.J.; Nidhiry, A.S.; Garner, E.R.; Filippakis, H.; Lam, H.C.; Shi, W.; Viswanathan, S.R.; et al. TSC2 regulates lysosome biogenesis via a non-canonical RAGC and TFEB-dependent mechanism. Nat. Commun. 2021, 12, 4245. [Google Scholar] [CrossRef]
- Dalle Pezze, P.; Ruf, S.; Sonntag, A.G.; Langelaar-Makkinje, M.; Hall, P.; Heberle, A.M.; Razquin Navas, P.; van Eunen, K.; Tolle, R.C.; Schwarz, J.J.; et al. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat. Commun. 2016, 7, 13254. [Google Scholar] [CrossRef]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Lievens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Hara-Kuge, S.; Yamashita, S.; Fujiki, Y. Peroxin Pex14p is the key component for coordinated autophagic degradation of mammalian peroxisomes by direct binding to LC3-II. Genes Cells 2015, 20, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Hara-Kuge, S.; Fujiki, Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp. Cell Res. 2008, 314, 3531–3541. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W. Mechanisms and Functions of Pexophagy in Mammalian Cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef] [PubMed]
- Huybrechts, S.J.; Van Veldhoven, P.P.; Brees, C.; Mannaerts, G.P.; Los, G.V.; Fransen, M. Peroxisome dynamics in cultured mammalian cells. Traffic 2009, 10, 1722–1733. [Google Scholar] [CrossRef]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef] [Green Version]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Antar, L.N.; Li, C.; Zhang, H.; Carroll, R.C.; Bassell, G.J. Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell Neurosci. 2006, 32, 37–48. [Google Scholar] [CrossRef]
- Didiot, M.C.; Tian, Z.; Schaeffer, C.; Subramanian, M.; Mandel, J.L.; Moine, H. The G-quartet containing FMRP binding site in FMR1 mRNA is a potent exonic splicing enhancer. Nucleic Acids Res. 2008, 36, 4902–4912. [Google Scholar] [CrossRef] [Green Version]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Lai, A.; Valdez-Sinon, A.N.; Bassell, G.J. Regulation of RNA granules by FMRP and implications for neurological diseases. Traffic 2020, 21, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.R.; Kronengold, J.; Gazula, V.R.; Chen, Y.; Strumbos, J.G.; Sigworth, F.J.; Navaratnam, D.; Kaczmarek, L.K. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 2010, 13, 819–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myrick, L.K.; Deng, P.Y.; Hashimoto, H.; Oh, Y.M.; Cho, Y.; Poidevin, M.J.; Suhl, J.A.; Visootsak, J.; Cavalli, V.; Jin, P.; et al. Independent role for presynaptic FMRP revealed by an FMR1 missense mutation associated with intellectual disability and seizures. Proc. Natl. Acad. Sci. USA 2015, 112, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Curia, G.; Gualtieri, F.; Bartolomeo, R.; Vezzali, R.; Biagini, G. Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front. Cell Neurosci. 2013, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Diaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Pampliega, O.; Cuervo, A.M. Autophagy and primary cilia: Dual interplay. Curr. Opin. Cell Biol. 2016, 39, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Marley, A.; von Zastrow, M. A simple cell-based assay reveals that diverse neuropsychiatric risk genes converge on primary cilia. PLoS ONE 2012, 7, e46647. [Google Scholar] [CrossRef]
- Migliavacca, E.; Golzio, C.; Mannik, K.; Blumenthal, I.; Oh, E.C.; Harewood, L.; Kosmicki, J.A.; Loviglio, M.N.; Giannuzzi, G.; Hippolyte, L.; et al. A Potential Contributory Role for Ciliary Dysfunction in the 16p11.2 600 kb BP4-BP5 Pathology. Am. J. Hum. Genet. 2015, 96, 784–796. [Google Scholar] [CrossRef] [Green Version]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Kumamoto, N.; Gu, Y.; Wang, J.; Janoschka, S.; Takemaru, K.; Levine, J.; Ge, S. A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nat. Neurosci. 2012, 15, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, M.; Zhao, X.; Li, J.; Yuan, W.; Yan, G.; Tong, M.; Guo, S.; Zhu, Y.; Jiang, Y.; Liu, Y.; et al. Tumor Suppressor Folliculin Regulates mTORC1 through Primary Cilia. J. Biol. Chem. 2016, 291, 11689–11697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Godel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.C.; Reiter, J.F.; Kim, D.S.; Kim, H.H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nardo, A.; Lenoel, I.; Winden, K.D.; Ruhmkorf, A.; Modi, M.E.; Barrett, L.; Ercan-Herbst, E.; Venugopal, P.; Behne, R.; Lopes, C.A.M.; et al. Phenotypic Screen with TSC-Deficient Neurons Reveals Heat-Shock Machinery as a Druggable Pathway for mTORC1 and Reduced Cilia. Cell Rep. 2020, 31, 107780. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; Garcia-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| Screening HEK293 Primer A | GCCTCGCCCCCCAGCCCAC |
| Screening HEK293 Primer B | CTCCGGCTCCGGGCGCTCCG |
| Screening HEK293 Primer C | TCGGGCCTTCCGGGCTGGGC |
| Genotyping HEK293 KO F | CATCTGTGAGCCTGGCTGTC |
| Genotyping HEK293 KO R | GAAGACTCGCCTGAGGTTGC |
| Genotyping MDCK KO F | CCCATCTCTTGCCAGGTGG |
| Genotyping MDCK KO R | AATGGCGACACCAATACTGGG |
| Antibody | Species | Source | Identifier |
|---|---|---|---|
| β-Actin | Mouse | Cell Signaling Technology | 3700S |
| Cp110 | Rabbit | Abcam | ab243696 |
| PCM-1 (Gln15) | Rabbit | Cell Signaling Technology | 5259S |
| ULK1 | Rabbit | Cell Signaling Technology | 8054 |
| phospho ULK1 (Ser757) | Rabbit | Cell Signaling Technology | 6888 |
| phospho p70S6K (Thr389) | Rabbit | Cell Signaling Technology | 9234 |
| p70S6K | Rabbit | Cell Signaling Technology | 9202S |
| p62 | Rabbit | Cell Signaling Technology | 5114 |
| LC3B | Rabbit | Cell Signaling Technology | 2775 |
| phospho S6 ribosomal Protein (Ser240/244) | Rabbit | Cell Signaling Technology | 2215 |
| S6 ribosomal Protein | Mouse | Cell Signaling Technology | 2317 |
| phospho 4E-BP1 (Thr37/46) | Rabbit | Cell Signaling Technology | 2855 P |
| FMRP | Rabbit | Cell Signaling Technology | 4317S |
| TFEB | Rabbit | Cell Signaling Technology | 4240 |
| TSC2 | Rabbit | Cell Signaling Technology | 4308 |
| RICTOR | Rabbit | Cell Signaling Technology | 2114Z |
| KPTN | Rabbit | Proteintech Group | 16094-1-AP |
| LAMP-1 | Mouse | Pharmingen | 34201A |
| SZT2 | Rabbit | Novus Biologicals | NBP1-89886 |
| IQGAP1 | Mouse | BD Biosciences | 610612 |
| α-Tubulin | Mouse | Developmental Studies Hybridoma Bank | 12G10 |
| RAPTOR | Rabbit | Cell Signaling Technology | 2280 |
| γ-Tubulin | Mouse | Sigma-Aldrich | T5326 |
| phospho AKT (Ser473) | Rabbit | Cell Signaling Technology | 4060S |
| phospho AKT (Thr309) | Rabbit | Cell Signaling Technology | 13038S |
| phospho AKT (Thr308) | Rabbit | Cell Signaling Technology | 9275 |
| HA.11 | Mouse | Biolegend | MMS-101R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cattelani, C.; Lesiak, D.; Liebscher, G.; Singer, I.I.; Stasyk, T.; Wallnöfer, M.H.; Heberle, A.M.; Corti, C.; Hess, M.W.; Pfaller, K.; et al. The SZT2 Interactome Unravels New Functions of the KICSTOR Complex. Cells 2021, 10, 2711. https://doi.org/10.3390/cells10102711
Cattelani C, Lesiak D, Liebscher G, Singer II, Stasyk T, Wallnöfer MH, Heberle AM, Corti C, Hess MW, Pfaller K, et al. The SZT2 Interactome Unravels New Functions of the KICSTOR Complex. Cells. 2021; 10(10):2711. https://doi.org/10.3390/cells10102711
Chicago/Turabian StyleCattelani, Cecilia, Dominik Lesiak, Gudrun Liebscher, Isabel I. Singer, Taras Stasyk, Moritz H. Wallnöfer, Alexander M. Heberle, Corrado Corti, Michael W. Hess, Kristian Pfaller, and et al. 2021. "The SZT2 Interactome Unravels New Functions of the KICSTOR Complex" Cells 10, no. 10: 2711. https://doi.org/10.3390/cells10102711
APA StyleCattelani, C., Lesiak, D., Liebscher, G., Singer, I. I., Stasyk, T., Wallnöfer, M. H., Heberle, A. M., Corti, C., Hess, M. W., Pfaller, K., Kwiatkowski, M., Pramstaller, P. P., Hicks, A. A., Thedieck, K., Müller, T., Huber, L. A., & Eca Guimaraes de Araujo, M. (2021). The SZT2 Interactome Unravels New Functions of the KICSTOR Complex. Cells, 10(10), 2711. https://doi.org/10.3390/cells10102711