
A Novel Pro-Inflammatory Mechanosensing Pathway Orchestrated by the Disintegrin Metalloproteinase ADAM15 in Synovial Fibroblasts

 ,
,
Abstract
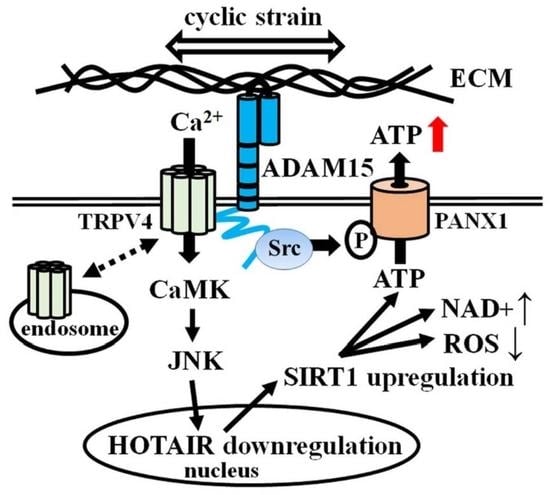
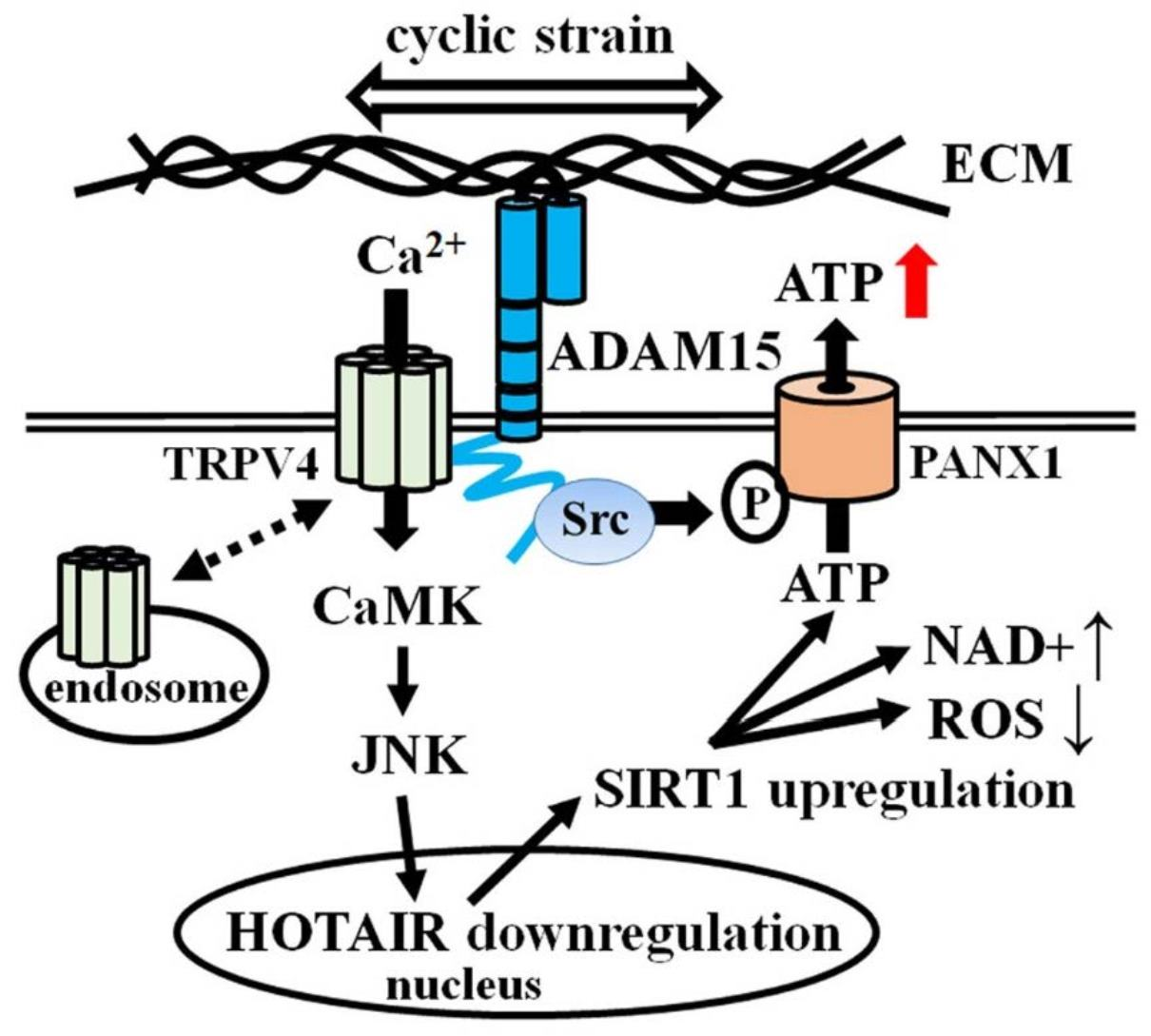{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell Culture
2.3. Cyclic Biaxial Tensile Strain
2.4. RNAi Silencing in SF
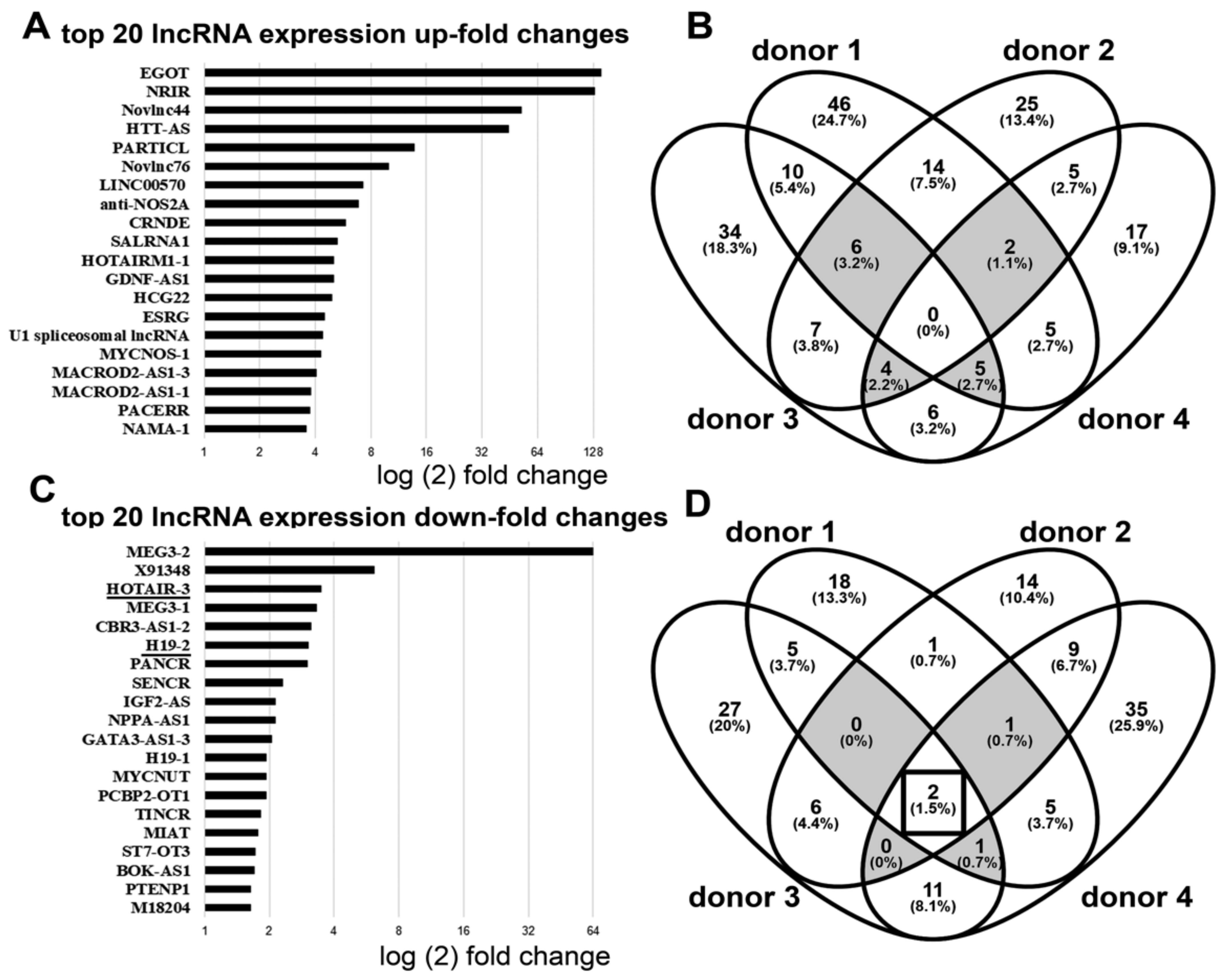
2.5. ArrayStar LncRNA Array
2.6. Inhibitor Assays
2.7. Semi-Quantitative qPCR
2.8. NAD+ Assays
2.9. Reactive Oxygen Species (ROS) Assays
2.10. ATP Assays
2.11. Preparation of Cell Lysates and Western Blotting
2.12. Preparation of Nuclear Fractions
2.13. Co-Immunoprecipitation
2.14. Enrichment of Plasma Membrane by Cell Surface Biotinylation
2.15. Immunofluorescence
2.16. Statistical Analysis
3. Results
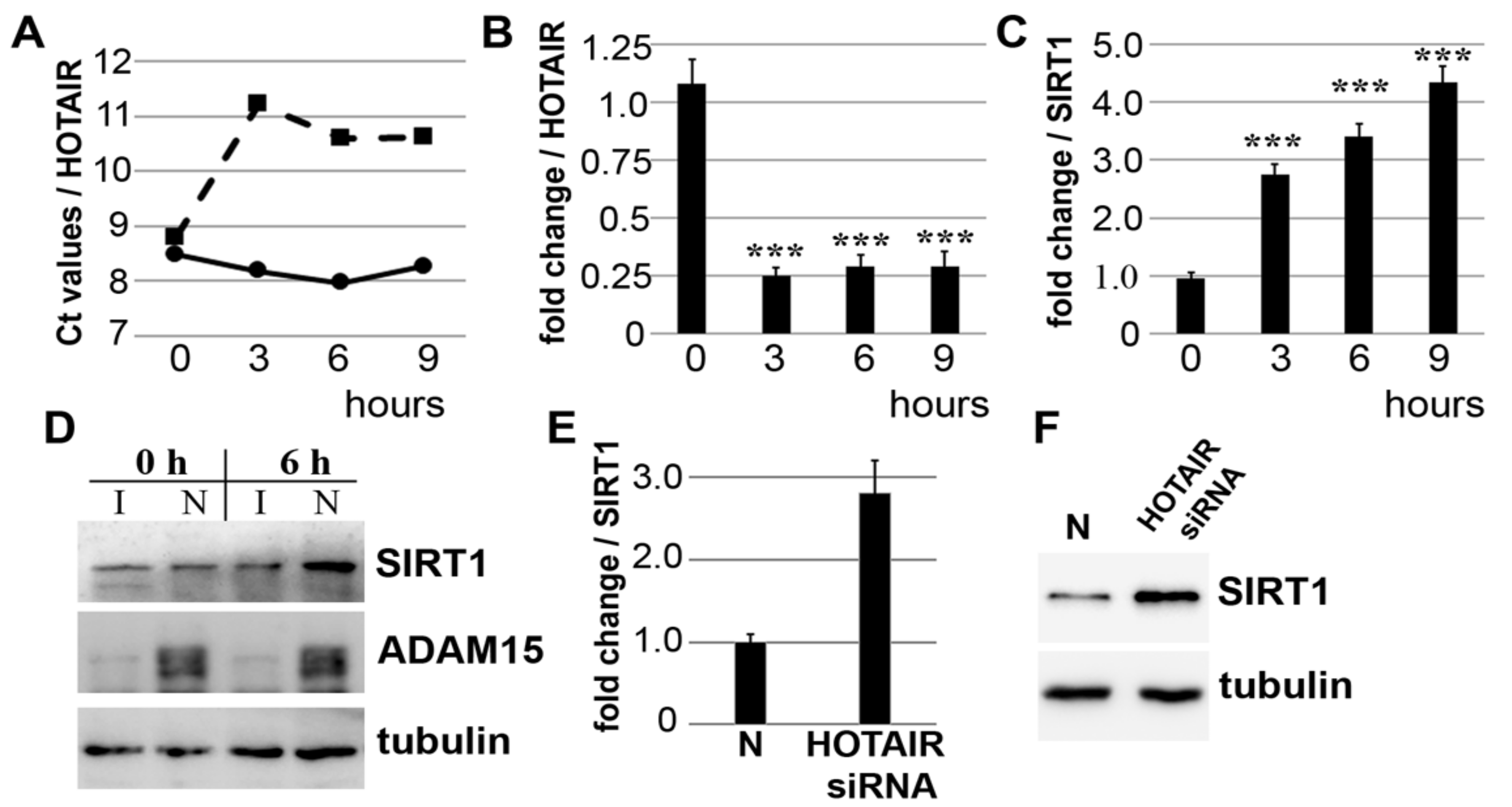
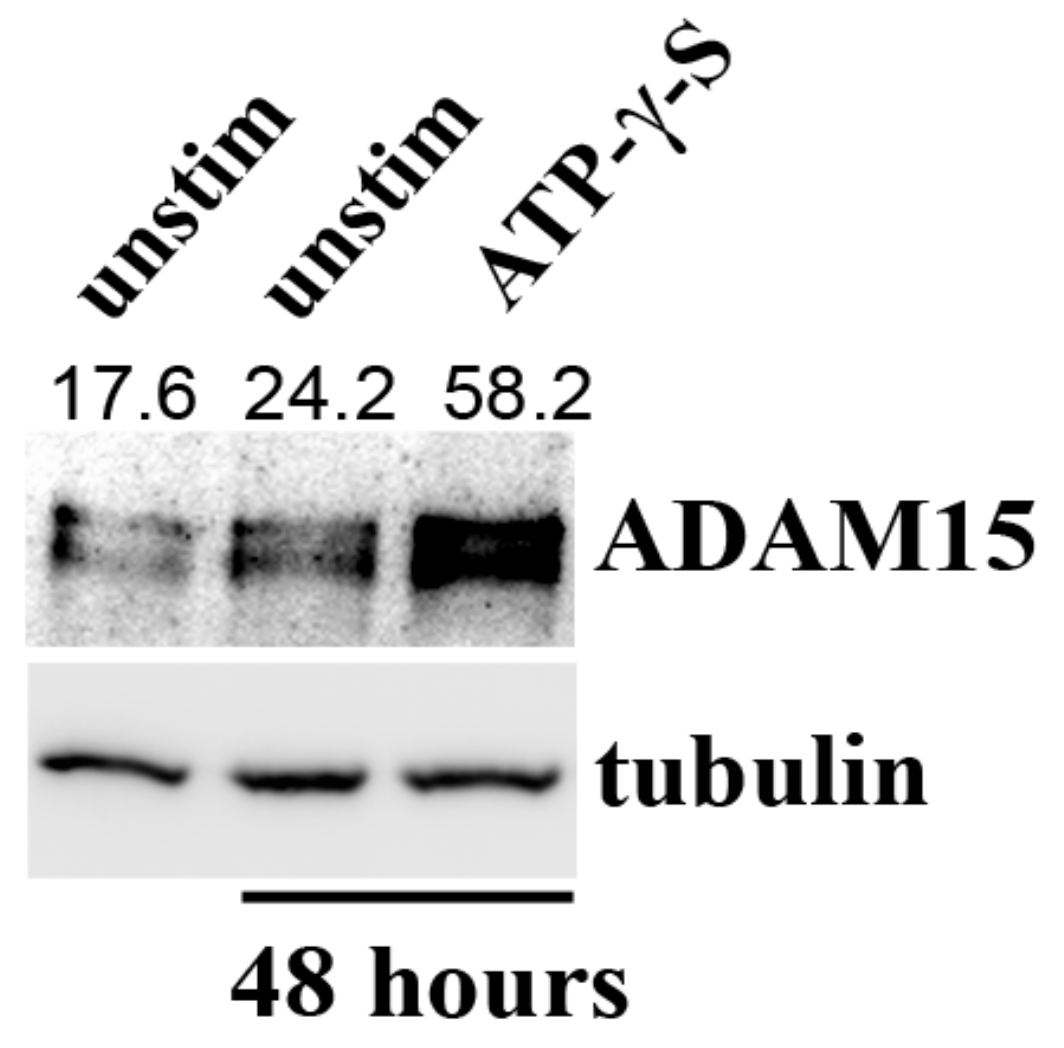
3.1. Downregulation of lncRNA HOTAIR by Mechanical Strain Is Critically Dependent on ADAM15
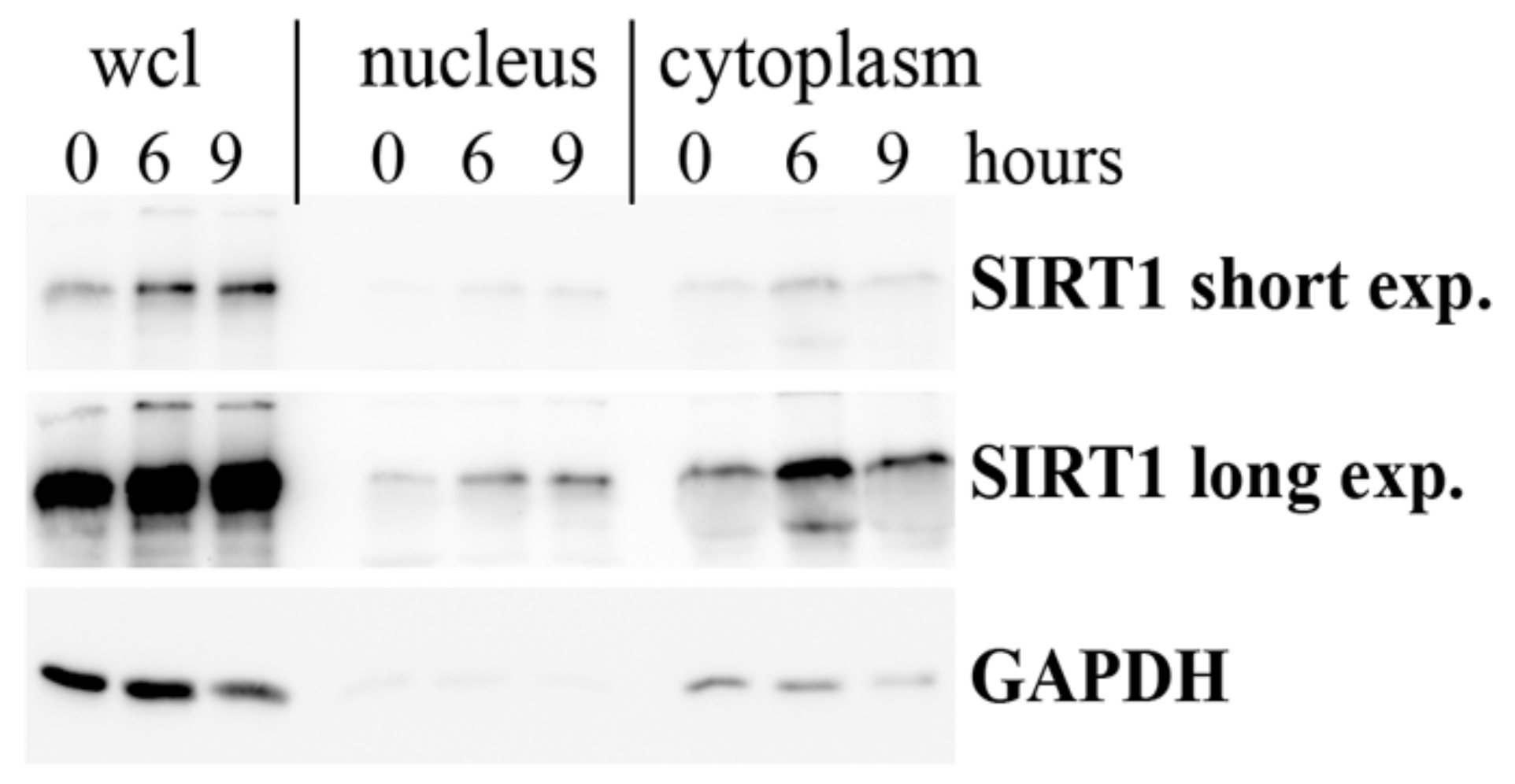
3.2. Strain-Induced SIRT1 Upregulation via ADAM15-Mediated Downregulation of HOTAIR
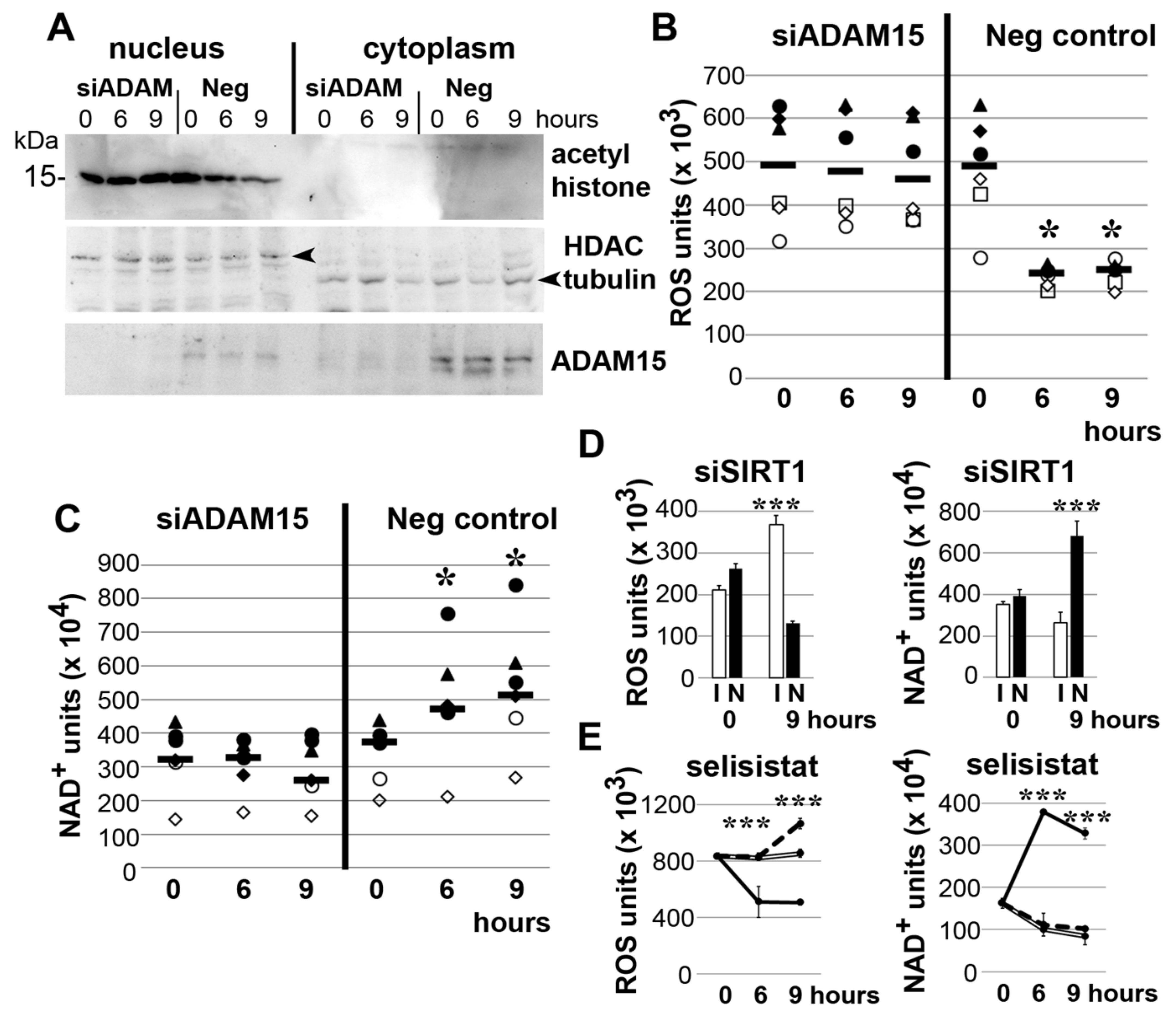
3.3. Impact of ADAM15 and SIRT1 on Histone Acetylation, ROS and NAD+
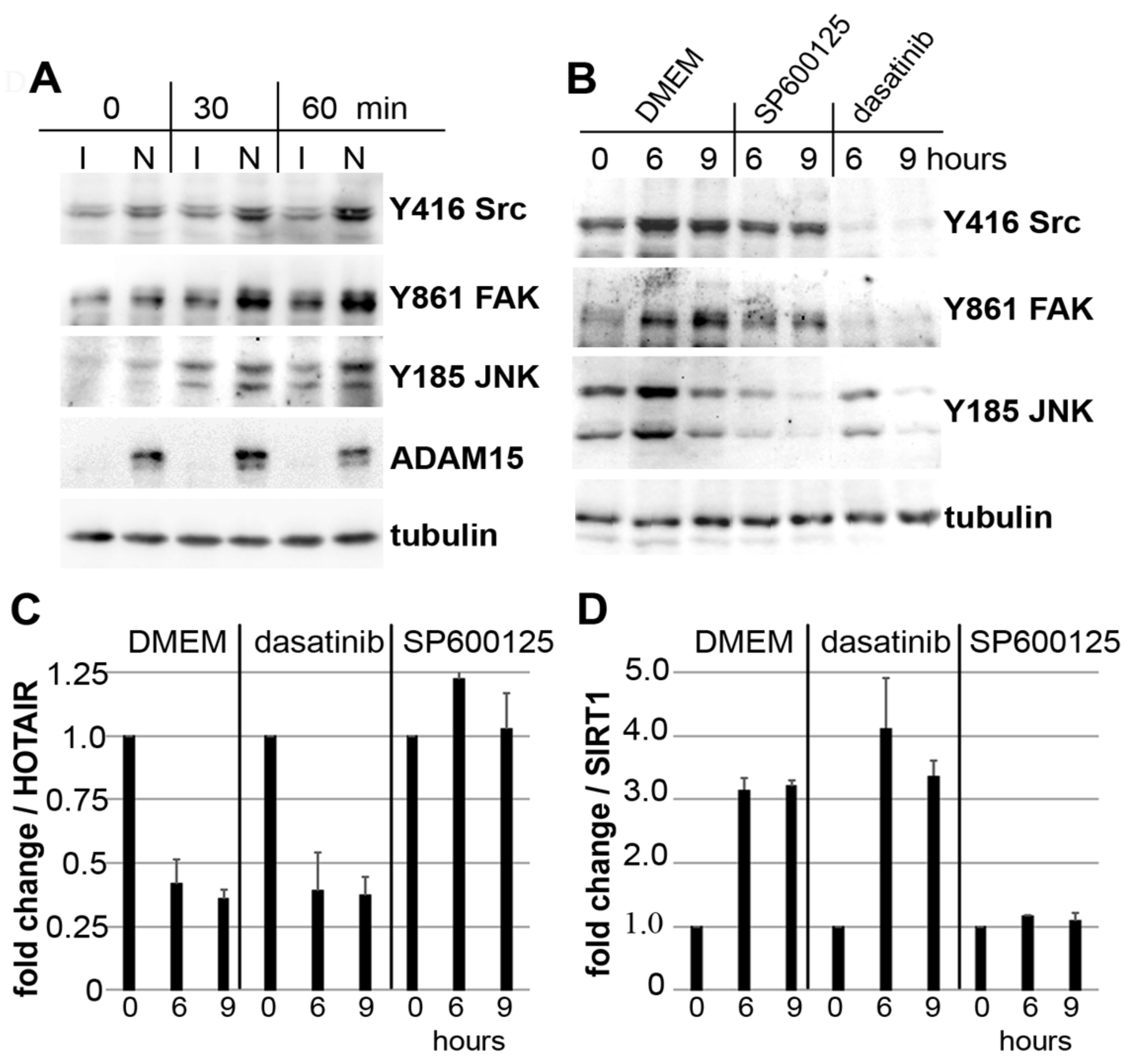
3.4. Impact of JNK on ADAM15-Dependent Mechano-Signaling in HOTAIR/SIRT1 Regulation
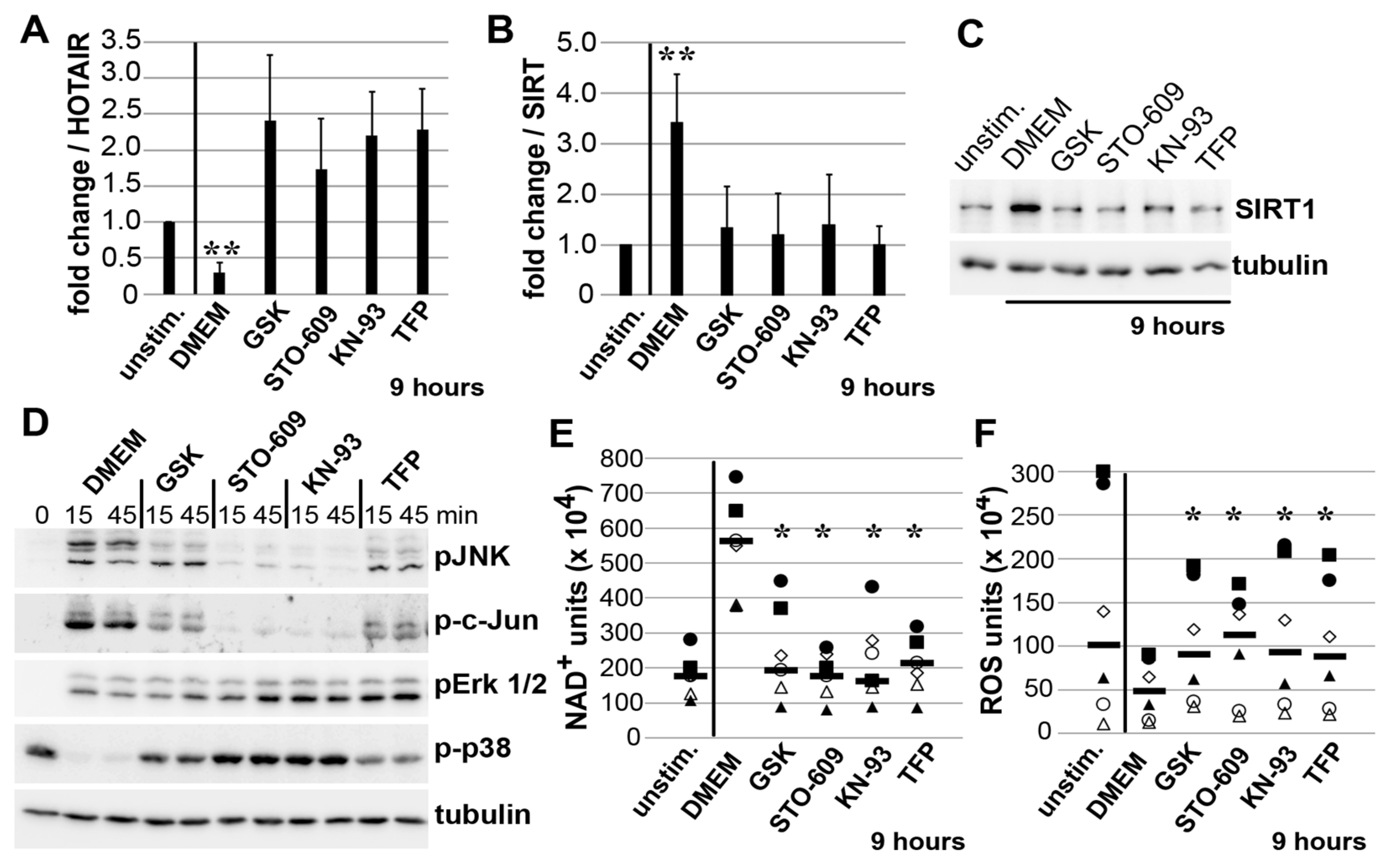
3.5. Mechano-Induced Activation of TRPV4 and CAMK Upstream of JNK
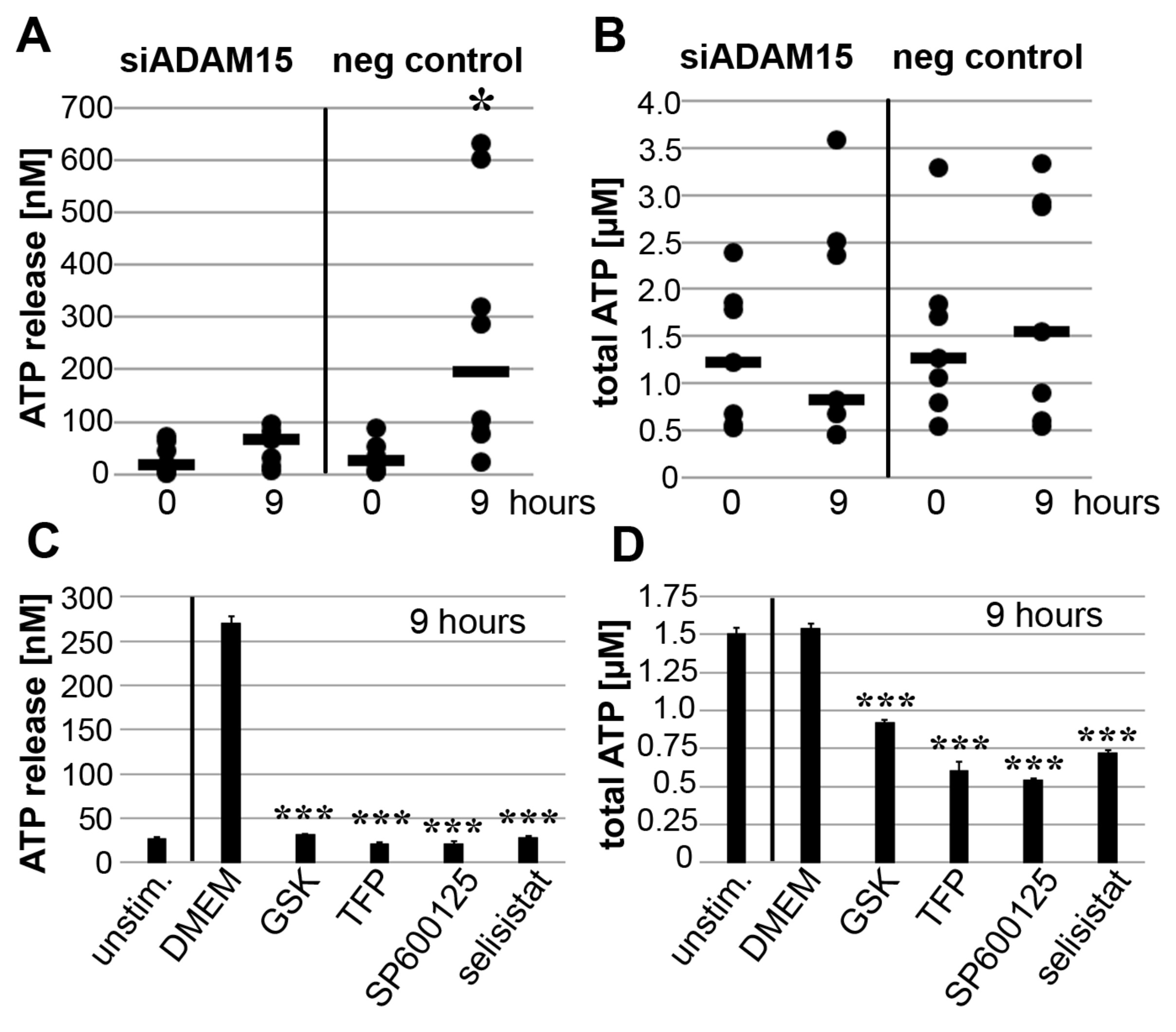
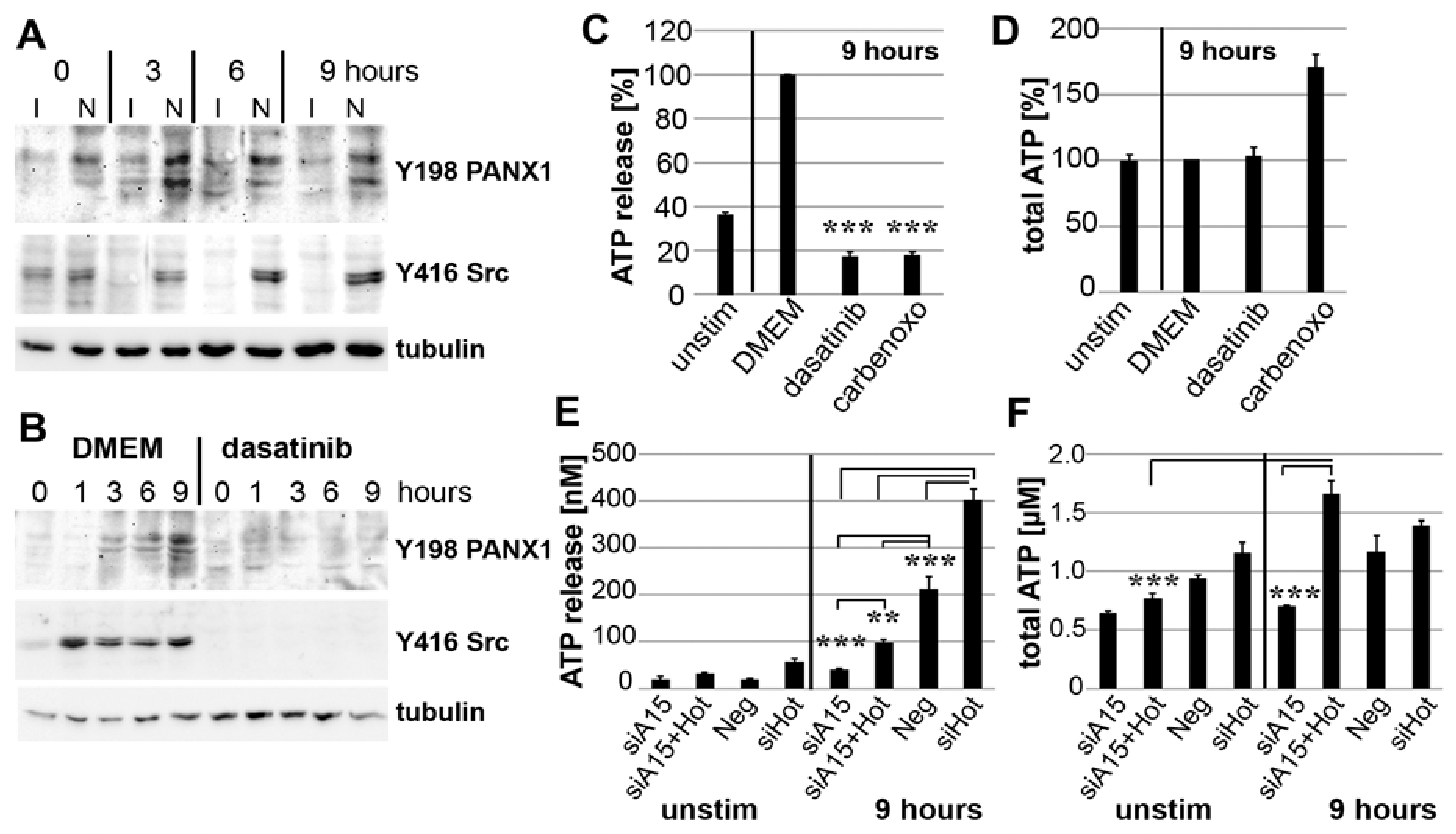
3.6. Impact of ADAM15 and Calcium Signaling on Strain-Induced ATP Release
3.7. PANX1 Activity Is Controlled by ADAM15
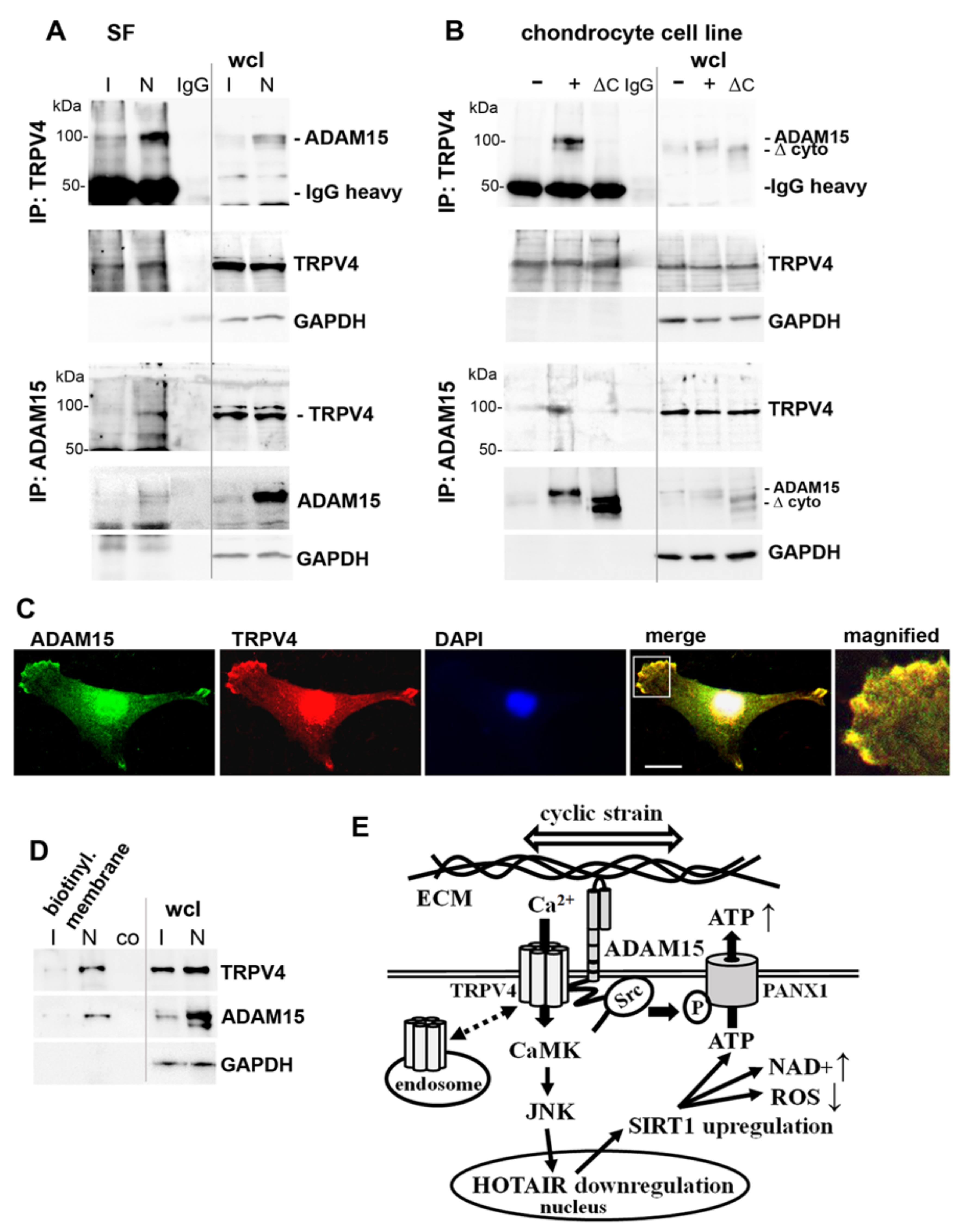
3.8. Binding of ADAM15 to TRPV4 Is Critical for Its Membrane Localization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Firestein, G.; McInnes, I.B. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef]
- Nygaard, G.; Firestein, G.S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef]
- Cambré, I.; Gaublomme, D.; Burssens, A.; Jacques, P.; Schryvers, N.; De Muynck, A.; Meuris, L.; Lambrecht, S.; Carter, S.; de Bleser, P.; et al. Mechanical strain determines the site-specific localization of inflammation and tissue damage in arthritis. Nat. Commun. 2018, 9, 4613. [Google Scholar] [CrossRef] [PubMed]
- Romani, P.; Valcarcel-Jimenez, L.; Frezza, C.; Dupont, S. Crosstalk between mechanotransduction and metabolism. Nat. Rev. Mol. Cell. Biol. 2021, 22, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, Y.T.; Li, T.F.; Hukkanen, M.; Ma, J.; Xu, J.W.; Virtanen, I. Fibroblast biology: Signals targeting the synovial fibroblast in arthritis. Arthritis Res. 2000, 2, 348–355. [Google Scholar] [CrossRef][Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell. Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Ringer, P.; Colo, G.; Fässler, R.; Grashoff, C. Sensing the mechano-chemical properties of the extracellular matrix. Matrix Biol. 2017, 64, 6–16. [Google Scholar] [CrossRef]
- Arnadóttir, J.; Chalfie, M. Eukaryotic mechanosensitive channels. Annu. Rev. Biophys. 2010, 39, 111–137. [Google Scholar] [CrossRef]
- Delco, M.L.; Bonassar, L.J. Targeting calcium-related mechanotransduction in early OA. Nat. Rev. Rheumatol. 2021, 17, 445–446. [Google Scholar] [CrossRef]
- Michalick, L.; Kuebler, W.M. TRPV4—A Missing Link Between Mechanosensation and Immunity. Front. Immunol. 2020, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.M.; Tudor, C.; Kanger, J.S.; Subramaniam, V.; Martin-Blanco, E. Integrin-Dependent Activation of the JNK Signaling Pathway by Mechanical Stress. PLoS ONE 2011, 6, e26182. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Richter, F.; Segond von Banchet, G.; Schaible, H.G. Transient Receptor Potential vanilloid 4 ion channel in C-fibres is involved in mechanonociception of the normal and inflamed joint. Sci. Rep. 2019, 9, 10928. [Google Scholar] [CrossRef] [PubMed]
- Böhm, B.B.; Aigner, T.; Blobel, C.P.; Kalden, J.R.; Burkhardt, H. Highly enhanced expression of the disintegrin metalloproteinase MDC15 (metargidin) in rheumatoid synovial tissue. Arthritis Rheum. 2001, 44, 2046–2054. [Google Scholar] [CrossRef]
- Lu, D.; Scully, M.; Kakkar, V.; Lu, X. ADAM-15 Disintegrin-Like Domain Structure and Function. Toxins 2010, 2, 2411–2427. [Google Scholar] [CrossRef]
- Fried, D.; Böhm, B.B.; Krause, K.; Burkhardt, H. ADAM15 protein amplifies focal adhesion kinase phosphorylation under genotoxic stress conditions. J. Biol. Chem. 2012, 287, 21214–21223. [Google Scholar] [CrossRef]
- Böhm, B.B.; Schirner, A.; Burkhardt, H. ADAM15 modulates outside-in signalling in chondrocyte–matrix interactions. J. Cell. Mol. Med. 2009, 13, 2634–2644. [Google Scholar] [CrossRef]
- Janczi, T.; Böhm, B.B.; Fehrl, Y.; DeGiacomo, P.; Kinne, R.W.; Burkhardt, H. ADAM15 in Apoptosis Resistance of Synovial Fibroblasts: Converting Fas/CD95 Death Signals into the Activation of Prosurvival Pathways by Calmodulin Recruitment. Arthritis Rheumatol. 2019, 71, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Wu, M.H.; Lee, E.S.; Yuan, S.Y. A disintegrin and metalloproteinase 15 contributes to atherosclerosis by mediating endothelial barrier dysfunction via Src family kinase activity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Jones, S.W. Review: Long Noncoding RNAs in the Regulation of Inflammatory Pathways in Rheumatoid Arthritis and Osteoarthritis. Arthritis Rheumatol. 2016, 68, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Sun, W.; Shi, Y.; Wang, Z.; Zhang, J.; Cai, H.; Zhang, J.; Huang, D. Interaction of long-chain non-coding RNAs and important signaling pathways on human cancers (Review). Int. J. Oncol. 2018, 53, 2343–2355. [Google Scholar] [CrossRef]
- Song, J.; Kim, D.; Han, J.; Kim, Y.; Lee, M.; Jin, E.J. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin. Exp. Med. 2015, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2.0: A Comprehensive Database for Target Genes of lncRNAs in Human and Mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Kupis, W.; Pałyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Niederer, F.; Ospelt, C.; Brentano, F.; Hottiger, M.O.; Gay, R.E.; Gay, S.; Detmar, M.; Kyburz, D. SIRT1 overexpression in the rheumatoid arthritis synovium contributes to proinflammatory cytokine production and apoptosis resistance. Ann. Rheum. Dis. 2011, 70, 1866–1873. [Google Scholar] [CrossRef] [PubMed]
- Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2018, 8, 11. [Google Scholar] [CrossRef]
- Mikolajewicz, N.; Mohammed, A.; Morris, M.; Komarova, S.V. Mechanically stimulated ATP release from mammalian cells: Systematic review and meta-analysis. J. Cell Sci. 2018, 131, jcs223354. [Google Scholar] [CrossRef]
- Makarenkova, H.P.; Shestopalov, V.I. The role of pannexin hemichannels in inflammation and regeneration. Front. Physiol. 2014, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Rogge, E.; Vandendriessche, B.; Shiva, S.; Brouckaert, P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014, 5, e1102. [Google Scholar] [CrossRef]
- Zimmermann, T.; Kunisch, E.; Pfeiffer, R.; Hirth, A.; Stahl, H.D.; Sack, U.; Laube, A.; Liesaus, E.; Roth, A.; Palombo-Kinne, E.; et al. Isolation and characterization of rheumatoid arthritis synovial fibroblasts from primary culture—Primary culture cells markedly differ from fourth-passage cells. Arthritis Res. 2001, 3, 72–76. [Google Scholar] [CrossRef]
- Cheung, M.; Bao, W.; Behm, D.J.; Brooks, C.A.; Bury, M.J.; Dowdell, S.E.; Eidam, H.S.; Fox, R.M.; Goodman, K.B.; Holt, D.A.; et al. Discovery of GSK2193874: An Orally Active, Potent, and Selective Blocker of Transient Receptor Potential Vanilloid 4. ACS Med. Chem. Lett. 2017, 8, 549–554. [Google Scholar] [CrossRef]
- O’Byrne, S.N.; Scott, J.W.; Pilotte, J.R.; da Santiago, S.A.; Langendorf, C.G.; Oakhill, J.S.; Eduful, B.J.; Couñago, R.M.; Wells, C.I.; Zuercher, W.J.; et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules 2020, 25, 325. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.H.; Samal, A.B.; Lee, M.; Vlach, J.; Novikov, N.; Niedziela-Majka, A.; Feng, J.Y.; Koltun, D.O.; Brendza, K.M.; Kwon, H.J.; et al. The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) Activity by Binding to Ca 2+/CaM. J. Mol. Biol. 2019, 431, 1440–1459. [Google Scholar] [CrossRef] [PubMed]
- Vandonselaar, M.; Hickie, R.A.; Quail, J.W.; Delbaere, L.T. Trifluoperazine-induced conformational change in Ca2+-calmodulin. Nat. Struct. Biol. 1994, 1, 795–801. [Google Scholar] [CrossRef] [PubMed]
- DeLalio, L.J.; Billaud, M.; Ruddiman, C.A.; Johnstone, S.R.; Butcher, J.T.; Wolpe, A.G.; Jin, X.; Keller, T.S.; Keller, A.S.; Rivière, T.; et al. Constitutive SRC-mediated phosphorylation of pannexin 1 at tyrosine 198 occurs at the plasma membrane. J. Biol. Chem. 2019, 294, 6940–6956. [Google Scholar] [CrossRef]
- Michalski, K.; Kawate, T. Carbenoxolone inhibits Pannexin1 channels through interactions in the first extracellular loop. J. Gen. Physiol. 2016, 147, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Dakin, S.G.; Coles, M.; Sherlock, J.P.; Powrie, F.; Carr, A.J.; Buckely, C.D. Pathogenic stromal cells as therapeutic targets in joint inflammation. Nat. Rev. Rheumatol. 2018, 14, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Bleuel, J.; Zaucke, F.; Brüggemann, G.P.; Niehoff, A. Effects of cyclic tensile strain on chondrocyte metabolism: A systematic review. PLoS ONE 2015, 10, e0119816. [Google Scholar] [CrossRef] [PubMed]
- Carrion, K.; Dyo, J.; Patel, V.; Sasik, R.; Mohamed, S.A.; Hardiman, G.; Nigam, V. The Long Non-Coding HOTAIR Is Modulated by Cyclic Stretch and WNT/β-CATENIN in Human Aortic Valve Cells and Is a Novel Repressor of Calcification Genes. PLoS ONE 2014, 9, e96577. [Google Scholar] [CrossRef]
- Li, Z.Q.; Gu, X.Y.; Hu, J.X.; Ping, Y.; Li, H.; Yan, J.Y.; Li, J.; Sun, R.; Yu, Z.J.; Zhang, Y. Hepatitis C virus core protein impairs metabolic disorder of liver cell via HOTAIR-Sirt1 signalling. Biosci. Rep. 2016, 36, e00336. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Guo, Y.; Wang, X.J.; Duan, B.H.; Li, L. HOTAIR participates in hepatic insulin resistance via regulating SIRT1. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7883–7890. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, X.; Guo, S.; Xiao, L.; Liang, C.; Wang, Z.; Li, Y.; Liu, Y.; Yao, R.; Liu, Y.; et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J. Cell Physiol. 2019, 234, 4944–4958. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Pap, T.; Aupperle, K.R.; Gay, S.; Firestein, G.S.; Gay, R.E. Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001, 44, 676–681. [Google Scholar] [CrossRef]
- MacKenna, D.A.; Dolfi, F.; Vuori, K.; Ruoslahti, E. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activation by mechanical stretch is integrin-dependent and matrix-specific in rat cardiac fibroblasts. J. Clin. Invest. 1998, 101, 301–310. [Google Scholar] [CrossRef]
- Kook, S.H.; Hwang, J.M.; Park, J.S.; Kim, E.M.; Heo, J.S.; Jeon, Y.M.; Lee, J.C. Mechanical force induces type I collagen expression in human periodontal ligament fibroblasts through activation of ERK/JNK and AP-1. J. Cell. Biochem. 2009, 106, 1060–1067. [Google Scholar] [CrossRef]
- Arnoczky, S.P.; Tian, T.; Lavagnino, M.; Gardner, K.; Schuler, P.; Morse, P. Activation of stress-activated protein kinases (SAPK) in tendon cells following cyclic strain: The effects of strain frequency, strain magnitude, and cytosolic calcium. J. Orthop. Res. 2002, 20, 947–952. [Google Scholar] [CrossRef]
- Rizzo, M.T.; Leaver, A.H.; Yu, W.M.; Kovacs, R.J. Arachidonic acid induces mobilization of calcium stores and c-jun gene expression: Evidence that intracellular calcium release is associated with c-jun activation. Prostaglandins Leukot. Essent. Fatty Acids 1999, 60, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Thodeti, C.K.; Matthews, B.; Ravi, A.; Mammoto, A.; Ghosh, K.; Bracha, A.L.; Ingber, D.E. TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 2009, 104, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.D.; Thodeti, C.K.; Tytell, J.D.; Mammoto, A.; Overby, D.R.; Ingber, D.E. Ultra-rapid activation of TRPV4 ion channels by mechanical forces applied to cell surface beta1 integrins. Integr. Biol. 2010, 2, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Cuajungco, M.P.; Grimm, C.; Oshima, K.; D’hoedt, D.; Nilius, B.; Mensenkamp, A.R.; Bindels, R.J.M.; Plomann, M.; Heller, S. PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J. Biol. Chem. 2006, 281, 18753–18762. [Google Scholar] [CrossRef]
- Grove, L.M.; Mohan, M.L.; Abraham, S.; Scheraga, R.G.; Southern, B.D.; Crish, J.F.; Prasad, S.V.N.; Olman, M.A. Translocation of TRPV4-PI3Kγ complexes to the plasma membrane drives myofibroblast transdifferentiation. Sci. Signal. 2019, 12, eaau1533. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Umaru, B.; Pyriochou, A.; Kotsikoris, V.; Papapetropoulos, A.; Topouzis, S. ATP-sensitive potassium channel activation induces angiogenesis in vitro and in vivo. J. Pharmacol. Exp. Ther. 2015, 354, 79–87. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Kucia, M.; Tse, W.; Ratajczak, J.; Wiktor-Jedrzejczak, W. The Emerging Link Between the Complement Cascade and Purinergic Signaling in Stress Hematopoiesis. Front. Immunol. 2018, 9, 1295. [Google Scholar] [CrossRef]
- Cambré, I.; Gaublomme, D.; Schryvers, N.; Lambrecht, S.; Lories, R.; Venken, K.; Elewaut, D. Running promotes chronicity of arthritis by local modulation of complement activators and impairing T regulatory feedback loops. Ann. Rheum. Dis. 2019, 78, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Babendreyer, A.; Molls, L.; Simons, I.M.; Dreymueller, D.; Biller, K.; Jahr, H.; Denecke, B.; Boon, R.A.; Bette, S.; Schnakenberg, U.; et al. The metalloproteinase ADAM15 is upregulated by shear stress and promotes survival of endothelial cells. J. Mol. Cell. Cardiol. 2019, 134, 51–61. [Google Scholar] [CrossRef]
- Taylor, K.A.; Wright, J.R.; Vial, C.; Evans, R.J.; Mahaut-Smith, M.P. Amplification of human platelet activation by surface pannexin-1 channels. J. Thromb. Haemost. 2014, 12, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Stoffels, M.; Zaal, R.; Kok, N.; van der Meer, J.W.M.; Dinarello, C.A.; Simon, A. ATP-Induced IL-1β Specific Secretion: True Under Stringent Conditions. Front. Immunol. 2015, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- Charrier-Hisamuddin, L.; Laboisse, C.L.; Merlin, D. ADAM-15: A metalloprotease that mediates inflammation. FASEB J. 2008, 22, 641–653. [Google Scholar] [CrossRef]
- Gao, J.; Zheng, W.; Wang, L.; Song, B. A disintegrin and metallproteinase 15 knockout decreases migration of fibroblast-like synoviocytes and inflammation in rheumatoid arthritis. Mol. Med. Rep. 2015, 11, 4389–4396. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; McKiernan, E.; O’Donovan, N.; McGowan, P.M. Role of ADAMs in cancer formation and progression. Clin. Cancer Res. 2009, 15, 1140–1144. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janczi, T.; Meier, F.; Fehrl, Y.; Kinne, R.W.; Böhm, B.; Burkhardt, H. A Novel Pro-Inflammatory Mechanosensing Pathway Orchestrated by the Disintegrin Metalloproteinase ADAM15 in Synovial Fibroblasts. Cells 2021, 10, 2705. https://doi.org/10.3390/cells10102705
Janczi T, Meier F, Fehrl Y, Kinne RW, Böhm B, Burkhardt H. A Novel Pro-Inflammatory Mechanosensing Pathway Orchestrated by the Disintegrin Metalloproteinase ADAM15 in Synovial Fibroblasts. Cells. 2021; 10(10):2705. https://doi.org/10.3390/cells10102705
Chicago/Turabian StyleJanczi, Tomasz, Florian Meier, Yuliya Fehrl, Raimund W. Kinne, Beate Böhm, and Harald Burkhardt. 2021. "A Novel Pro-Inflammatory Mechanosensing Pathway Orchestrated by the Disintegrin Metalloproteinase ADAM15 in Synovial Fibroblasts" Cells 10, no. 10: 2705. https://doi.org/10.3390/cells10102705
APA StyleJanczi, T., Meier, F., Fehrl, Y., Kinne, R. W., Böhm, B., & Burkhardt, H. (2021). A Novel Pro-Inflammatory Mechanosensing Pathway Orchestrated by the Disintegrin Metalloproteinase ADAM15 in Synovial Fibroblasts. Cells, 10(10), 2705. https://doi.org/10.3390/cells10102705