
Myricetin 3-O-β-D-Galactopyranoside Exhibits Potential Anti-Osteoporotic Properties in Human Bone Marrow-Derived Mesenchymal Stromal Cells via Stimulation of Osteoblastogenesis and Suppression of Adipogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
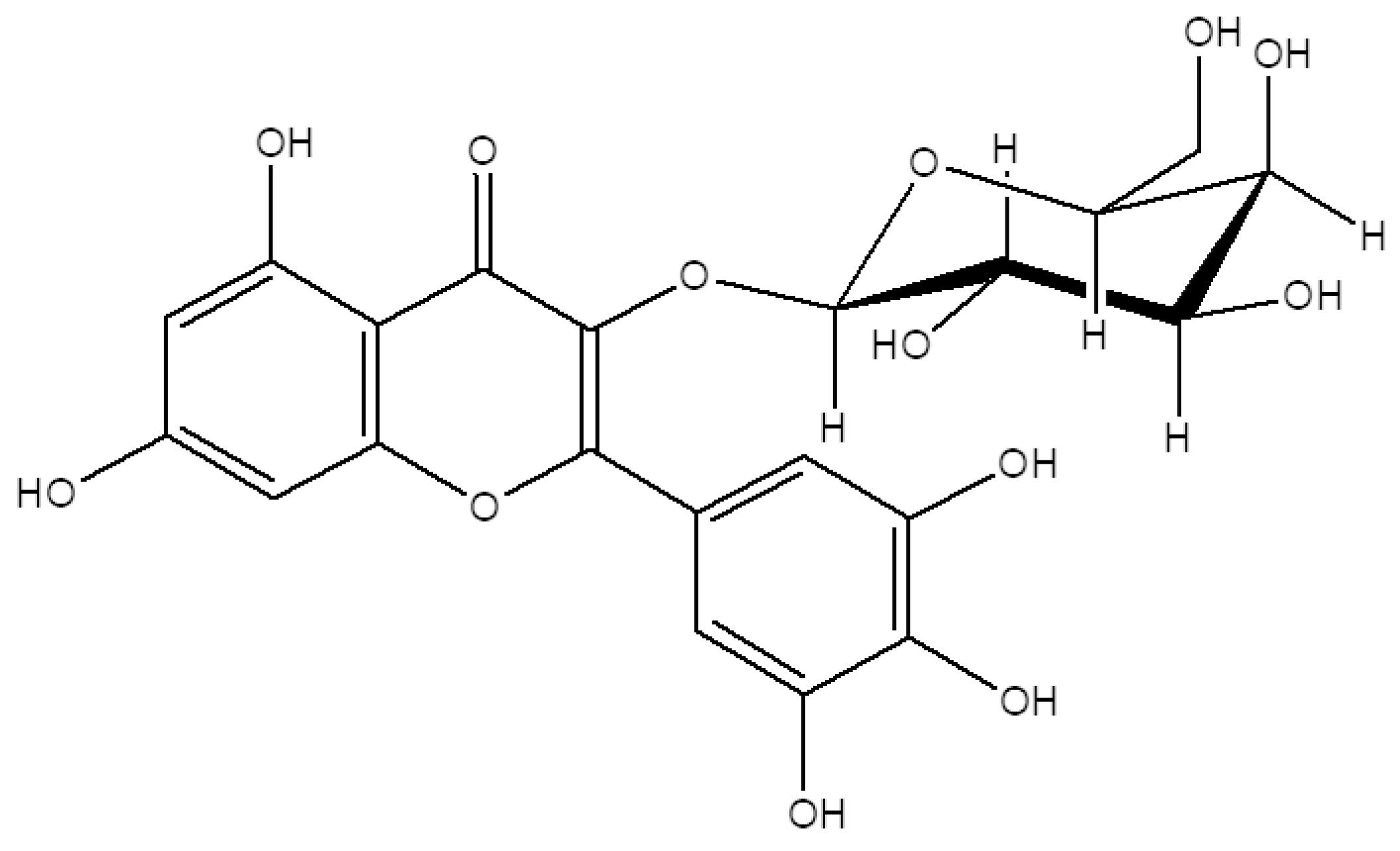
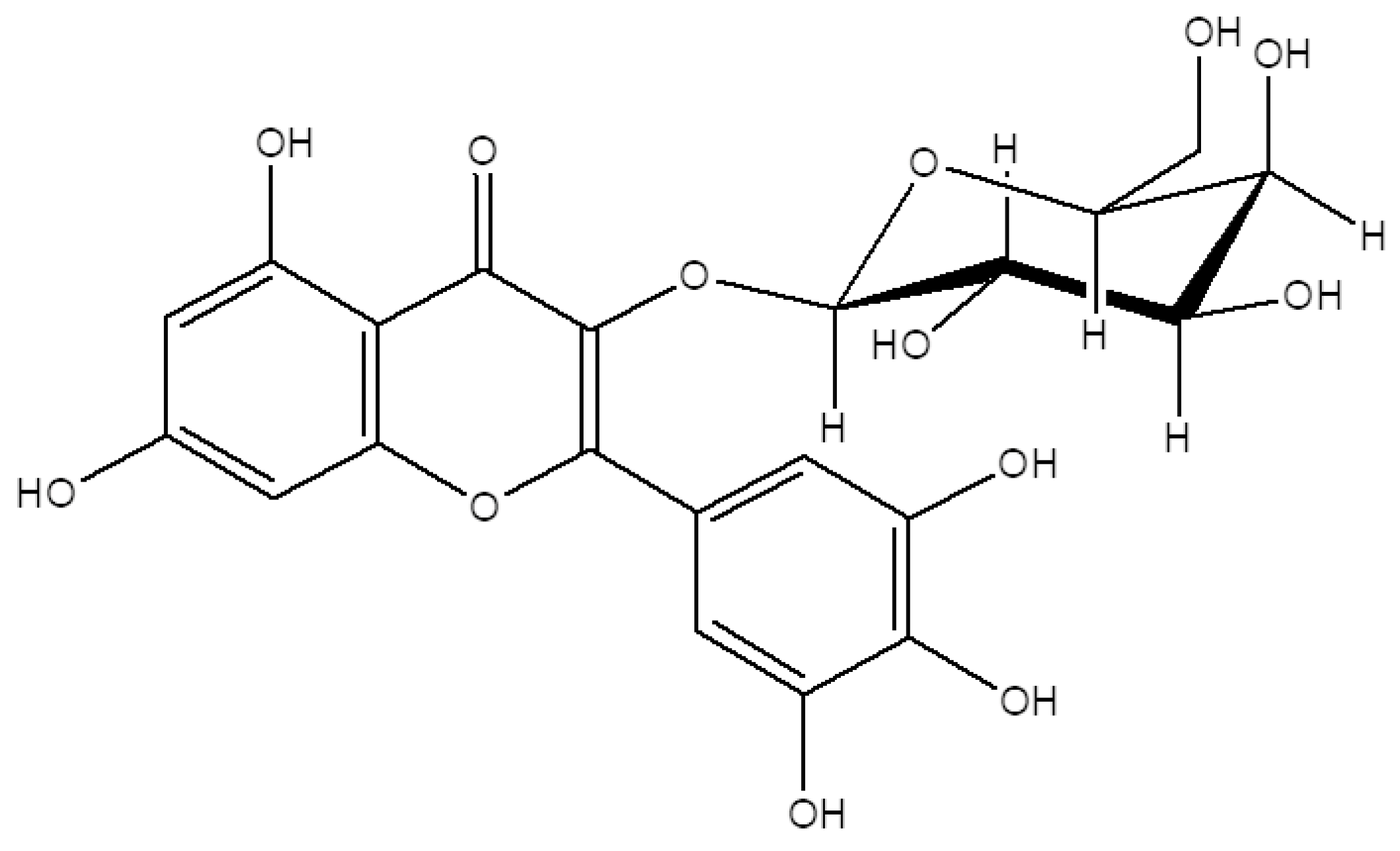
2.1. M3G Isolation and Characterization
2.2. Cell Culture and Differentiation
2.3. Cell Viability Assay
2.4. Staining of Lipid Droplets in Adipocytes by Oil Red O Staining
2.5. mRNA Expression Analysis
2.6. Protein Level Analysis
2.7. Cellular Alkaline Phosphatase (ALP) Activity
2.8. Alizarin Red Staining
2.9. Immunohistochemical Staining
2.10. Statistical Analysis
3. Results
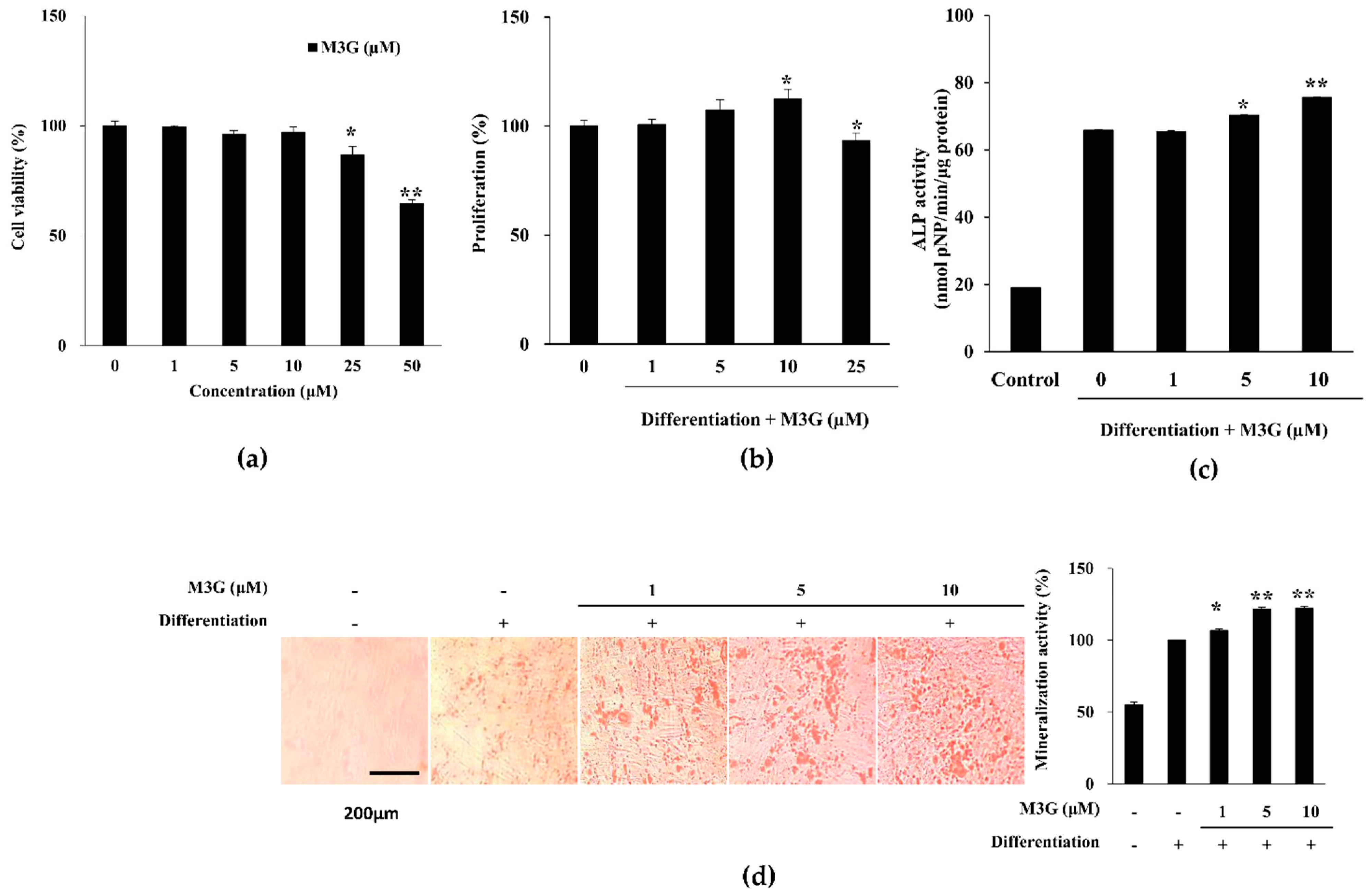
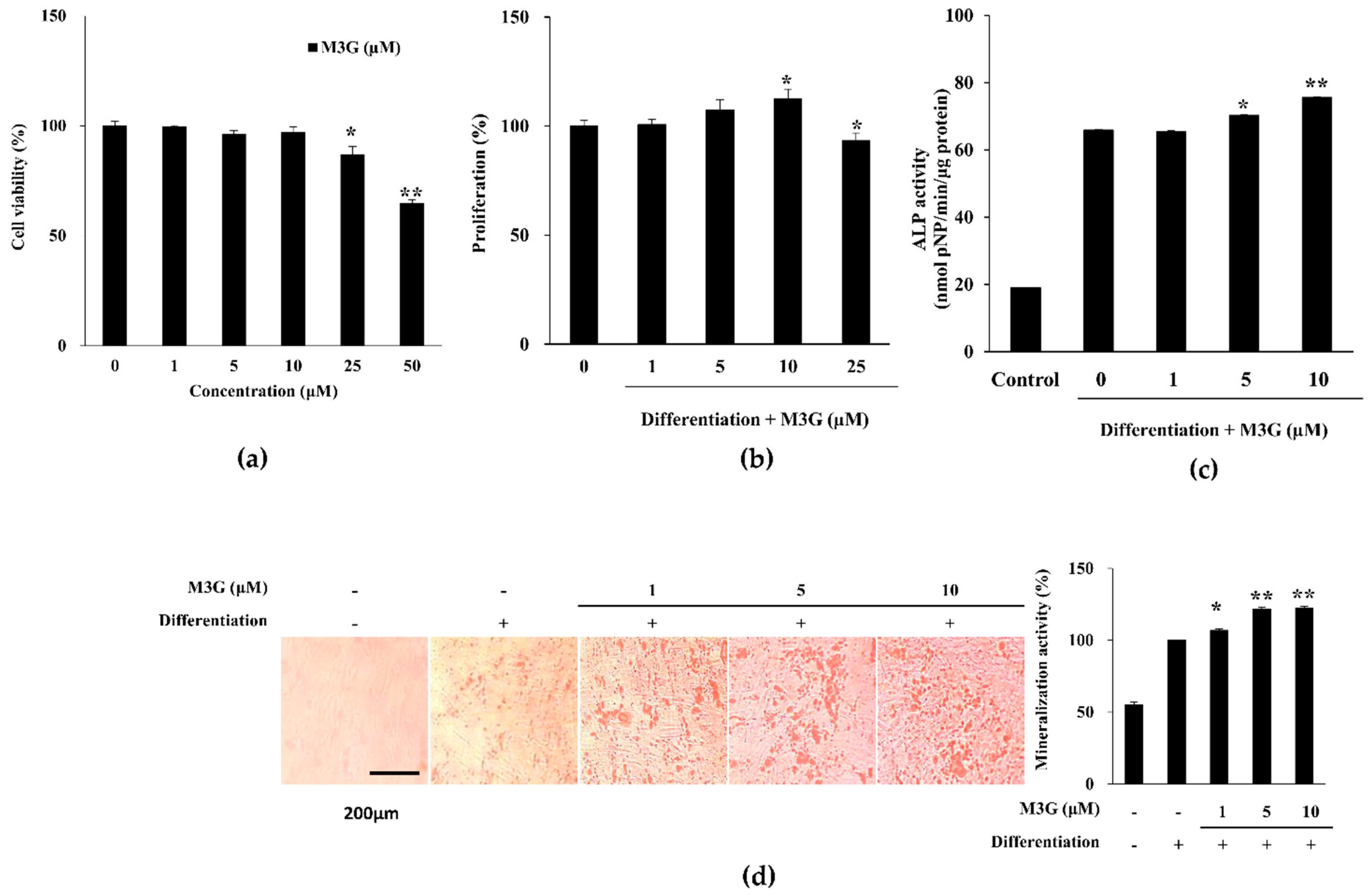
3.1. Proliferation, ALP Activity and Extracellular Mineralization of Osteo-Induced hBM-MSCs
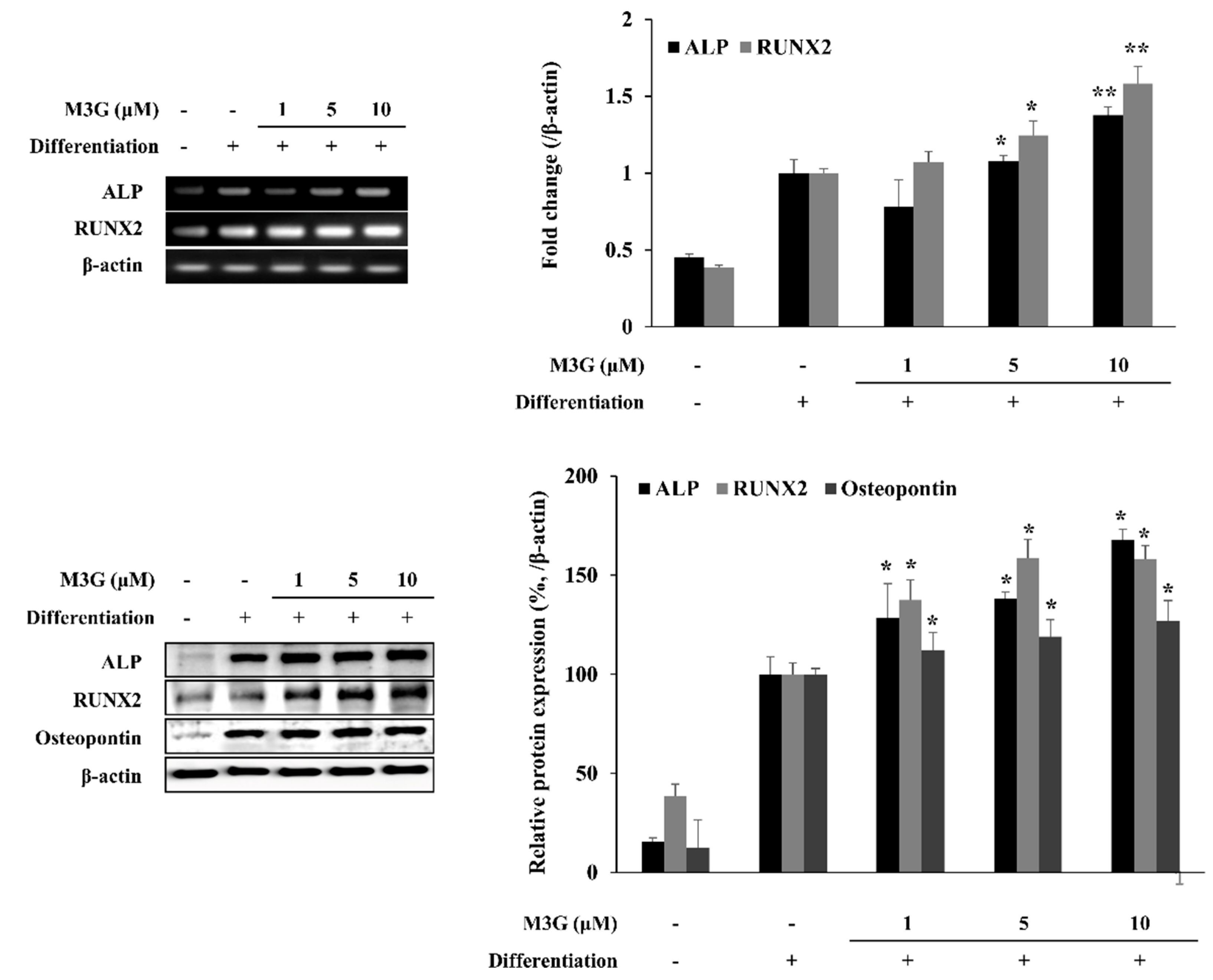
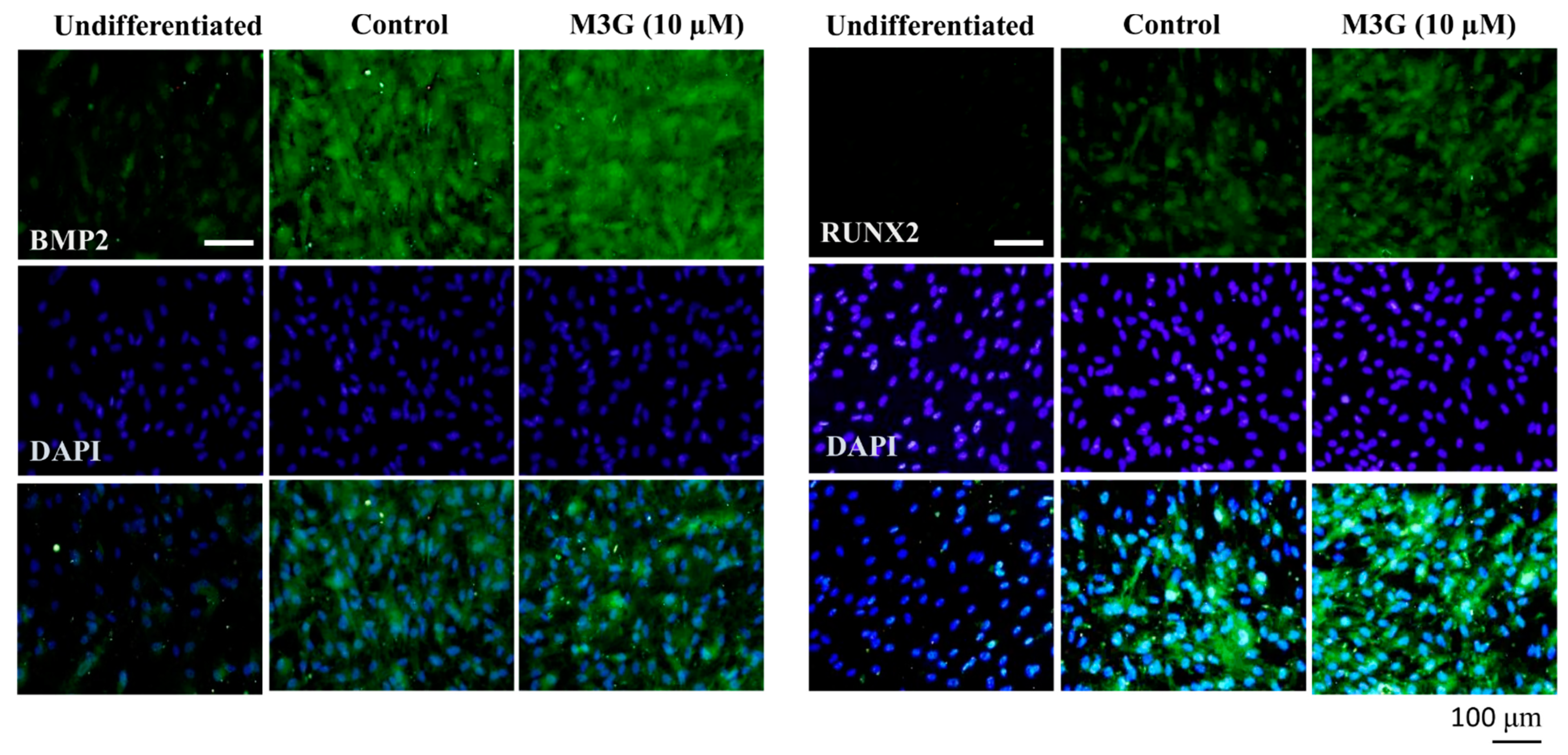
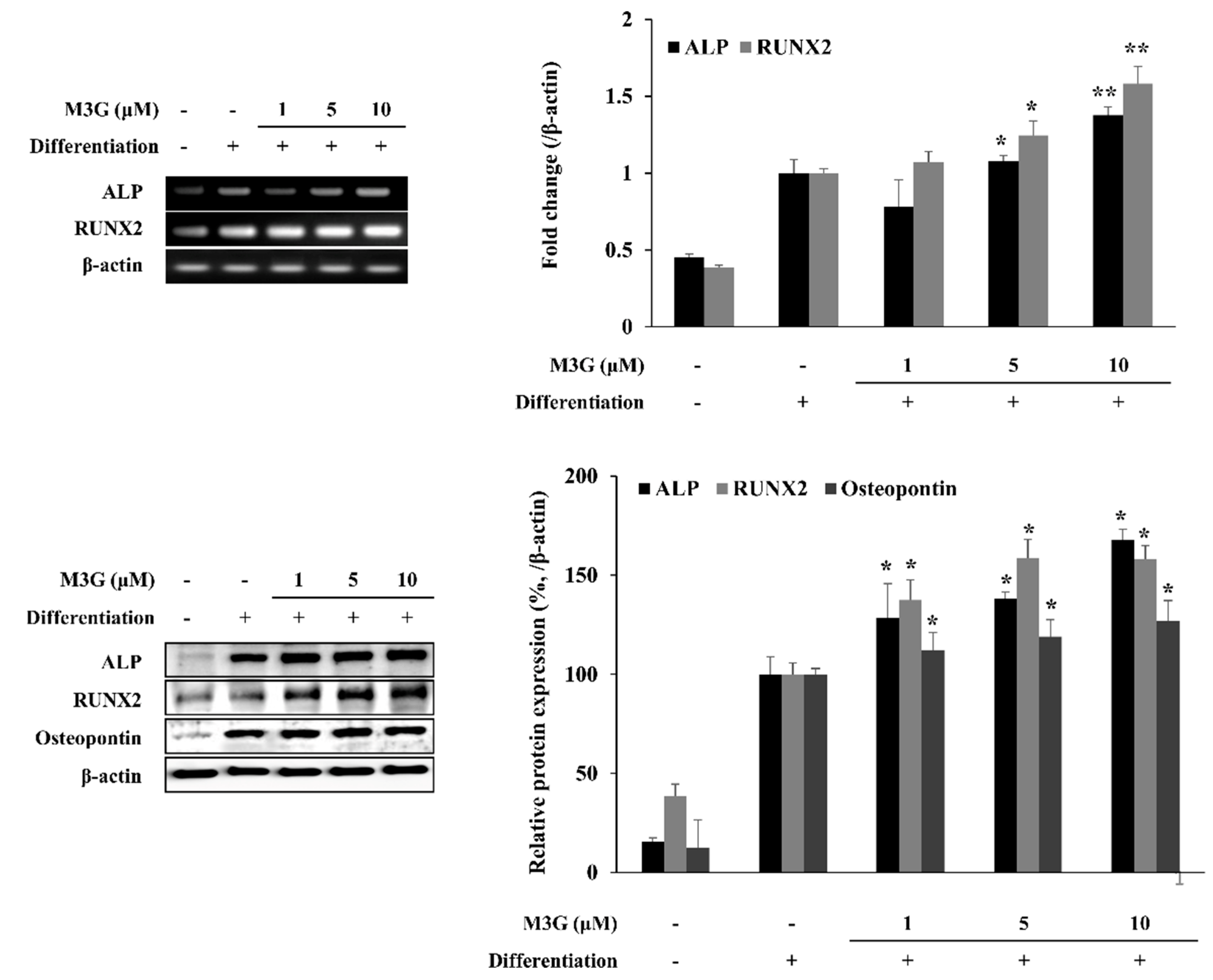
3.2. Osteoblast Marker Gene and Protein Expression
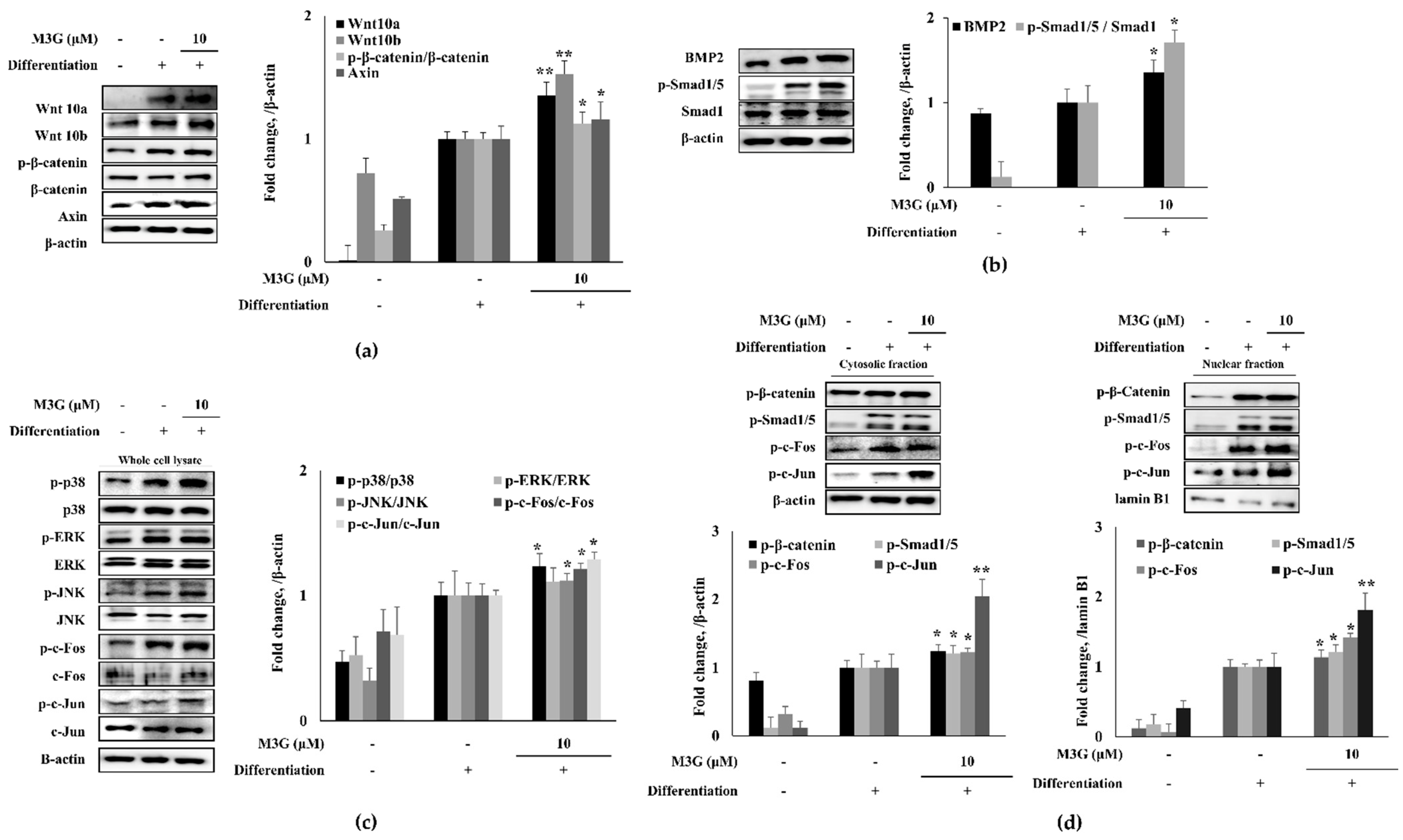
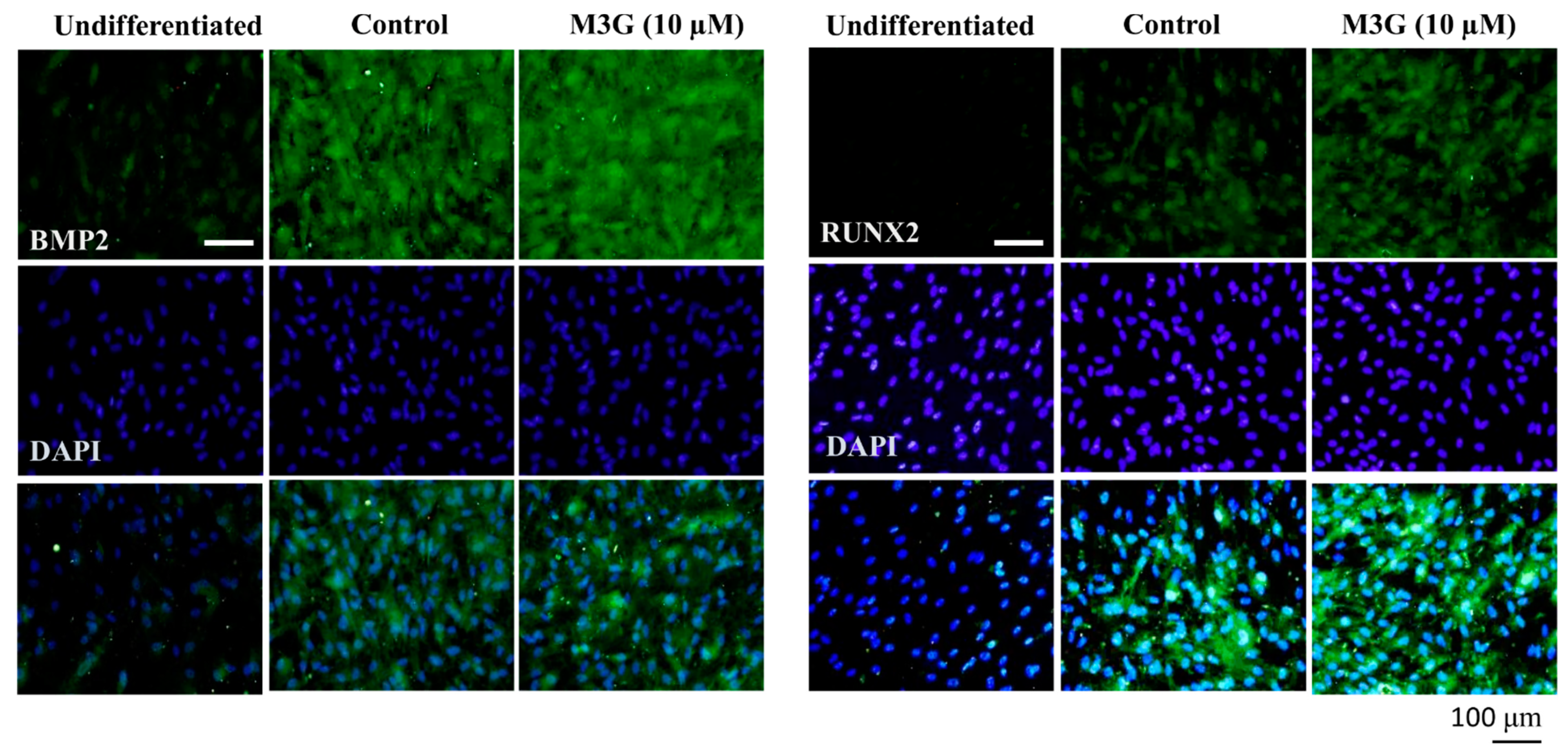
3.3. Effect of M3G on Canonical Wnt/BMP Signaling during Osteoblastogenesis
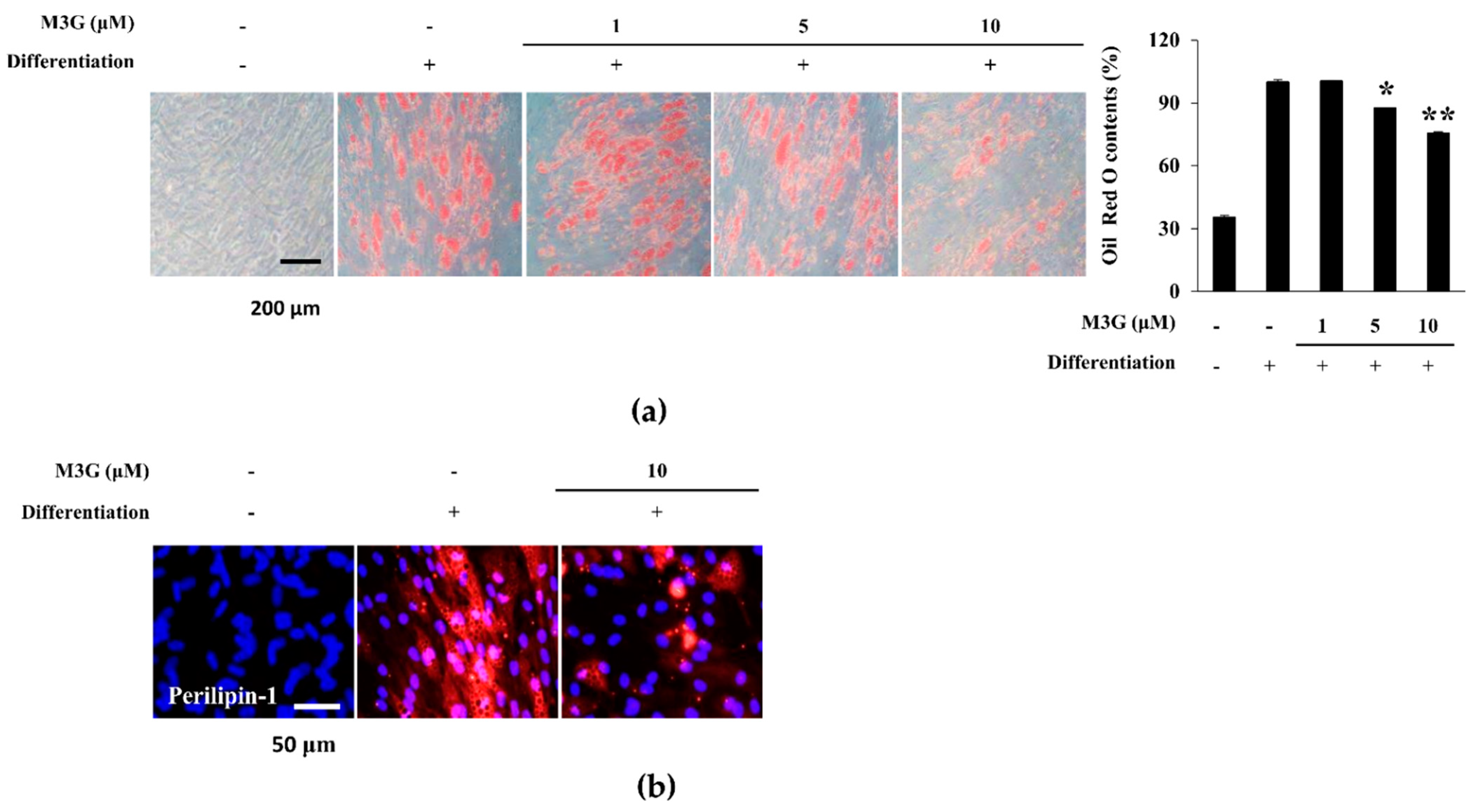
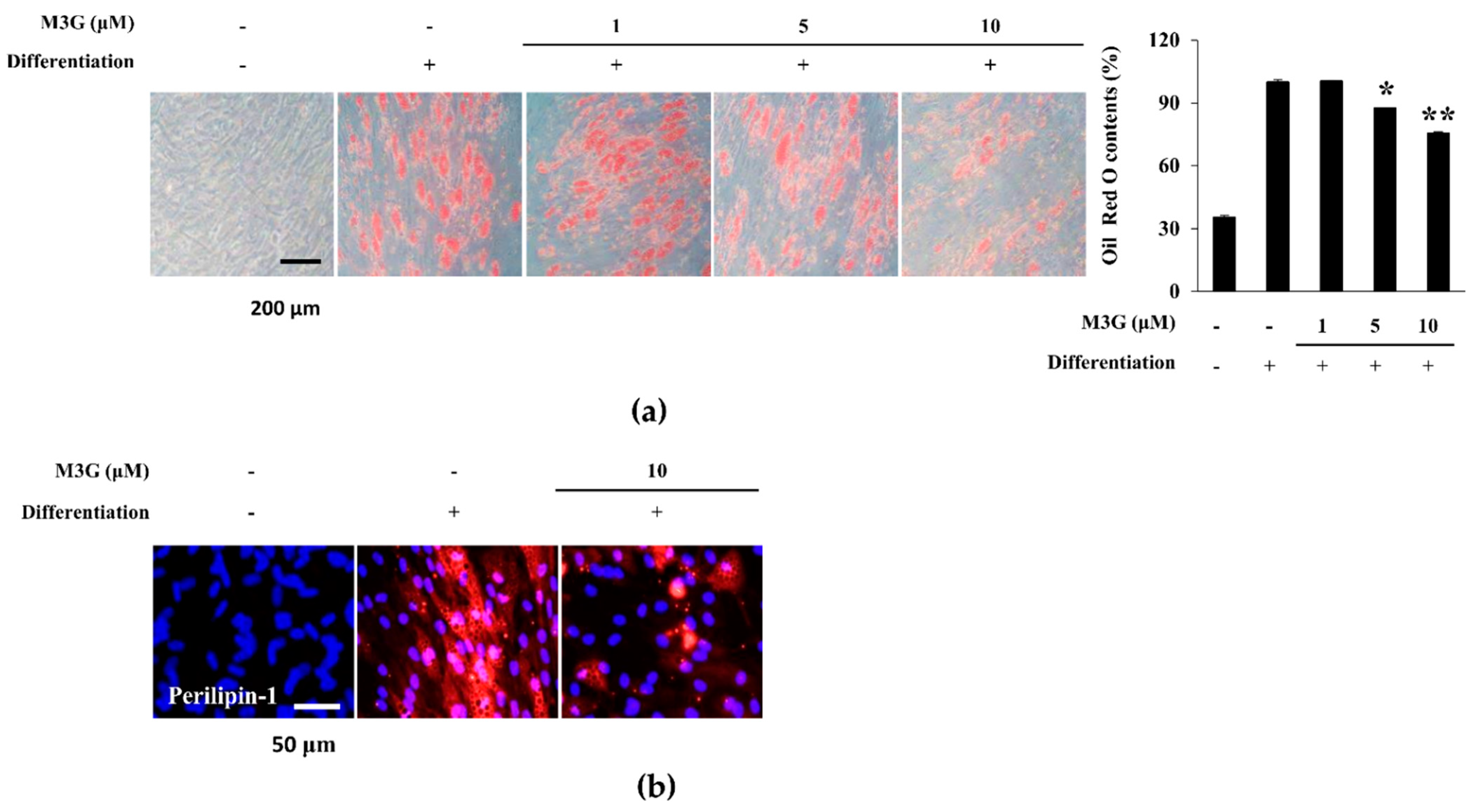
3.4. Effect of M3G on the Lipid Accumulation of hBM-MSC Adipocytes
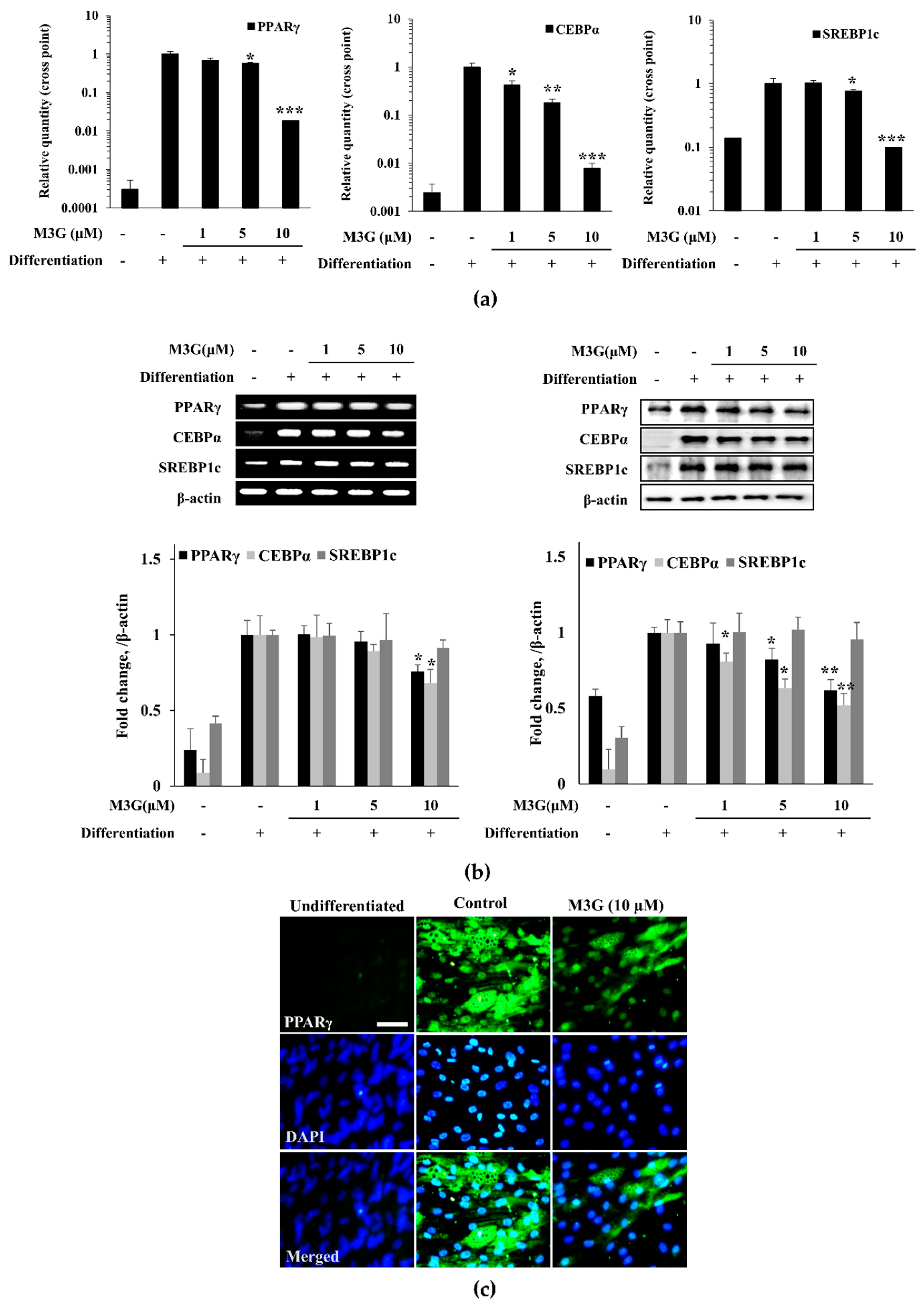
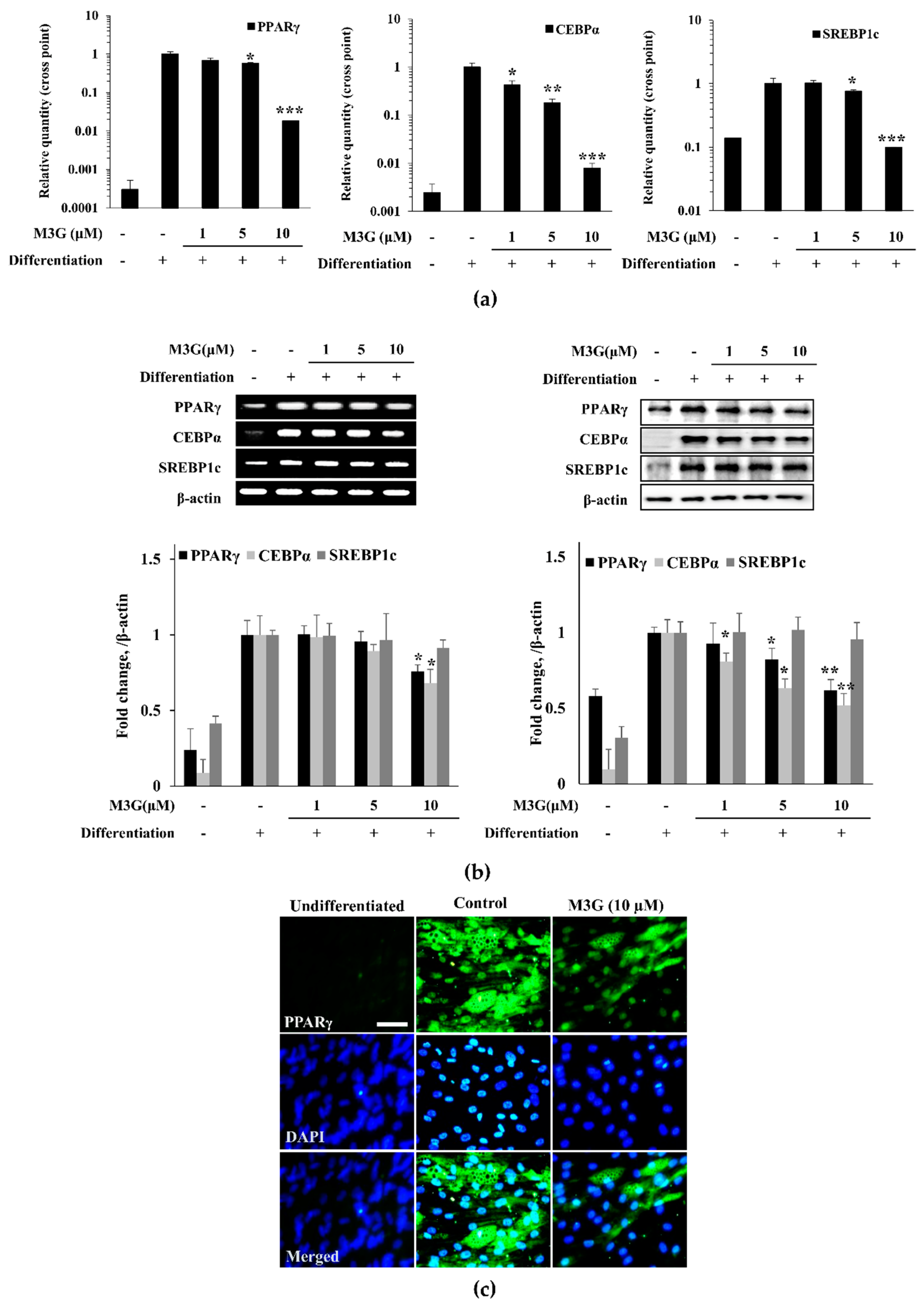
3.5. Effect of M3G on the Expression of Adipogenesis Marker Genes and Proteins
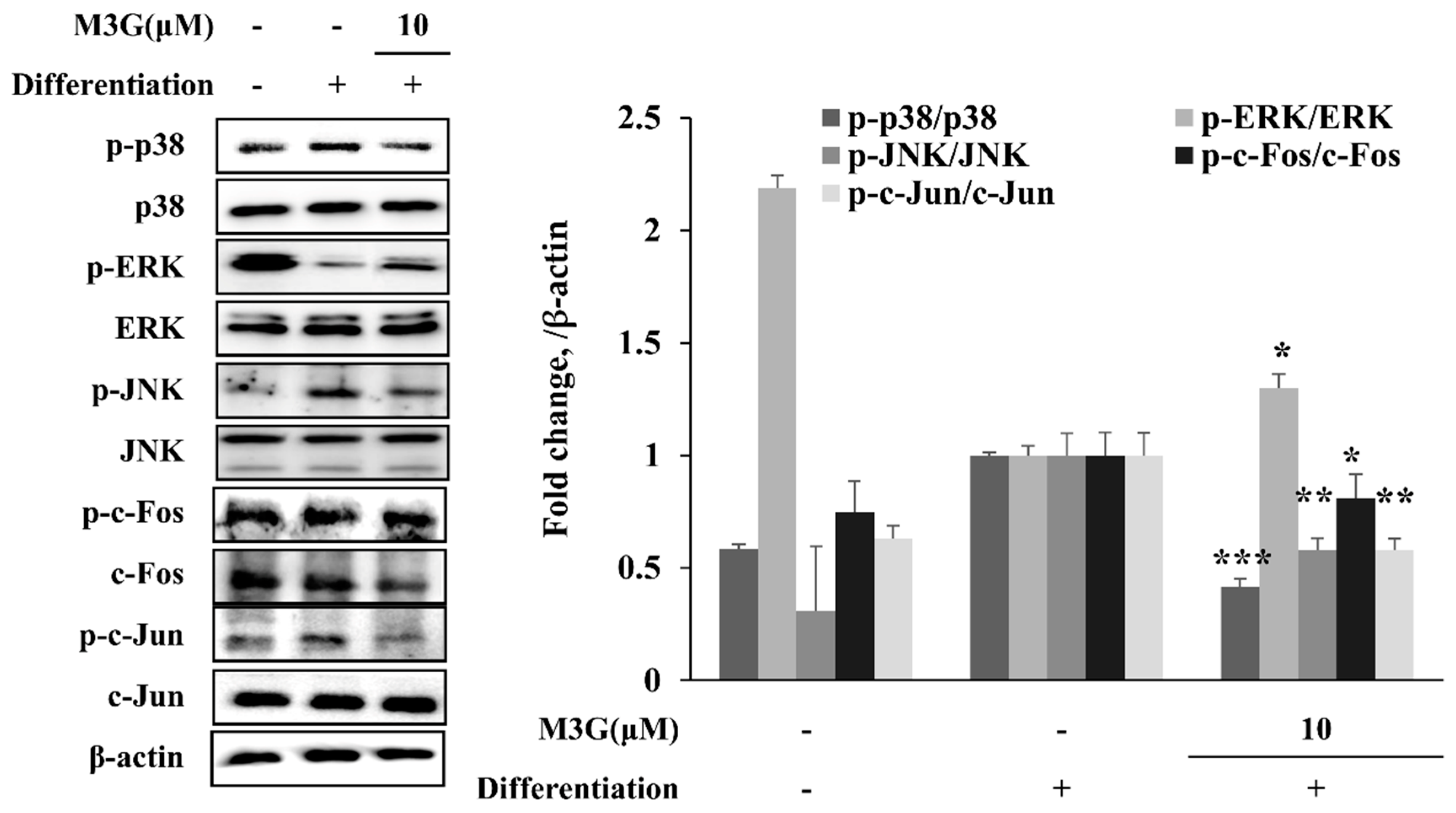
3.6. Effect of M3G on MAPK/AP-1 Signaling in Adipo-Induced hBM-MSCs
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pino, A.M.; Rosen, C.J.; Pablo Rodríguez, J. In Osteoporosis, differentiation of mesenchymal stem cells (MSCs) improves bone marrow adipogenesis. Biol. Res. 2012, 45, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Meng, H.; Wang, X.; Zhao, C.; Peng, J.; Wang, Y. Differentiation of bone marrow mesenchymal stem cells in osteoblasts and adipocytes and its role in treatment of osteoporosis. Med. Sci. Monit. 2016, 22, 226–233. [Google Scholar] [CrossRef]
- Cook, D.; Genever, P. Regulation of mesenchymal stem cell differentiation. In Transcriptional and Translational Regulation of Stem Cells; Hime, G., Abud, H., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 213–229. [Google Scholar]
- Xu, C.; Wang, J.; Zhu, T.; Shen, Y.; Tang, X.; Fang, L.; Xu, Y. Cross-talking between PPAR and WNT signaling and its regulation in mesenchymal stem cell differentiation. Curr. Stem Cell Res. Ther. 2016, 11, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Del Bo, C.; Bernardi, S.; Marino, M.; Porrini, M.; Tucci, M.; Guglielmetti, S.; Cherubini, A.; Carrieri, B.; Kirkup, B.; Kroon, P.; et al. Systematic review on polyphenol intake and health outcomes: Is there sufficient evidence to define a health-promoting polyphenol-rich dietary pattern? Nutrients 2019, 11, 1355. [Google Scholar]
- Chang, S.K.; Alasalvar, C.; Shahidi, F. Superfruits: Phytochemicals, antioxidant efficacies, and health effects—A comprehensive review. Crit. Rev. Food Sci. Nutr. 2019, 59, 1580–1604. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Kay, C.D.; Crozier, A. The bioavailability, transport, and bioactivity of dietary flavonoids: A review from a historical perspective. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1054–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Barrado, M.J.; Iglesias-Osma, M.C.; Pérez-García, E.; Carrero, S.; Blanco, E.J.; Carretero-Hernández, M.; Carretero, J. Role of flavonoids in the interactions among obesity, inflammation, and autophagy. Pharmaceuticals 2020, 13, 342. [Google Scholar] [CrossRef]
- Bellavia, D.; Dimarco, E.; Costa, V.; Carina, V.; De Luca, A.; Raimondi, L.; Fini, M.; Gentile, C.; Caradonna, F.; Giavaresi, G. Flavonoids in bone erosive diseases: Perspectives in osteoporosis treatment. Trends Endocrinol. Metab. 2021, 32, 76–94. [Google Scholar] [CrossRef]
- Song, X.; Tan, L.; Wang, M.; Ren, C.; Guo, C.; Yang, B.; Ren, Y.; Cao, Z.; Li, Y.; Pei, J. Myricetin: A review of the most recent research. Biomed. Pharmacother. 2021, 134, 111017. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Su, S.; Chen, M.; He, J.; Liu, L.; He, M.; Wang, H.; Xue, W. Synthesis and antibacterial and antiviral activities of myricetin derivatives containing a 1,2,4-triazole Schiff base. RSC Adv. 2019, 9, 23045–23052. [Google Scholar] [CrossRef] [Green Version]
- Nallappan, D.; Chua, K.H.; Ong, K.C.; Chong, C.W.; Teh, C.S.J.; Palanisamy, U.D.; Kuppusamy, U.R. Amelioration of high-fat diet-induced obesity and its associated complications by a myricetin derivative-rich fraction from Syzygium malaccense in C57BL/6J mice. Food Funct. 2021, 12, 5876–5891. [Google Scholar] [CrossRef] [PubMed]
- Büchter, C.; Ackermann, D.; Honnen, S.; Arnold, N.; Havermann, S.; Koch, K.; Wätjen, W. Methylated derivatives of myricetin enhance life span in Caenorhabditis elegans dependent on the transcription factor DAF-16. Food Funct. 2015, 6, 3383–3392. [Google Scholar] [CrossRef] [PubMed]
- Arroo, R.R.J.; Şöhretoğlu, D.; Spandidos, D.A.; Androutsopoulos, V.P. Anticancer potential of flavones. Proceedings 2017, 1, 975. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.I.; Kong, C.-S.; Jung, M.E.; Hong, J.W.; Lim, S.Y.; Seo, Y. Antioxidant activity of the halophyte Limonium tetragonum and its major active components. Biotechnol. Bioprocess Eng. 2011, 16, 992–999. [Google Scholar] [CrossRef]
- Oh, J.H.; Karadeniz, F.; Seo, Y.; Kong, C.S. Effect of quercetin 3-o-β-d-galactopyranoside on the adipogenic and osteoblastogenic differentiation of human bone marrow-derived mesenchymal stromal cells. Int. J. Mol. Sci. 2020, 21, 8044. [Google Scholar] [CrossRef]
- Taheri, Y.; Suleria, H.A.R.; Martins, N.; Sytar, O.; Beyatli, A.; Yeskaliyeva, B.; Seitimova, G.; Salehi, B.; Semwal, P.; Painuli, S.; et al. Myricetin bioactive effects: Moving from preclinical evidence to potential clinical applications. BMC Complement. Med. Ther. 2020, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Siddiqui, M.A.; Khan, M.M.; Ajmal, M.; Ahsan, R.; Rahaman, M.A.; Ahmad, M.A.; Arshad, M.; Khushtar, M. Current pharmacological trends on myricetin. Drug Res. (Stuttg.) 2020, 70, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Chen, X.; Feng, Y.; Xu, H.Z.; Chen, H.; Yu, K.; Cheng, S.; Peng, L. Myricetin enhances osteogenic differentiation through the activation of canonical Wnt/β-catenin signaling in human bone marrow stromal cells. Eur. J. Pharmacol. 2014, 738, 22–30. [Google Scholar] [CrossRef]
- Ying, X.; Chen, X.; Wang, T.; Zheng, W.; Chen, L.; Xu, Y. Possible osteoprotective effects of myricetin in STZ induced diabetic osteoporosis in rats. Eur. J. Pharmacol. 2020, 866, 172805. [Google Scholar] [CrossRef]
- Bin, H.-S.; Choi, U.-K. Myricetin inhibits adipogenesis in human adipose tissue-derived mesenchymal stem cells. Food Sci. Biotechnol. 2012, 21, 1391–1396. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Chang, J.K.; Tsai, C.H.; Chien, T.T.C.; Kuo, P.L. Myricetin induces human osteoblast differentiation through bone morphogenetic protein-2/p38 mitogen-activated protein kinase pathway. Biochem. Pharmacol. 2007, 73, 504–514. [Google Scholar] [CrossRef]
- Kim, H.Y.; Park, S.Y.; Choung, S.Y. Enhancing effects of myricetin on the osteogenic differentiation of human periodontal ligament stem cells via BMP-2/Smad and ERK/JNK/p38 mitogen-activated protein kinase signaling pathway. Eur. J. Pharmacol. 2018, 834, 84–91. [Google Scholar] [CrossRef]
- Colaianni, G.; Brunetti, G.; Faienza, M.F.; Colucci, S.; Grano, M. Osteoporosis and obesity: Role of Wnt pathway in human and murine models. World J. Orthop. 2014, 5, 242–246. [Google Scholar] [CrossRef]
- He, H.P.; Gu, S. The PPAR-γ/SFRP5/Wnt/β-catenin signal axis regulates the dexamethasone-induced osteoporosis. Cytokine 2021, 143, 155488. [Google Scholar] [CrossRef]
- Cawthorn, W.P.; Bree, A.J.; Yao, Y.; Du, B.; Hemati, N.; Martinez-Santibañez, G.; MacDougald, O.A. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β-catenin-dependent mechanism. Bone 2012, 50, 477–489. [Google Scholar] [CrossRef] [Green Version]
- Mbalaviele, G.; Sheikh, S.; Stains, J.P.; Salazar, V.S.; Cheng, S.-L.; Chen, D.; Civitelli, R. β-Catenin and BMP-2 synergize to promote osteoblast differentiation and new bone formation. J. Cell. Biochem. 2005, 94, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawadi, G.; Vayssière, B.; Dunn, F.; Baron, R.; Roman-Roman, S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Miner. Res. 2003, 18, 1842–1853. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Yin, C.; Zhao, F.; Ali, A.; Ma, J.; Qian, A. Mesenchymal stem cells: Cell fate decision to osteoblast or adipocyte and application in osteoporosis treatment. Int. J. Mol. Sci. 2018, 19, 360. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, X.; Lu, L.; Yu, X. The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. 2020, 52, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Payne, V.A.; Au, W.-S.; Lowe, C.E.; Rahman, S.M.; Friedman, J.E.; O’Rahilly, S.; Rochford, J.J. C/EBP transcription factors regulate SREBP1c gene expression during adipogenesis. Biochem. J. 2010, 425, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Chen, H.; Su, H.; Wu, X.; Xie, Z.; Wu, Y.; Shen, H. IL6 receptor facilitates adipogenesis differentiation of human mesenchymal stem cells through activating p38 pathway. Int. J. Stem Cells 2020, 13, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Luther, J.; Ubieta, K.; Hannemann, N.; Jimenez, M.; Garcia, M.; Zech, C.; Schett, G.; Wagner, E.F.; Bozec, A. Fra-2/AP-1 controls adipocyte differentiation and survival by regulating PPARγ and hypoxia. Cell Death Differ. 2014, 21, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, Y.; Koga, M.; Saito, M.; Matsunaga, Y.; Nakayama, K. Inhibition of adipocyte differentiation by mechanical stretching through ERK-mediated downregulation of PPARγ2. J. Cell Sci. 2004, 117, 3605–3614. [Google Scholar] [CrossRef] [Green Version]
- Aranaz, P.; Navarro-Herrera, D.; Zabala, M.; Miguéliz, I.; Romo-Hualde, A.; López-Yoldi, M.; Alfredo Martínez, J.; Vizmanos, J.L.; Milagro, F.I.; González-Navarro, C.J. Phenolic compounds inhibit 3T3-L1 adipogenesis depending on the stage of differentiation and their binding affinity to PPARγ. Molecules 2019, 24, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.K.; Dayem, A.A.; Han, J.; Yin, Y.; Kim, K.; Saha, S.K.; Yang, G.M.; Choi, H.Y.; Cho, S.G. Molecular mechanisms of the anti-obesity and anti-diabetic properties of flavonoids. Int. J. Mol. Sci. 2016, 17, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossini, M.; Gatti, D.; Adami, S. Involvement of WNT/β-catenin signaling in the treatment of osteoporosis. Calcif. Tissue Int. 2013, 93, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Liao, L.; An, Y.; Su, X.; Liu, S.; Shuai, Y.; Zhang, X.; Jin, Y. Suppression of EZH2 prevents the shift of osteoporotic MSC fate to adipocyte and enhances bone formation during osteoporosis. Mol. Ther. 2016, 24, 217–229. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Gao, X.; Chen, P.; Li, X. Myricetin ameliorates glucocorticoid-induced osteoporosis through the ERK signaling pathway. Life Sci. 2018, 207, 205–211. [Google Scholar] [CrossRef]
- Su, H.; Feng, L.; Zheng, X.; Chen, W. Myricetin protects against diet-induced obesity and ameliorates oxidative stress in C57BL/6 mice. J. Zhejiang Univ.—Sci. B 2016, 17, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, G.; Gong, L.; Sun, G.; Shi, B.; Bao, H.; Duan, Y. Isopsoralen regulates PPAR-γ/WNT to inhibit oxidative stress in osteoporosis. Mol. Med. Rep. 2017, 17, 1125–1131. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karadeniz, F.; Oh, J.H.; Jo, H.J.; Seo, Y.; Kong, C.-S. Myricetin 3-O-β-D-Galactopyranoside Exhibits Potential Anti-Osteoporotic Properties in Human Bone Marrow-Derived Mesenchymal Stromal Cells via Stimulation of Osteoblastogenesis and Suppression of Adipogenesis. Cells 2021, 10, 2690. https://doi.org/10.3390/cells10102690
Karadeniz F, Oh JH, Jo HJ, Seo Y, Kong C-S. Myricetin 3-O-β-D-Galactopyranoside Exhibits Potential Anti-Osteoporotic Properties in Human Bone Marrow-Derived Mesenchymal Stromal Cells via Stimulation of Osteoblastogenesis and Suppression of Adipogenesis. Cells. 2021; 10(10):2690. https://doi.org/10.3390/cells10102690
Chicago/Turabian StyleKaradeniz, Fatih, Jung Hwan Oh, Hyun Jin Jo, Youngwan Seo, and Chang-Suk Kong. 2021. "Myricetin 3-O-β-D-Galactopyranoside Exhibits Potential Anti-Osteoporotic Properties in Human Bone Marrow-Derived Mesenchymal Stromal Cells via Stimulation of Osteoblastogenesis and Suppression of Adipogenesis" Cells 10, no. 10: 2690. https://doi.org/10.3390/cells10102690
APA StyleKaradeniz, F., Oh, J. H., Jo, H. J., Seo, Y., & Kong, C.-S. (2021). Myricetin 3-O-β-D-Galactopyranoside Exhibits Potential Anti-Osteoporotic Properties in Human Bone Marrow-Derived Mesenchymal Stromal Cells via Stimulation of Osteoblastogenesis and Suppression of Adipogenesis. Cells, 10(10), 2690. https://doi.org/10.3390/cells10102690