
The Inhibitory Receptor GPR56 (Adgrg1) Is Specifically Expressed by Tissue-Resident Memory T Cells in Mice But Dispensable for Their Differentiation and Function In Vivo



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Mice
2.2. Assessment of CRE Recombinase Activity at Adgrg1 Locus
2.3. Lymphocytic Choriomeningitis Virus (LCMV) and Listeria Monocytogenes Bacterial Infection
2.4. Tissue Preparation and Flow-Cytometric Analysis
2.5. In Vitro CD8 T Cell Stimulation
2.6. Quantitative RT-PCR
2.7. Statistical Analysis
3. Results
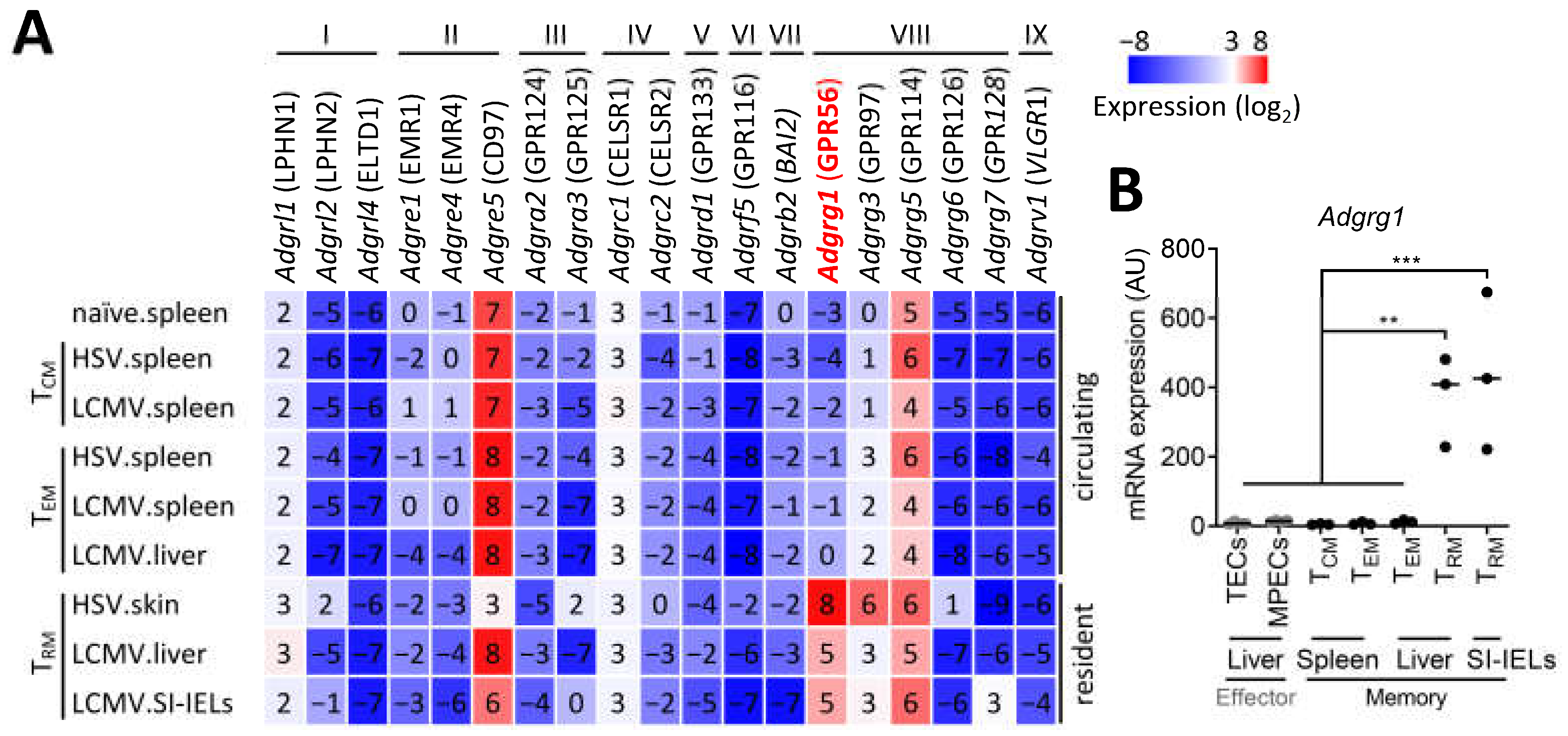
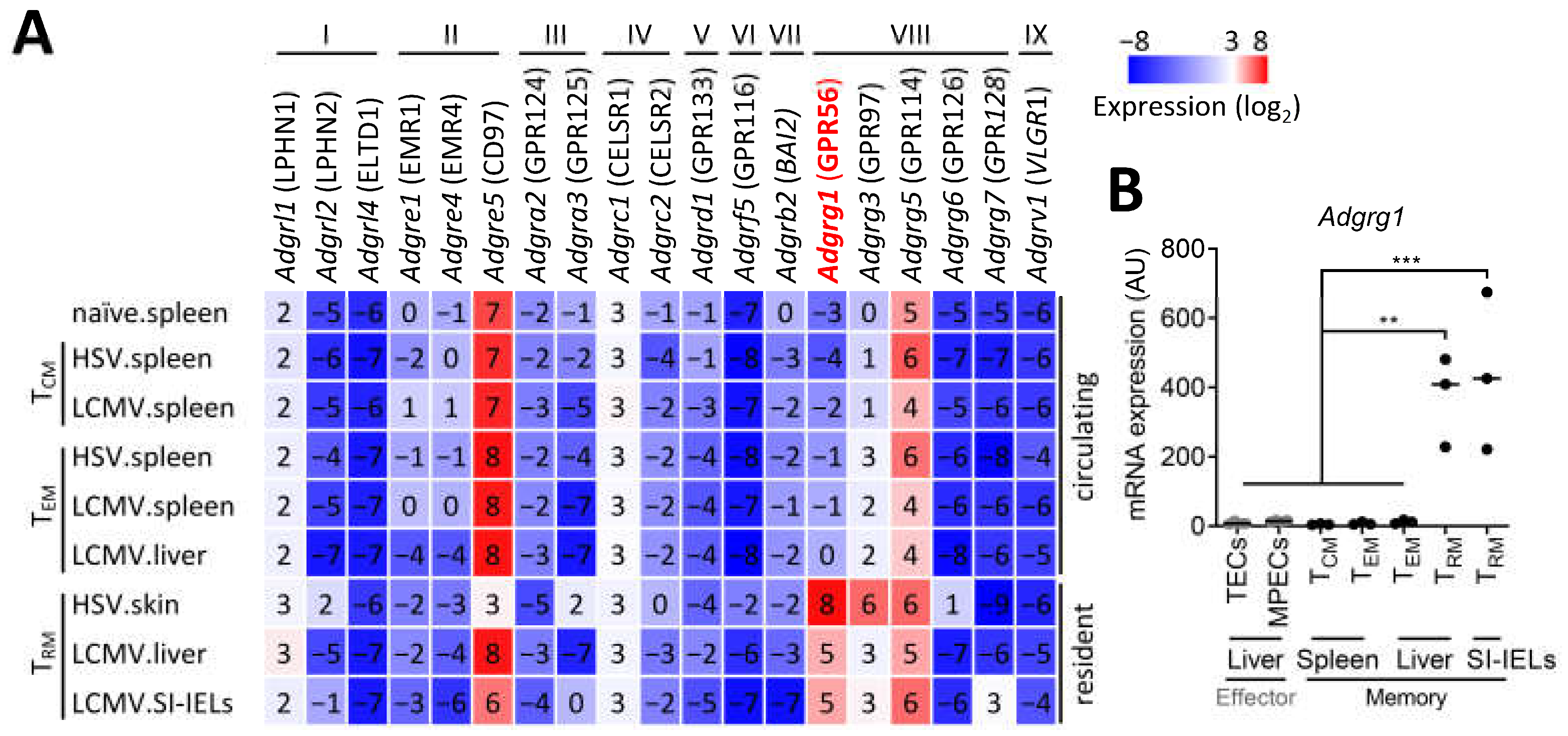
3.1. CD8+ TRM Cells Specifically Upregulate Adgrg1/GPR56
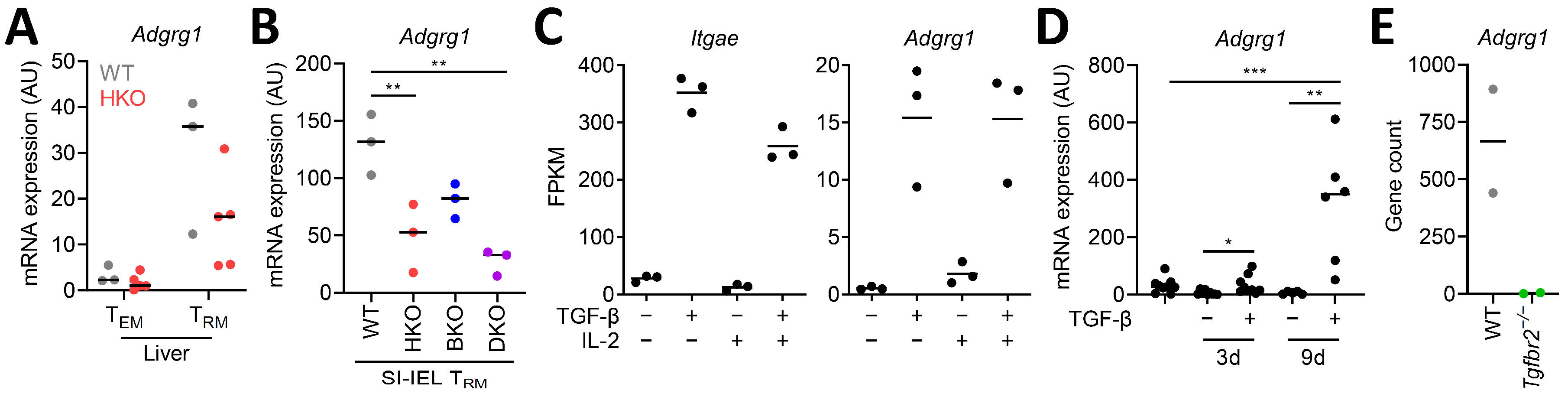
3.2. CD8+ TRM Cell-Inducing Factors Regulate Adgrg1/GPR56 Expression
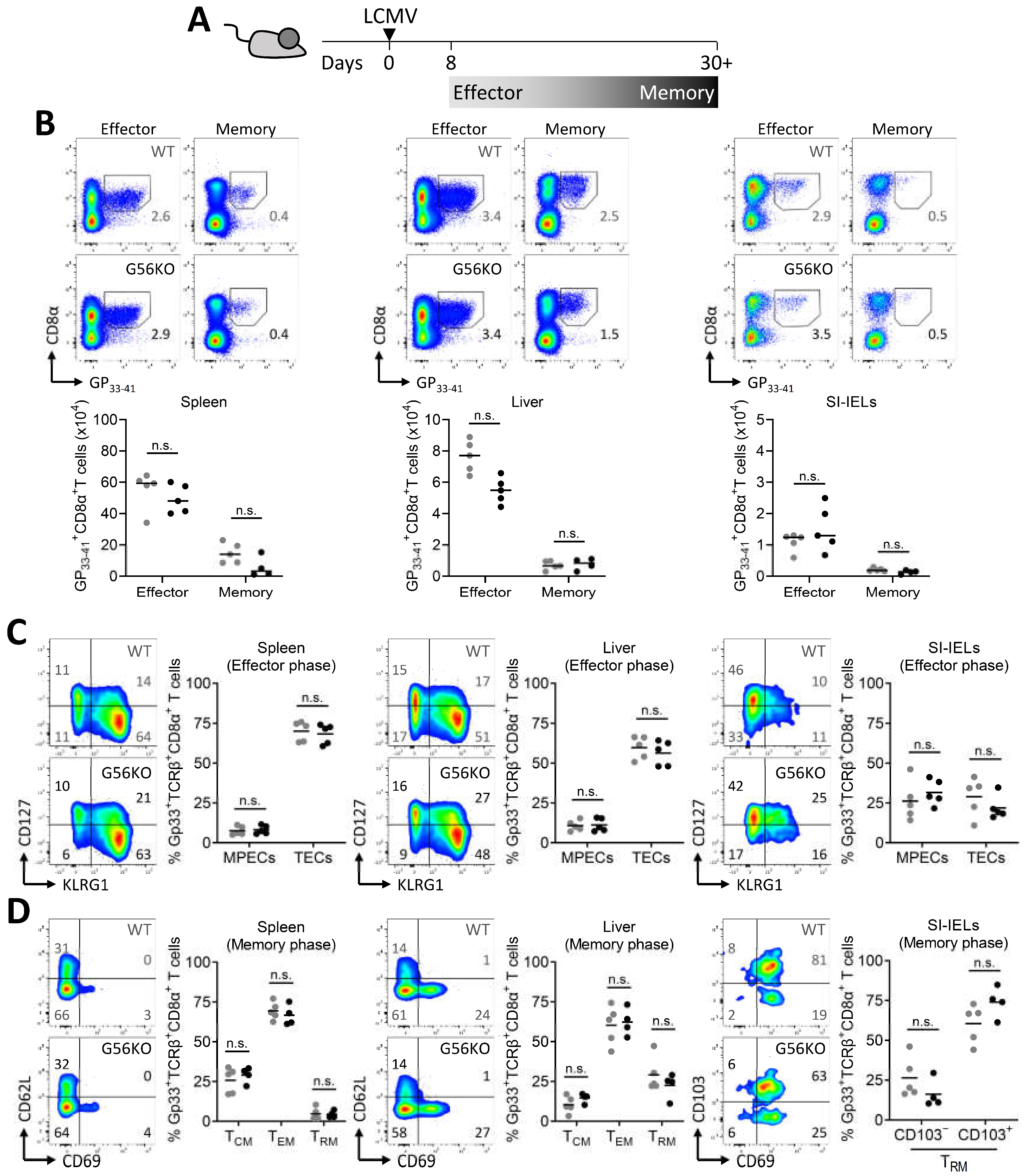
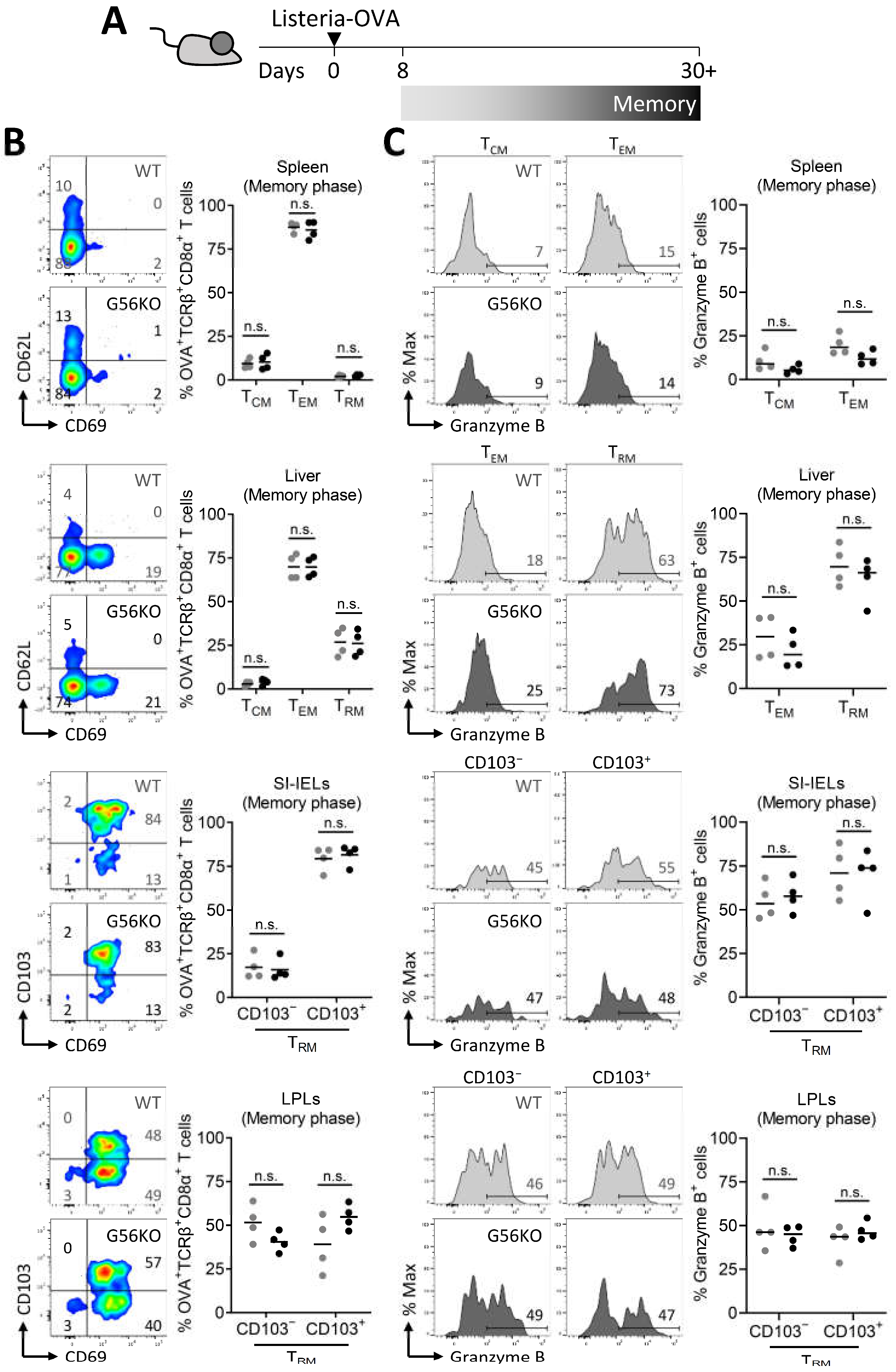
3.3. Adgrg1/GPR56 Is Dispensable for the Development of CD8+ TRM Cells
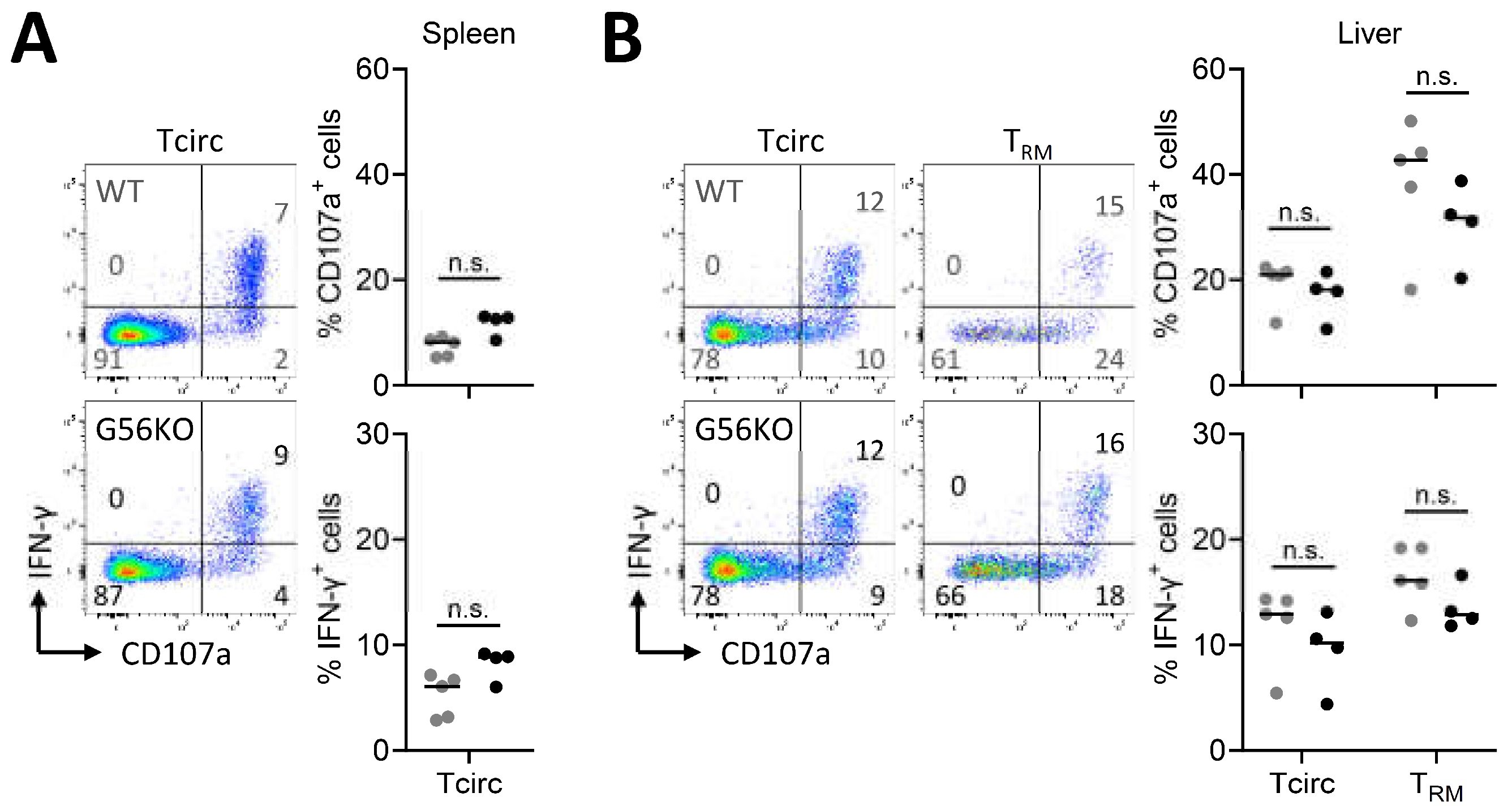
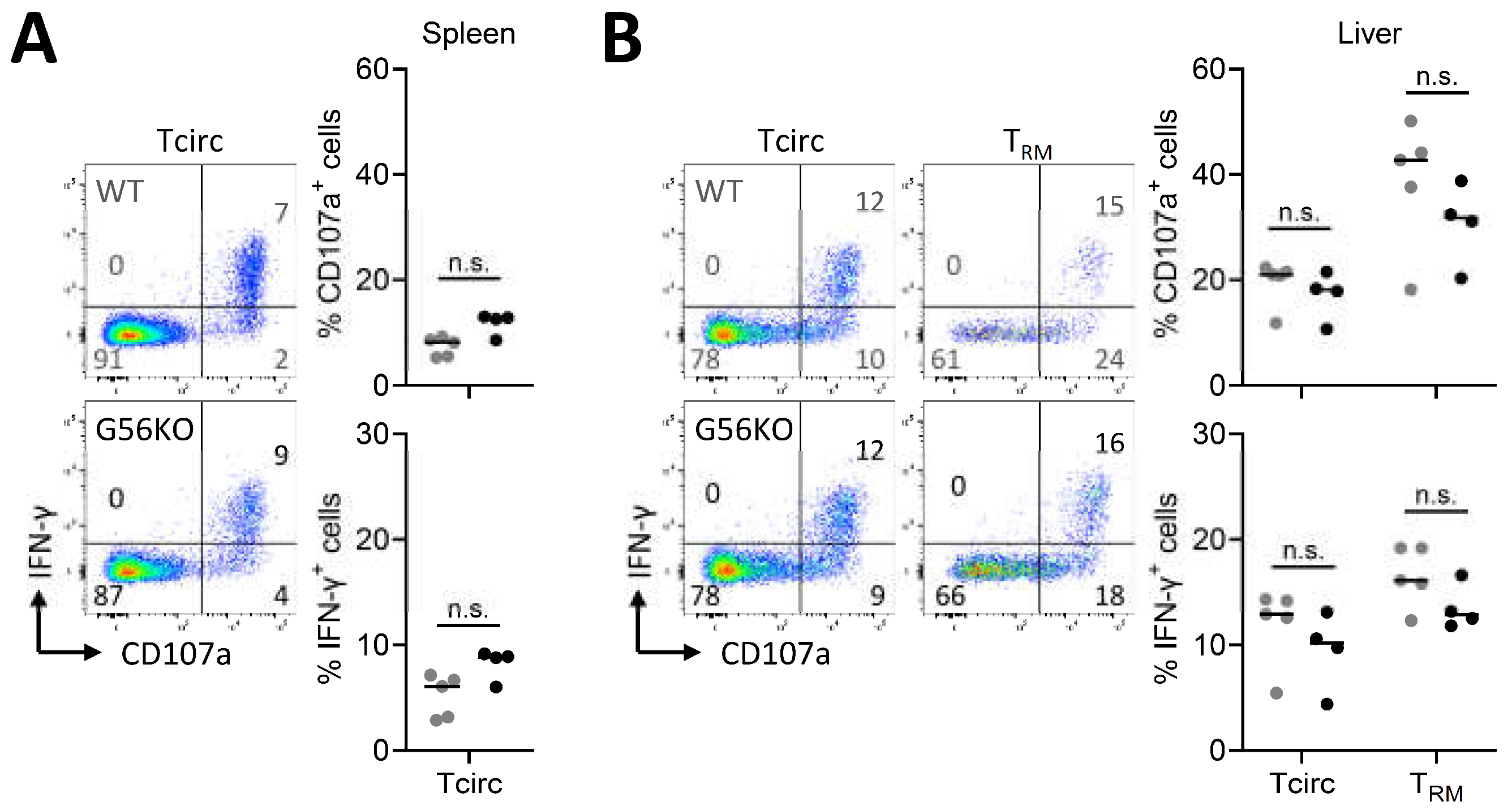
3.4. Adgrg1/GPR56 Does Not Regulate Cytokine Production and Release of CD8+ TRM Cells
3.5. Adgrg1/GPR56 Does Not Regulate Cytotoxic Function of CD8+ TRM Cells
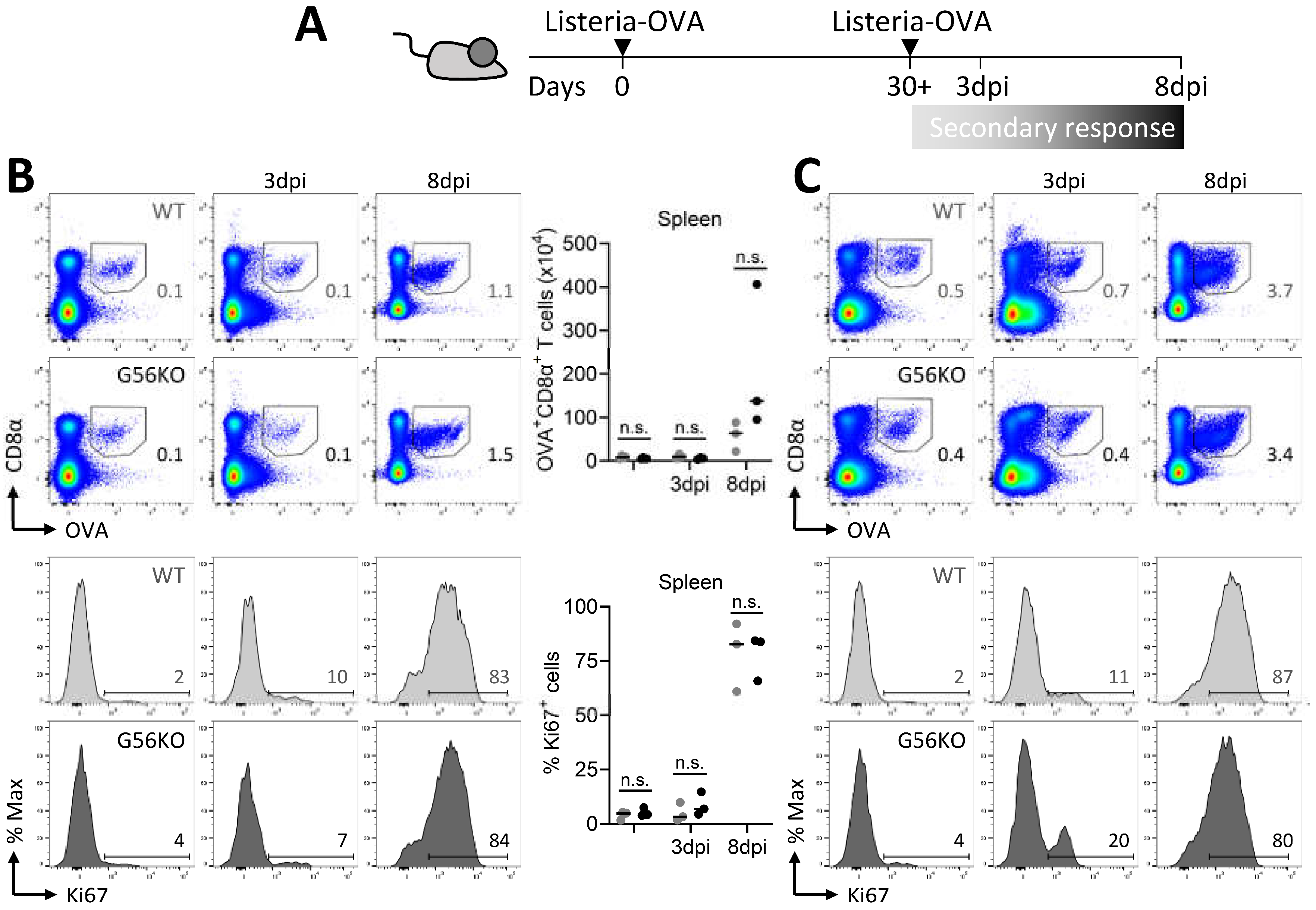
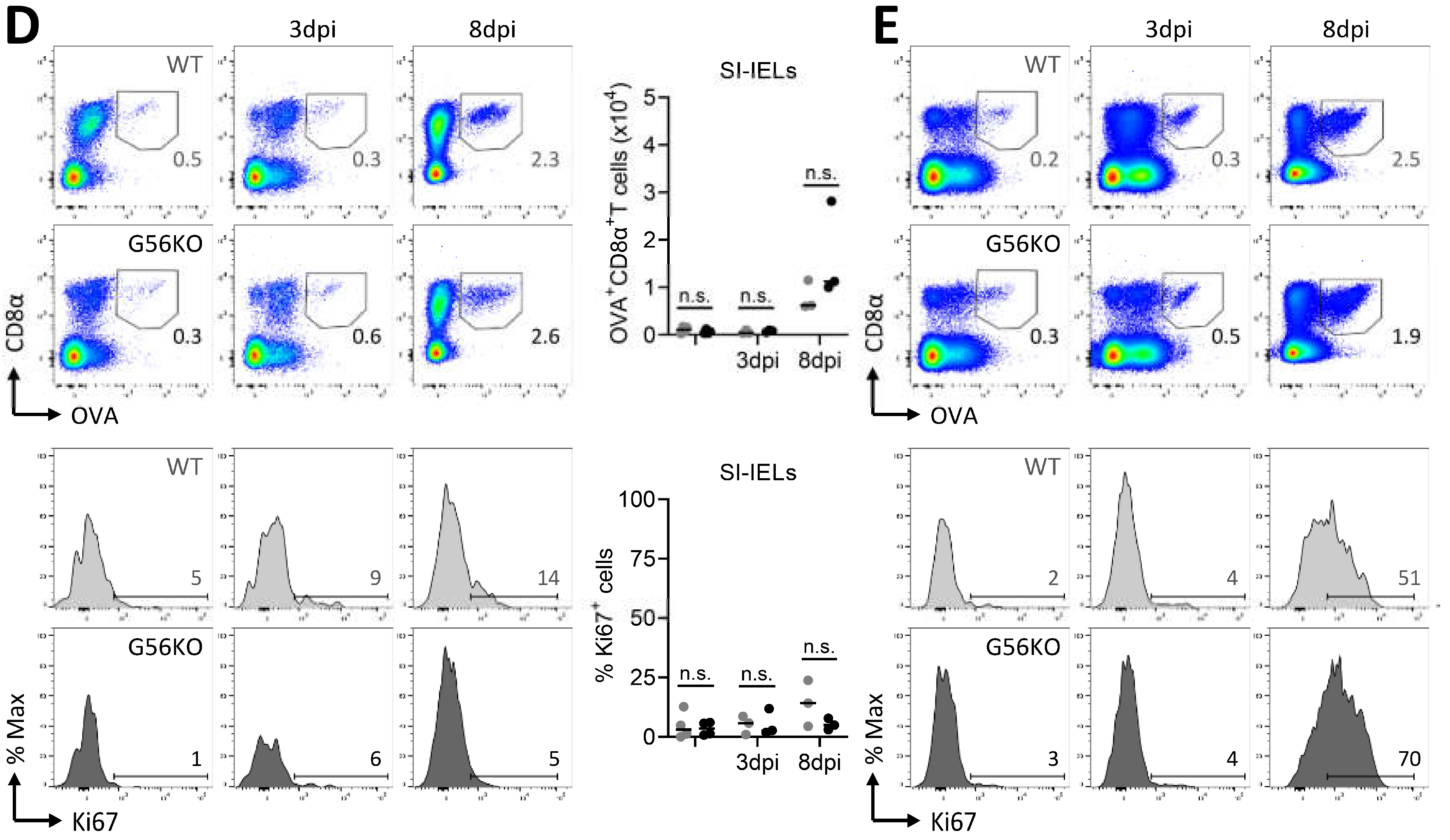
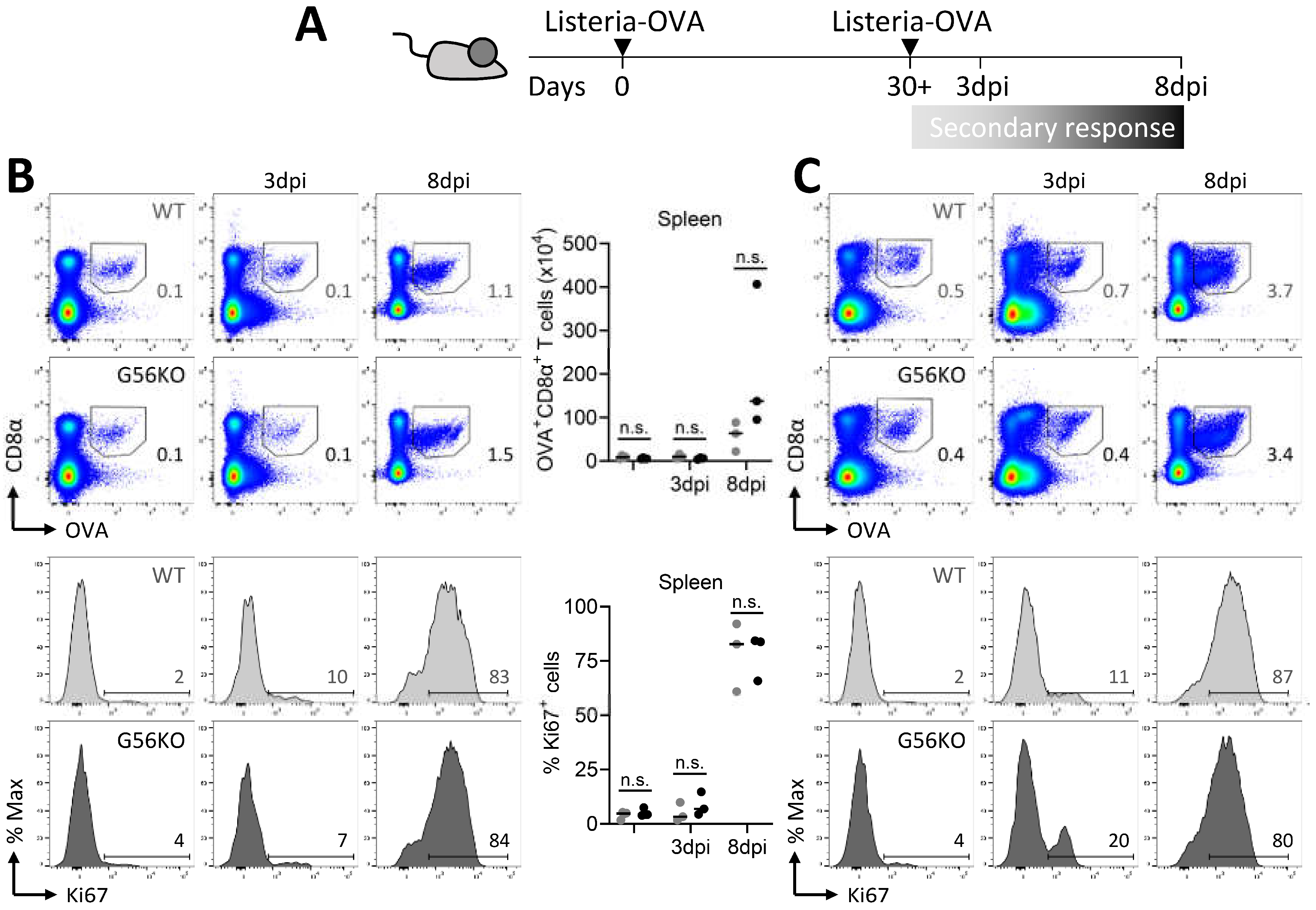
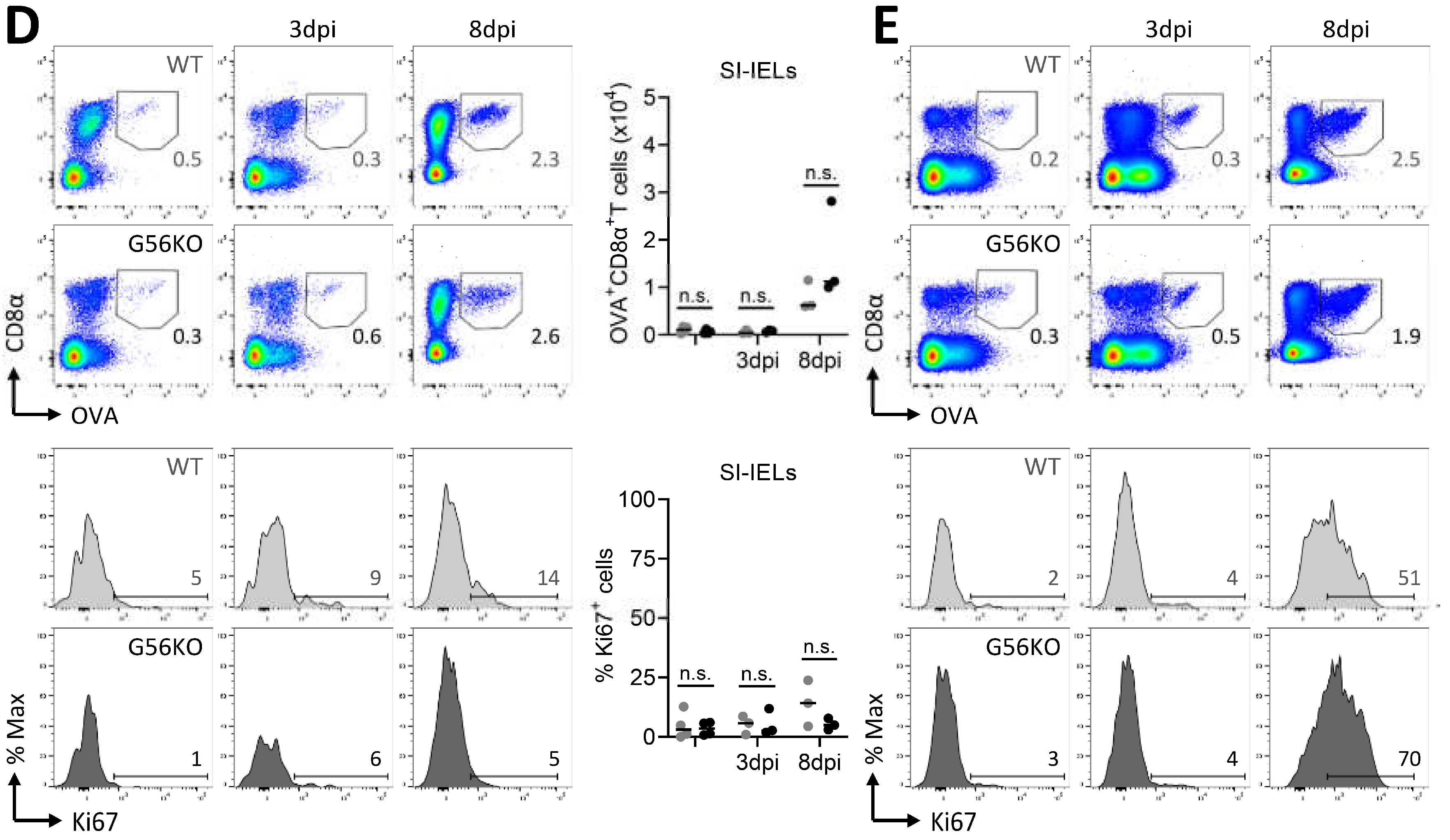
3.6. Secondary CD8+ T Cell Responses Are Not Regulated by Adgrg1/GPR56
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Falco, M.; Parolini, S.; Bellora, F.; Petretto, A.; Romeo, E.; Balsamo, M.; Gambarotti, M.; Scordamaglia, F.; Tabellini, G.; et al. GPR56 as a novel marker identifying the CD56dull CD16+ NK cell subset both in blood stream and in inflamed peripheral tissues. Int. Immunol. 2009, 22, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.-M.; van de Garde, M.D.B.; Cheng, K.-F.; Baars, P.A.; Remmerswaal, E.B.M.; van Lier, R.A.W.; Mackay, C.R.; Lin, H.-H.; Hamann, J. Specific expression of GPR56 by human cytotoxic lymphocytes. J. Leukoc. Biol. 2011, 90, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Babor, M.; Lane, J.; Schulten, V.; Patil, V.S.; Seumois, G.; Rosales, S.L.; Fu, Z.; Picarda, G.; Burel, J.; et al. Unique phenotypes and clonal expansions of human CD4 effector memory T cells re-expressing CD45RA. Nat. Commun. 2017, 8, 1473. [Google Scholar] [CrossRef] [PubMed]
- Truong, K.L.; Schlickeiser, S.; Vogt, K.; Boës, D.; Stanko, K.; Appelt, C.; Streitz, M.; Grütz, G.; Stobutzki, N.; Meisel, C.; et al. Killer-like receptors and GPR56 progressive expression defines cytokine production of human CD4+ memory T cells. Nat. Commun. 2019, 10, 2263. [Google Scholar] [CrossRef] [Green Version]
- Piao, X.; Hill, S.S.; Bodell, A.; Chang, B.S.; Basel-Vanagaite, L.; Straussberg, R.; Dobyns, W.B.; Qasrawi, B.; Winter, R.M.; Innes, A.M.; et al. G Protein-Coupled Receptor-Dependent Development of Human Frontal Cortex. Science 2004, 303, 2033–2036. [Google Scholar] [CrossRef] [Green Version]
- Hamann, J.; Aust, G.; Araç, D.; Engel, F.B.; Formstone, C.; Fredriksson, R.; Hall, R.A.; Harty, B.L.; Kirchhoff, C.; Knapp, B.; et al. International union of basic and clinical pharmacology. XCIV. adhesion G protein-coupled receptors. Pharmacol. Rev. 2015, 67, 338–367. [Google Scholar] [CrossRef]
- Lin, H.H.; Hsiao, C.C.; Pabst, C.; Hébert, J.; Schöneberg, T.; Hamann, J. Adhesion GPCRs in Regulating Immune Responses and Inflammation. Adv. Immunol. 2017, 136, 163–201. [Google Scholar] [CrossRef]
- Chang, G.W.; Hsiao, C.C.; Peng, Y.M.; Vieira Braga, F.A.; Kragten, N.A.M.; Remmerswaal, E.B.M.; van de Garde, M.D.B.; Straussberg, R.; König, G.M.; Kostenis, E.; et al. The Adhesion G Protein-Coupled Receptor GPR56/ADGRG1 Is an Inhibitory Receptor on Human NK Cells. Cell Rep. 2016, 15, 1757–1770. [Google Scholar] [CrossRef] [Green Version]
- Vieira Braga, F.A.; Hertoghs, K.M.L.; Kragten, N.A.M.; Doody, G.M.; Barnes, N.A.; Remmerswaal, E.B.M.; Hsiao, C.C.; Moerland, P.D.; Wouters, D.; Derks, I.A.M.; et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur. J. Immunol. 2015, 45, 2945–2958. [Google Scholar] [CrossRef] [Green Version]
- Oja, A.E.; Vieira Braga, F.A.; Remmerswaal, E.B.M.; Kragten, N.A.M.; Hertoghs, K.M.L.; Zuo, J.; Moss, P.A.; van Lier, R.A.W.; van Gisbergen, K.P.J.M.; Hombrink, P. The transcription factor hobit identifies human cytotoxic CD4+ T cells. Front. Immunol. 2017, 8, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisbergen, K.P.J.M.; Zens, K.D.; Münz, C. T cell memory in tissues. Eur. J. Immunol. 2021, 51, 1310–1324. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef]
- Gebhardt, T.; Whitney, P.G.; Zaid, A.; MacKay, L.K.; Brooks, A.G.; Heath, W.R.; Carbone, F.R.; Mueller, S.N. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 2011, 477, 216–219. [Google Scholar] [CrossRef]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8 + T RM cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, S.; Goplen, N.P.; Li, C.; Cheon, I.S.; Dai, Q.; Huang, S.; Shan, J.; Ma, C.; Ye, Z.; et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 2019, 4, eaaw1217. [Google Scholar] [CrossRef]
- Weisberg, S.P.; Carpenter, D.J.; Chait, M.; Dogra, P.; Gartrell-Corrado, R.D.; Chen, A.X.; Campbell, S.; Liu, W.; Saraf, P.; Snyder, M.E.; et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 2019, 29, 3916–3932.e5. [Google Scholar] [CrossRef] [Green Version]
- Sasson, S.C.; Slevin, S.M.; Cheung, V.T.; Nassiri, I.; Olsson-Brown, A.; Fryer, E.; Ferreira, R.C.; Trzupek, D.; Gupta, T.; Al-Hillawi, L.; et al. IFNγ-producing CD8+ tissue resident memory T cells are a targetable hallmark of immune checkpoint inhibitor-colitis. Gastroenterology 2021, 161, 1229–1244.e9. [Google Scholar] [CrossRef]
- Giera, S.; Deng, Y.; Luo, R.; Ackerman, S.D.; Mogha, A.; Monk, K.R.; Ying, Y.; Jeong, S.J.; Makinodan, M.; Bialas, A.R.; et al. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 2015, 6, 6121. [Google Scholar] [CrossRef] [Green Version]
- Van Gisbergen, K.P.J.M.; Kragten, N.A.M.; Hertoghs, K.M.L.; Wensveen, F.M.; Jonjic, S.; Hamann, J.; Nolte, M.A.; Van Lier, R.A.W. Mouse Hobit is a homolog of the transcriptional repressor Blimp-1 that regulates NKT cell effector differentiation. Nat. Immunol. 2012, 13, 864–871. [Google Scholar] [CrossRef]
- Behr, F.M.; Parga-Vidal, L.; Kragten, N.A.M.; van Dam, T.J.P.; Wesselink, T.H.; Sheridan, B.S.; Arens, R.; van Lier, R.A.W.; Stark, R.; van Gisbergen, K.P.J.M. Tissue-resident memory CD8+ T cells shape local and systemic secondary T cell responses. Nat. Immunol. 2020, 21, 1070–1081. [Google Scholar] [CrossRef]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 Transcription Factor Is Required for the Differentiation of Effector CD8+ T Cells and Memory Responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Kragten, N.A.M.; Behr, F.M.; Vieira Braga, F.A.; Remmerswaal, E.B.M.; Wesselink, T.H.; Oja, A.E.; Hombrink, P.; Kallies, A.; van Lier, R.A.W.; Stark, R.; et al. Blimp-1 induces and Hobit maintains the cytotoxic mediator granzyme B in CD8 T cells. Eur. J. Immunol. 2018, 48, 1644–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J.D.; Moss, P.A.H.; Goulder, P.J.R.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Rötzschke, O.; Falk, K.; Stevanovic, S.; Jung, G.; Walden, P.; Rammensee, H.-G. Exact prediction of a natural T cell epitope. Eur. J. Immunol. 1991, 21, 2891–2894. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.M.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Bevan, M.J. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 2013, 39, 687–696. [Google Scholar] [CrossRef] [Green Version]
- Christo, S.N.; Evrard, M.; Park, S.L.; Gandolfo, L.C.; Burn, T.N.; Fonseca, R.; Newman, D.M.; Alexandre, Y.O.; Collins, N.; Zamudio, N.M.; et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat. Immunol. 2021, 22, 1140–1151. [Google Scholar] [CrossRef]
- Nath, A.P.; Braun, A.; Ritchie, S.C.; Carbone, F.R.; Mackay, L.K.; Gebhardt, T.; Inouye, M. Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE 2019, 14, e0210495. [Google Scholar] [CrossRef]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation Directs Memory Precursor and Short-Lived Effector CD8+ T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [Green Version]
- MacKay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103+ CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, B.S.; Pham, Q.M.; Lee, Y.T.; Cauley, L.S.; Puddington, L.; Lefrançois, L. Oral infection drives a distinct population of intestinal resident memory cd8+ t cells with enhanced protective function. Immunity 2014, 40, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglitto, A.; Mariani, S.A.; de Pater, E.; Rodriguez-Seoane, C.; Vink, C.S.; Piao, X.; Lukke, M.L.; Dzierzak, E. Unexpected redundancy of Gpr56 and Gpr97 during hematopoietic cell development and differentiation. Blood Adv. 2021, 5, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Fransen, N.L.; Hsiao, C.C.; Van Der Poel, M.; Engelenburg, H.J.; Verdaasdonk, K.; Vincenten, M.C.J.; Remmerswaal, E.B.M.; Kuhlmann, T.; Mason, M.R.J.; Hamann, J.; et al. Tissue-resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain 2020, 143, 1714–1730. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.J.; Toma, C.; He, Z.; Kurd, N.S.; Nguyen, Q.P.; McDonald, B.; Quezada, L.; Widjaja, C.E.; Witherden, D.A.; Crowl, J.T.; et al. Heterogenous Populations of Tissue-Resident CD8+ T Cells Are Generated in Response to Infection and Malignancy. Immunity 2020, 52, 808–824.e7. [Google Scholar] [CrossRef] [PubMed]
- Kurd, N.S.; He, Z.; Louis, T.L.; Milner, J.J.; Omilusik, K.D.; Jin, W.; Tsai, M.S.; Widjaja, C.E.; Kanbar, J.N.; Olvera, J.G.; et al. Early precursors and molecular determinants of tissue-resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. 2020, 5, eaaz6894. [Google Scholar] [CrossRef] [PubMed]
- Kok, L.; Dijkgraaf, F.E.; Urbanus, J.; Bresser, K.; Vredevoogd, D.W.; Cardoso, R.F.; Perié, L.; Beltman, J.B.; Schumacher, T.N. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J. Exp. Med. 2020, 217, e20191711. [Google Scholar] [CrossRef]
- Beura, L.K.; Fares-Frederickson, N.J.; Steinert, E.M.; Scott, M.C.; Thompson, E.A.; Fraser, K.A.; Schenkel, J.M.; Vezys, V.; Masopust, D. CD4+ resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J. Exp. Med. 2019, 216, 1214–1229. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.; Wesselink, T.H.; Behr, F.M.; Kragten, N.A.M.; Arens, R.; Koch-Nolte, F.; van Gisbergen, K.P.J.M.; van Lier, R.A.W. TRM maintenance is regulated by tissue damage via P2RX7. Sci. Immunol. 2018, 3, eaau1022. [Google Scholar] [CrossRef] [Green Version]
- Mackay, L.K.; Wynne-Jones, E.; Freestone, D.; Pellicci, D.G.; Mielke, L.A.; Newman, D.M.; Braun, A.; Masson, F.; Kallies, A.; Belz, G.T.; et al. T-box Transcription Factors Combine with the Cytokines TGF-β and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 2015, 43, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Adachi, T.; Kobayashi, T.; Sugihara, E.; Yamada, T.; Ikuta, K.; Pittaluga, S.; Saya, H.; Amagai, M.; Nagao, K. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 2015, 21, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15–Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.L.; Buzzai, A.; Rautela, J.; Hor, J.L.; Hochheiser, K.; Effern, M.; McBain, N.; Wagner, T.; Edwards, J.; McConville, R.; et al. Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 2019, 565, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpréville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+ CD103+ Tumor–Infiltrating Lymphocytes Are Tumor-Specific Tissue-Resident Memory T Cells and a Prognostic Factor for Survival in Lung Cancer Patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, A.P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, C.-C.; Kragten, N.A.M.; Piao, X.; Hamann, J.; van Gisbergen, K.P.J.M. The Inhibitory Receptor GPR56 (Adgrg1) Is Specifically Expressed by Tissue-Resident Memory T Cells in Mice But Dispensable for Their Differentiation and Function In Vivo. Cells 2021, 10, 2675. https://doi.org/10.3390/cells10102675
Hsiao C-C, Kragten NAM, Piao X, Hamann J, van Gisbergen KPJM. The Inhibitory Receptor GPR56 (Adgrg1) Is Specifically Expressed by Tissue-Resident Memory T Cells in Mice But Dispensable for Their Differentiation and Function In Vivo. Cells. 2021; 10(10):2675. https://doi.org/10.3390/cells10102675
Chicago/Turabian StyleHsiao, Cheng-Chih, Natasja A. M. Kragten, Xianhua Piao, Jörg Hamann, and Klaas P. J. M. van Gisbergen. 2021. "The Inhibitory Receptor GPR56 (Adgrg1) Is Specifically Expressed by Tissue-Resident Memory T Cells in Mice But Dispensable for Their Differentiation and Function In Vivo" Cells 10, no. 10: 2675. https://doi.org/10.3390/cells10102675
APA StyleHsiao, C.-C., Kragten, N. A. M., Piao, X., Hamann, J., & van Gisbergen, K. P. J. M. (2021). The Inhibitory Receptor GPR56 (Adgrg1) Is Specifically Expressed by Tissue-Resident Memory T Cells in Mice But Dispensable for Their Differentiation and Function In Vivo. Cells, 10(10), 2675. https://doi.org/10.3390/cells10102675