
Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer

 , ,
, ,  and
and
Abstract
:1. Introduction
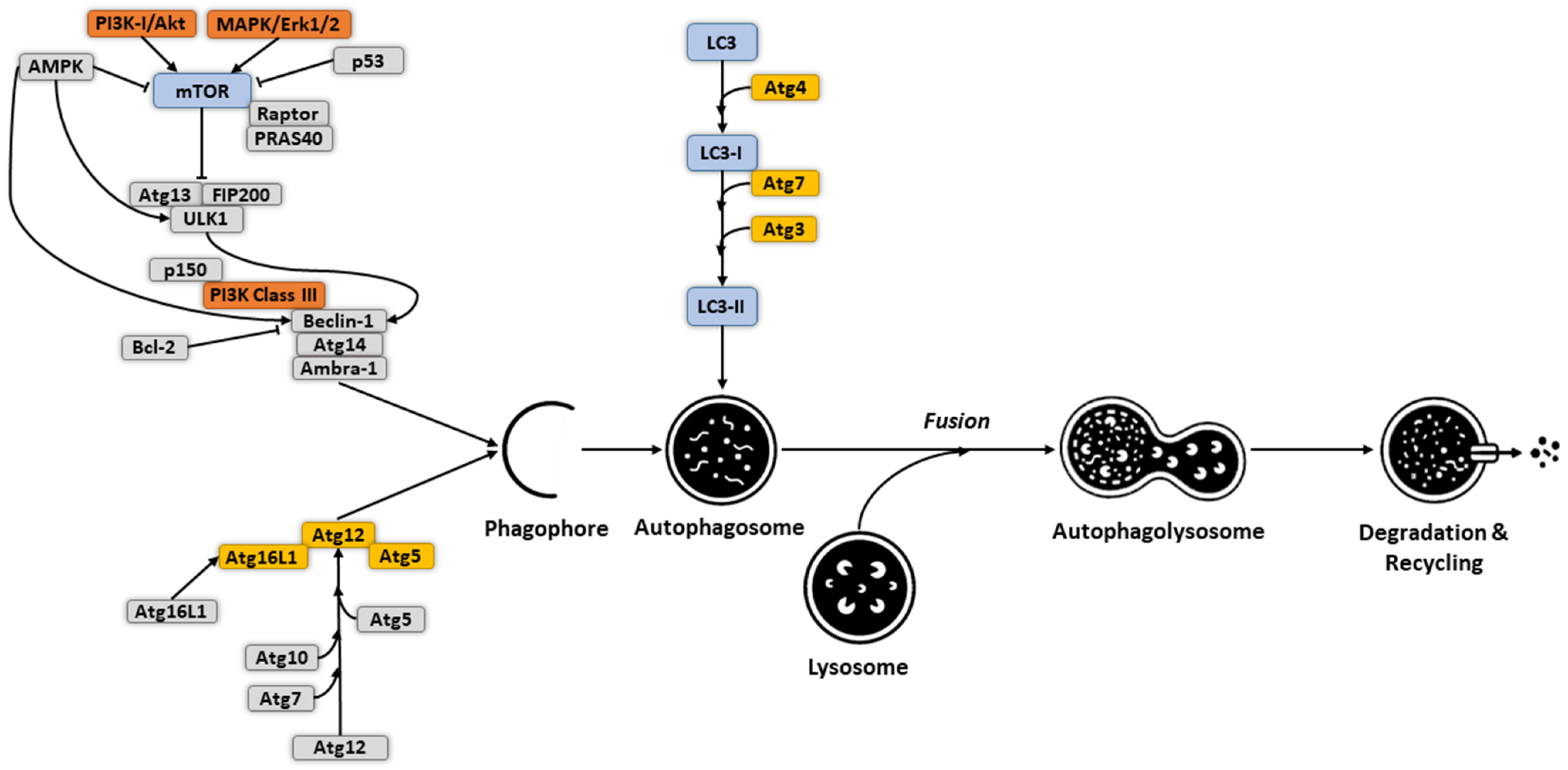
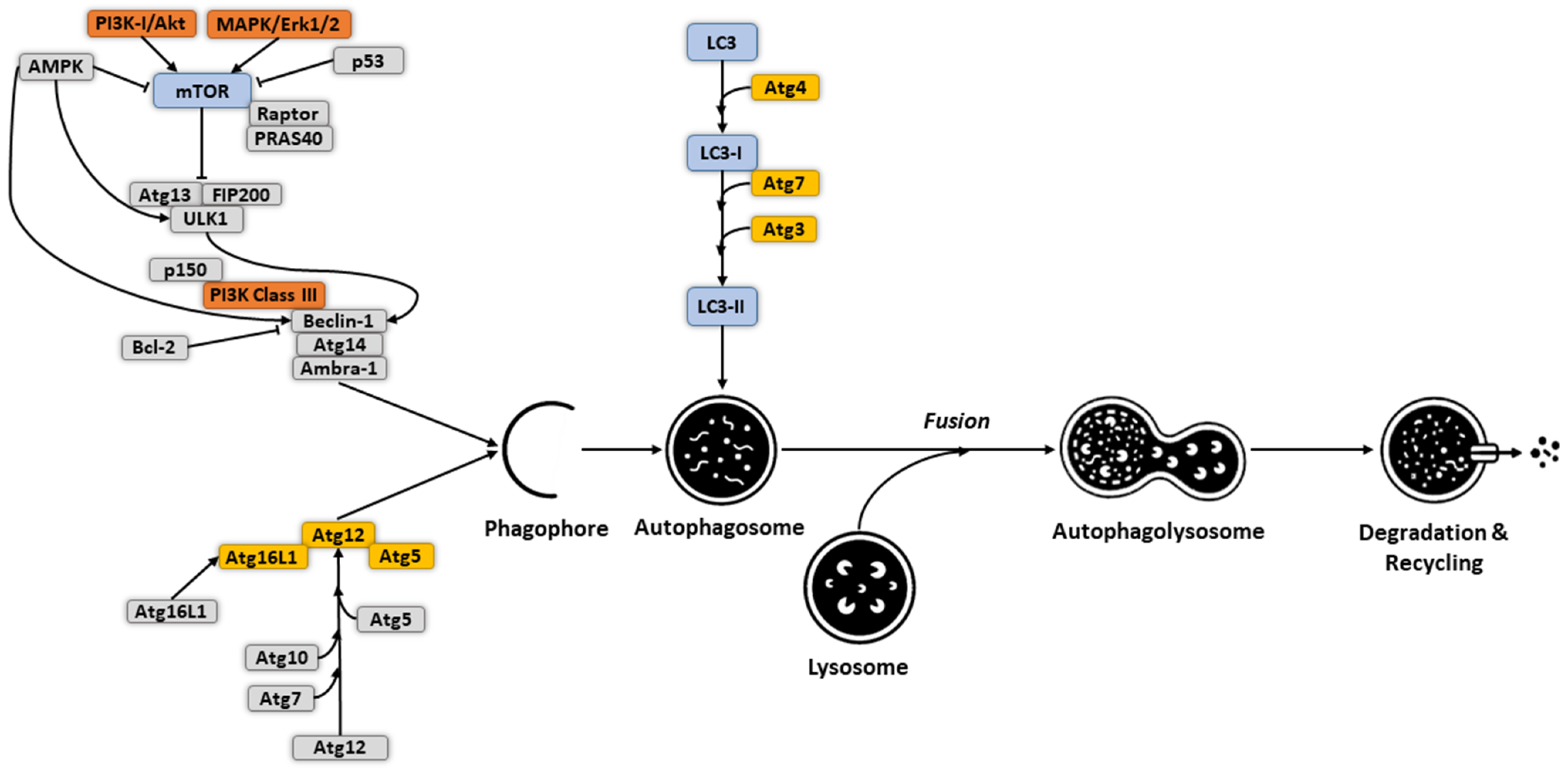
2. Molecular Mechanism and Regulation of Autophagy
3. Autophagy in Tumor Initiation and Development
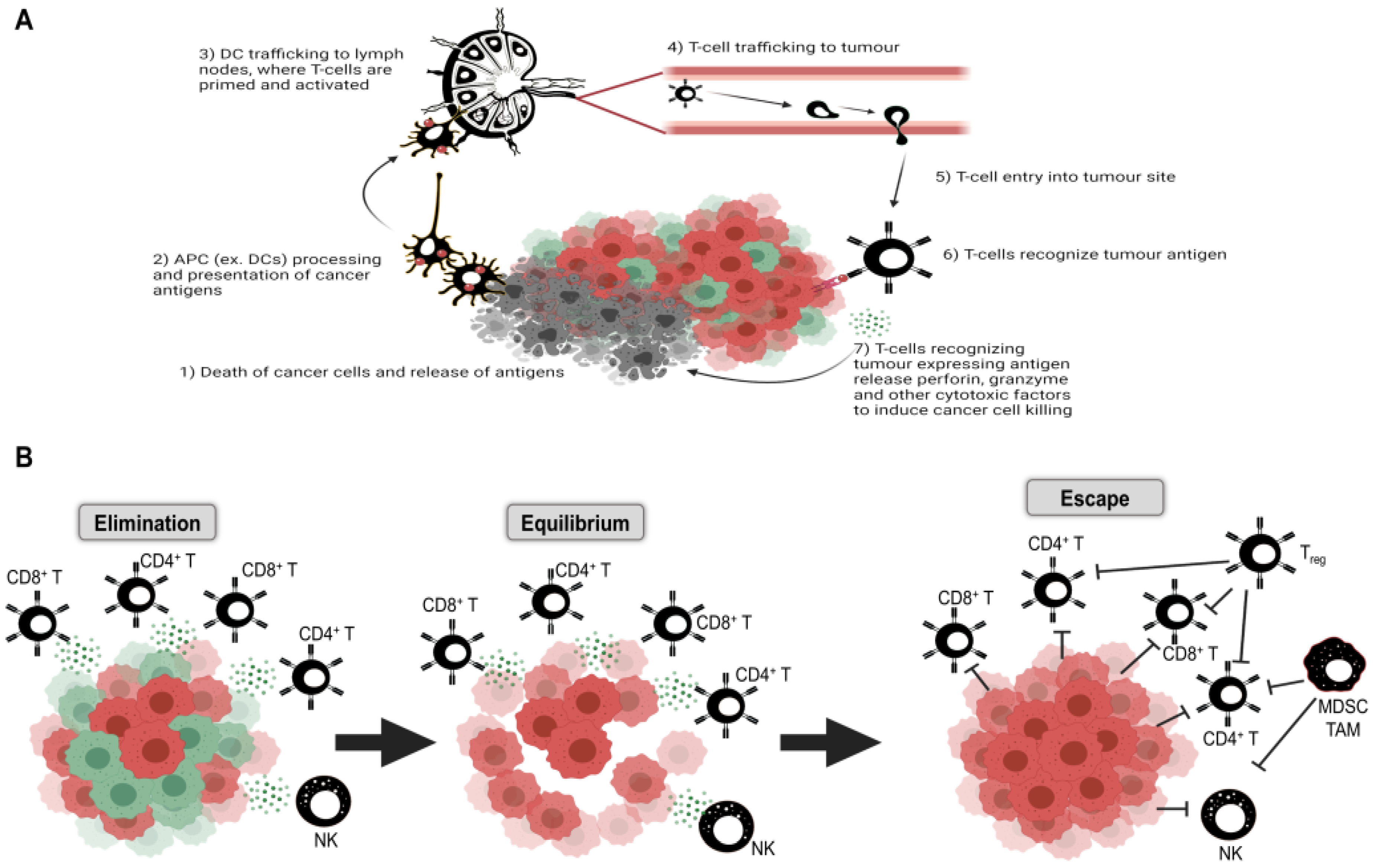
4. Autophagy in Tumor Immunity
5. Targeting Autophagy for Immunotherapy
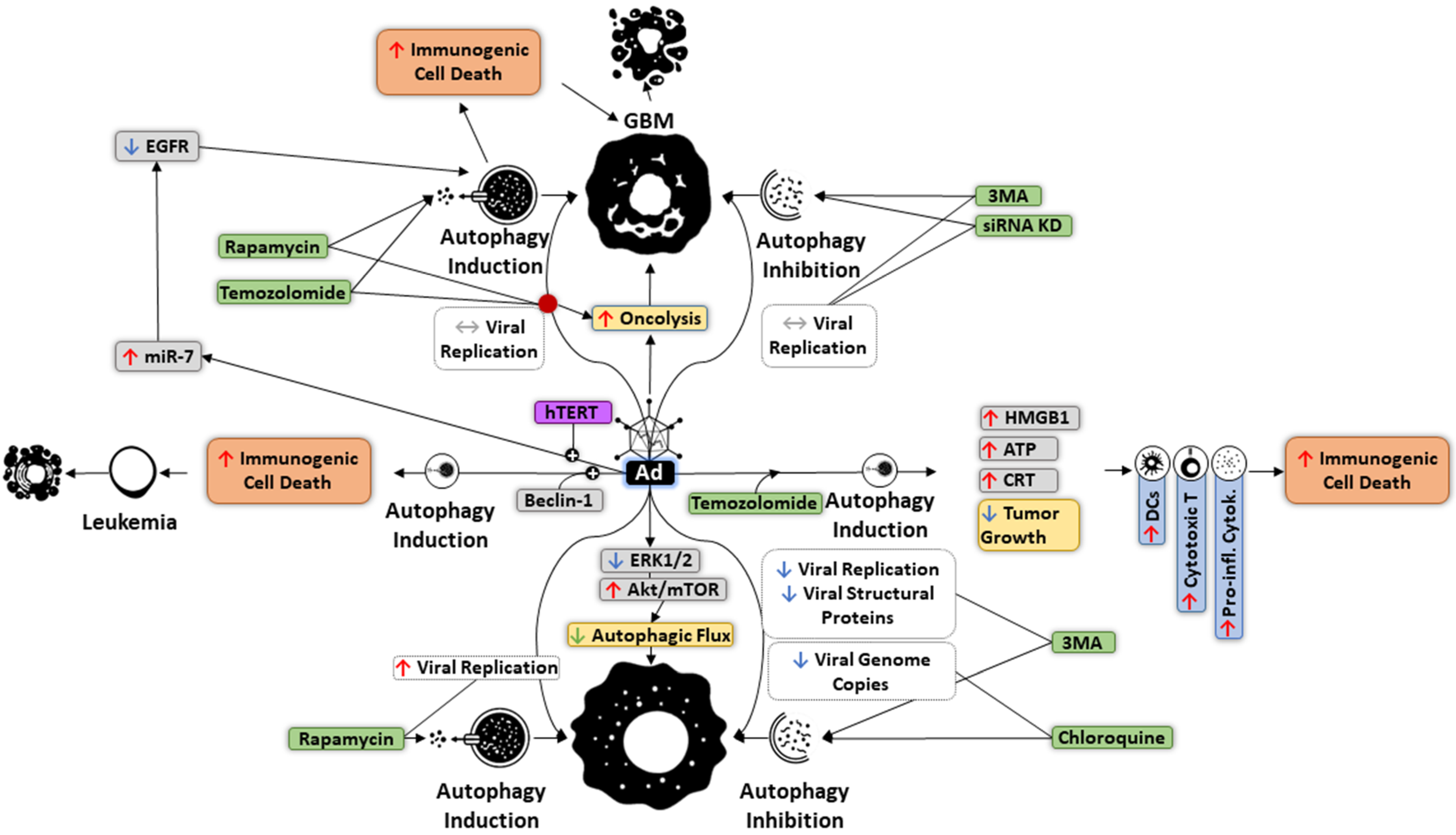
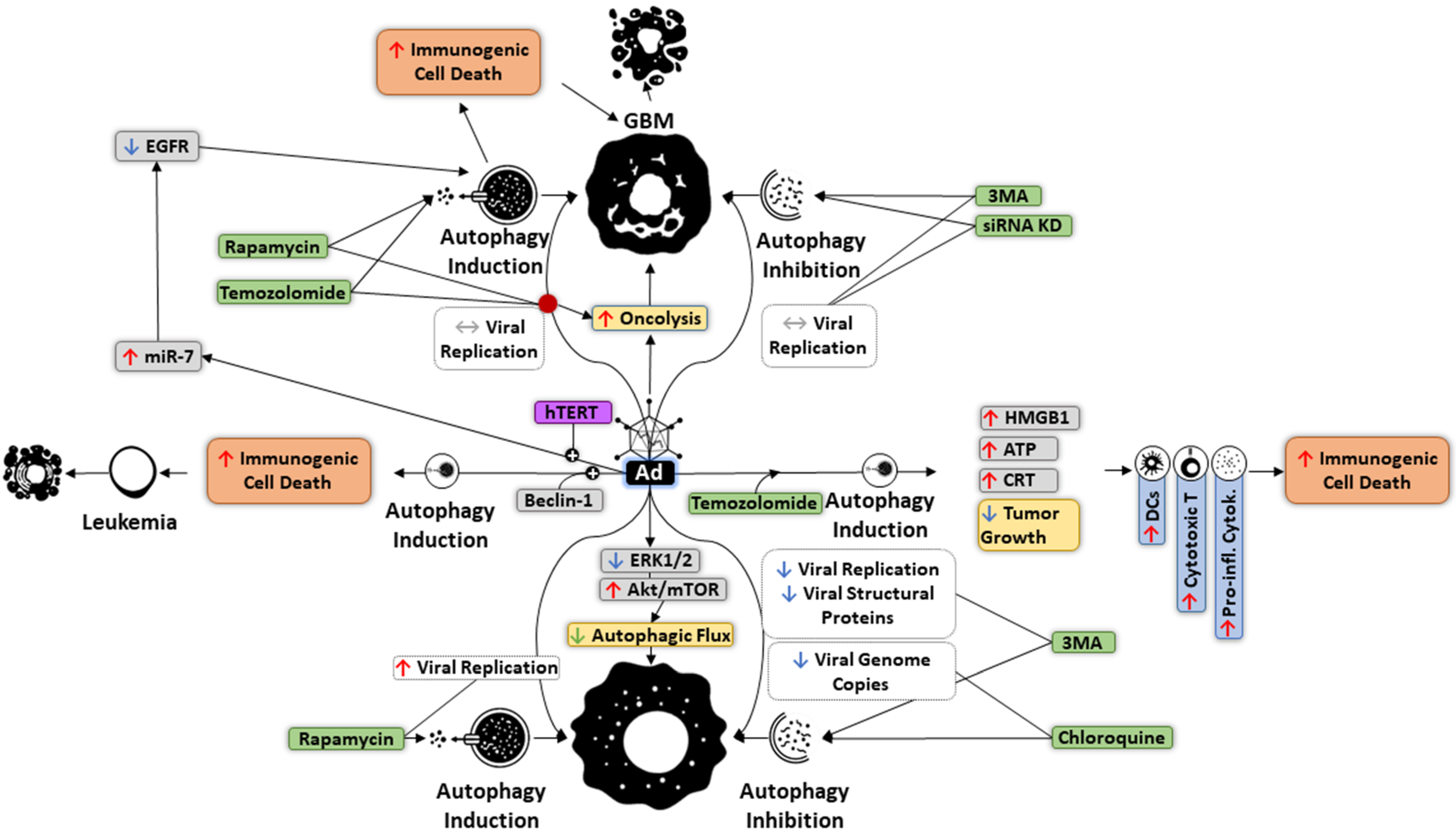
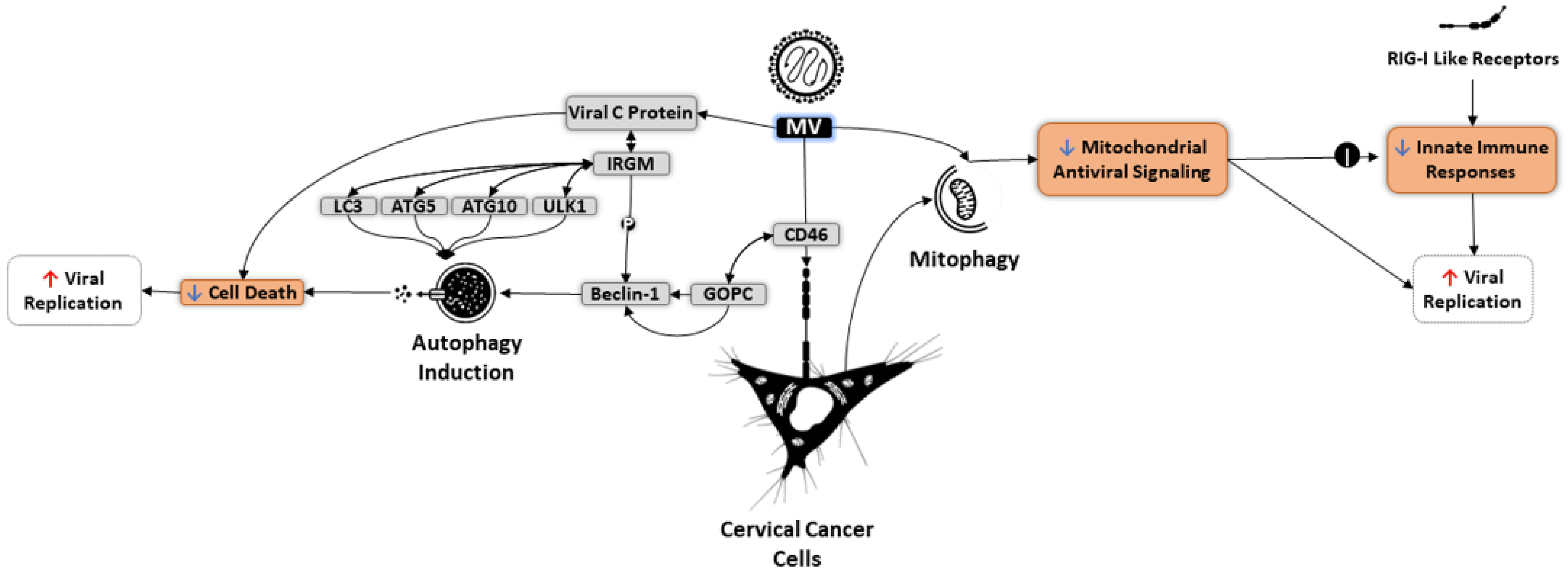
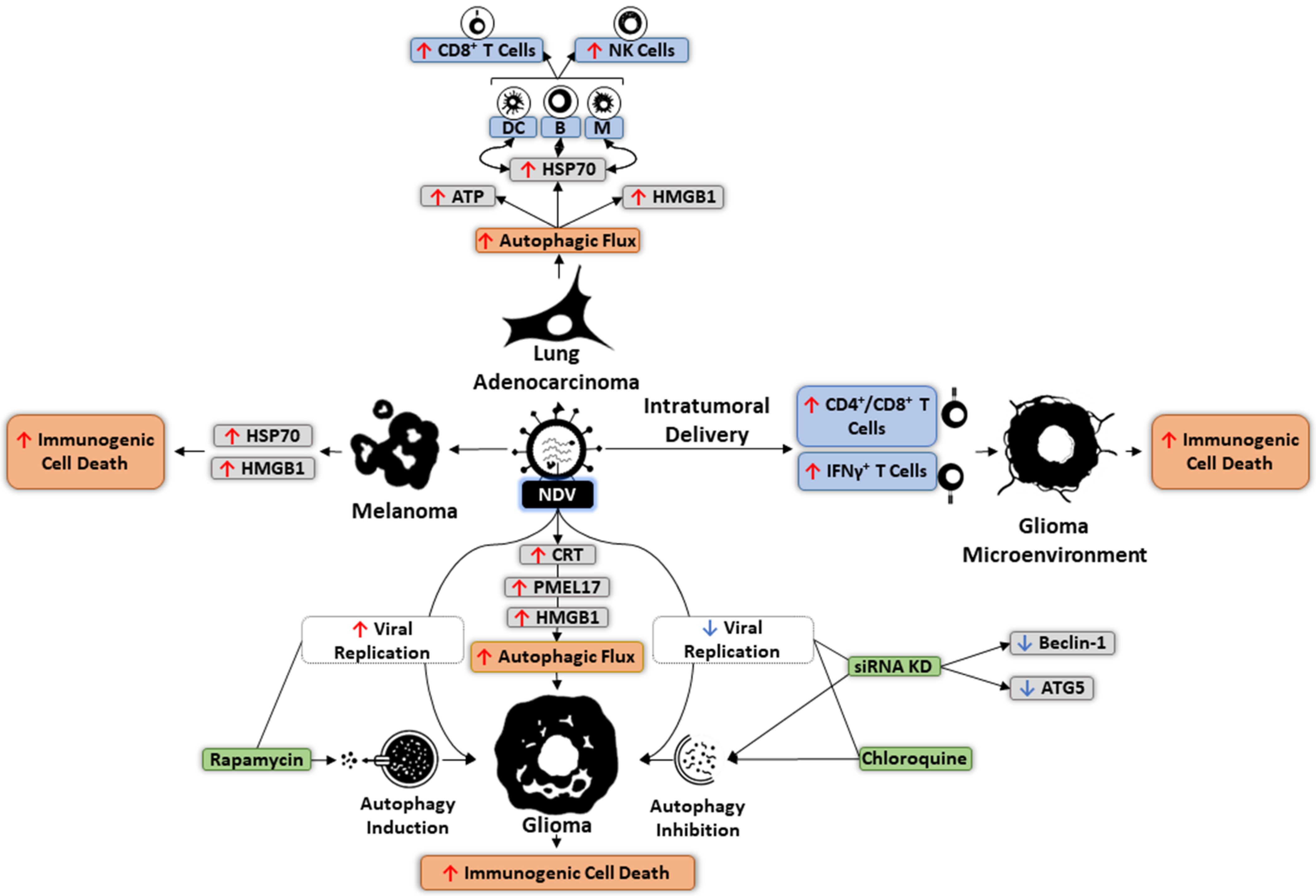
6. Autophagy and OVs
7. Targeting Autophagy in Oncolytic Immunotherapy
8. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Ad | Adenovirus |
| AMPK | AMP-activated protein kinase |
| APCs | Antigen-presenting cells |
| CRT | Calreticulin |
| cDCs | Conventional DCs |
| CSCs | Cancer stem cells |
| CSF-1 | Colony-stimulating factor 1 |
| DAMPs | Damage-associated molecular patterns |
| DCs | Dendritic cells |
| EAT | Early autophagy targeting |
| ER | Endoplasmic reticulum |
| GBM | Glioblastoma multiform |
| HIF | Hypoxia-inducible factor |
| HMGB1 | High mobility group box protein 1 |
| HSV | Herpes simplex virus |
| IFN | Interferon |
| iNOS | Inducible nitric oxide synthase |
| IRE1α | Inositol-requiring enzyme 1α |
| IRGM | Immunity-related GTPase family M |
| KIRs | Killer-cell immunoglobulin-like receptors |
| lncRNAs | Long noncoding RNAs |
| MAVS | Mitochondrial antiviral signaling |
| MDSCs | Myeloid-derived suppressor cells |
| miRNAs | MicroRNAs |
| mTOR | Mamalian target of rapamycin |
| MV | Measles virus |
| MYXV | Myxoma virus |
| NDV | Newcastle disease virus |
| NK | Natural killer |
| OVs | Oncolytic viruses |
| PAMPs | Pathogen-associated molecular patterns |
| pDCs | Plasmacytoid DCs |
| PE | Phosphatidylethanolamine |
| PERK | Pancreatic endoplasmic reticulum kinase |
| PAS | Phagophore assembly site |
| ROS | Reactive oxygen species |
| SVV | Seneca Valley virus |
| TAAs | Tumor-associated antigens |
| TAMs | Tumor-associated macrophages |
| TLRs | Toll-like receptors |
| TME | Tumor microenvironment |
| Treg | Regulatory T cell |
| UPR | Unfolded protein response |
| UPS | Ubiquitin-proteasome system |
| VSV | Vesicular stomatitis virus |
References
- Aurelian, L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. OncoTargets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Jiang, K.; Ding, C.; Meng, S. Targeting Autophagy for Oncolytic Immunotherapy. Biomedicines 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nature 2018, 20, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Richetta, C.; Grégoire, I.P.; Verlhac, P.; Azocar, O.; Baguet, J.; Flacher, M.; Tangy, F.; Rabourdin-Combe, C.; Faure, M. Sustained Autophagy Contributes to Measles Virus Infectivity. PLOS Pathog. 2013, 9, e1003599. [Google Scholar] [CrossRef]
- Furukawa, Y.; Takasu, A.; Yura, Y. Role of autophagy in oncolytic herpes simplex virus type 1-induced cell death in squamous cell carcinoma cells. Cancer Gene Ther. 2017, 24, 393–400. [Google Scholar] [CrossRef]
- Rodriguez-Rocha, H.; Gomez-Gutierrez, J.G.; Garcia-Garcia, A.; Rao, X.-M.; Chen, L.; McMasters, K.M.; Zhou, H.S. Adenoviruses induce autophagy to promote virus replication and oncolysis. Virology 2011, 416, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.-M.; Beltinger, C.; Wei, J. Mitophagy Enhances Oncolytic Measles Virus Replication by Mitigating DDX58/RIG-I-Like Receptor Signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, T.; Iwado, E.; Kondo, Y.; Aoki, H.; Hayashi, Y.; Georgescu, M.M.; Sawaya, R.; Hess, K.R.; Mills, G.B.; Kawamura, H.; et al. Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther. 2008, 15, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Ye, T.; Jiang, K.; Wei, L.; Barr, M.; Xu, Q.; Zhang, G.; Ding, C.; Meng, S.; Piao, H. Oncolytic Newcastle disease virus induces autophagy-dependent immunogenic cell death in lung cancer cells. Am. J. Cancer Res. 2018, 8. [Google Scholar]
- Jin, K.-T.; Lu, Z.-B.; Lv, J.-Q.; Zhang, J.-G. The role of long non-coding RNAs in mediating chemoresistance by modulating autophagy in cancer. RNA Biol. 2020, 17, 1727–1740. [Google Scholar] [CrossRef]
- Guo, J.Y.; Xia, B.; White, E. Autophagy-Mediated Tumor Promotion. Cell 2013, 155, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the Immune System. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Schoor, O.; Fischer, R.; Reich, M.; Kraus, M.; Muller, M.; Kreymborg, K.; Altenberend, F.; Brandenburg, J.; Kalbacher, H.; et al. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 7922–7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauvrit, A.; Brandler, S.; Sapede-Peroz, C.; Boisgerault, N.; Tangy, F.; Gregoire, M. Measles Virus Induces Oncolysis of Mesothelioma Cells and Allows Dendritic Cells to Cross-Prime Tumor-Specific CD8 Response. Cancer Res. 2008, 68, 4882–4892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, C.; Kaliappan, A.; Dennis, P.B. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009, 5, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Takamura, A.; Kishi, C.; Iemura, S.-I.; Natsume, T.; Guan, J.-L.; Mizushima, N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 2008, 181, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, M.B.; Novotny, C.J.; Shokat, K.M. Structure of the Human Autophagy Initiating Kinase ULK1 in Complex with Potent Inhibitors. ACS Chem. Biol. 2014, 10, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK 1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Liu, J.-J.; Li, Y.; Huang, Y.; Ta, N.; Chen, Y.; Fu, H.; Ye, M.-D.; Ding, Y.; Huang, W.; et al. Cryo-EM structure and biochemical analysis reveal the basis of the functional difference between human PI3KC3-C1 and -C2. Cell Res. 2017, 27, 989–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.N.; Cho, K.; Lawrence, R.; Zoncu, R.; Hurley, J.H. Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy. Proc. Natl. Acad. Sci. USA 2016, 113, 8224–8229. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, Y.; Soler, N.; García Ortegón, M.; Zhang, L.; Kirsten, M.L.; Perisic, O.; Masson, G.R.; Burke, J.E.; Jakobi, A.J.; Apostolakis, A.A.; et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy 2016, 12, 2129–2144. [Google Scholar] [CrossRef]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Mizushima, N.; Noda, T.; Ohsumi, Y. Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999, 18, 3888–3896. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit crosstalk in the Rag GTPase heterodimer enables mTORC1 to respond rapidly to amino acid availability. Mol. Cell 2017, 68, 552–565. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef]
- Chen, S.; Han, Q.; Wang, X.; Yang, M.; Zhang, Z.; Li, P.; Chen, A.; Hu, C.; Li, S. IBP-mediated suppression of autophagy promotes growth and metastasis of breast cancer cells via activating mTORC2/Akt/FOXO3a signaling pathway. Cell Death Dis. 2013, 4, e842. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Saeki, K.; Yuo, A.; Okuma, E.; Yazaki, Y.; Susin, S.A.; Kroemer, G.; Takaku, F. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000, 7, 1263–1269. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 Binding to a Linear Peptide Motif in MAP1B Stimulates Autophagy and Membrane Blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorimitsu, T.; Nair, U.; Yang, Z.; Klionsky, D.J. Endoplasmic Reticulum Stress Triggers Autophagy. J. Biol. Chem. 2006, 281, 30299–30304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.-C.; Chang, J.-M.; Chen, C.-A.; Chen, H.-C. Autophagy modulates endoplasmic reticulum stress-induced cell death in podocytes: A protective role. Exp. Biol. Med. 2014, 240, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2 α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Portal-Núñez, S.; Esbrit, P.; Alcaraz, M.J.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2015, 108, 1–10. [Google Scholar] [CrossRef]
- Shiomi, M.; Miyamae, M.; Takemura, G.; Kaneda, K.; Inamura, Y.; Onishi, A.; Koshinuma, S.; Momota, Y.; Minami, T.; Figueredo, V.M. Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J. Anesth. 2013, 28, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2013, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-Y.; Jiang, X.-X.; Zhu, X.; He, W.-Y.; Kuang, Y.-L.; Ren, K.; Lin, Y.; Gou, X. ROS activates JNK-mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta Pharmacol. Sin. 2015, 36, 1473–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wang, F.; Hu, S.; Yin, C.; Li, X.; Zhao, S.; Wang, J.; Yan, X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell. Signal. 2012, 24, 2179–2186. [Google Scholar] [CrossRef]
- Zhang, J.-A.; Zhou, B.-R.; Xu, Y.; Chen, X.; Liu, J.; Gozali, M.; Wu, D.; Yin, Z.-Q.; Luo, D. MiR-23a-depressed autophagy is a participant in PUVA- and UVB-induced premature senescence. Oncotarget 2016, 7, 37420–37435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, L.; Han, B.; Zhang, Y.; Bai, Y.; Chao, J.; Hu, G.; Yao, H. Engagement of circular RNA HECW2 in the nonautophagic role of ATG5 implicated in the endothelial-mesenchymal transition. Autophagy 2018, 14, 404–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Koster, S.; Sakowski, E.; Ramkhelawon, B.; Van Solingen, C.; Oldebeken, S.; Karunakaran, D.; Portal-Celhay, C.; Sheedy, F.; Ray, T.D.; et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 2016, 17, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.-M.; Jiang, M.-T.; Lin, P.; Yang, H.; Dang, Y.-W.; Yu, Q.; Liao, D.-Y.; Luo, D.-Z.; Chen, G. Potential ceRNA networks involved in autophagy suppression of pancreatic cancer caused by chloroquine diphosphate: A study based on differentially-expressed circRNAs, lncRNAs, miRNAs and mRNAs. Int. J. Oncol. 2018, 54, 600–626. [Google Scholar]
- Baek, S.H.; Kim, K.I. Epigenetic Control of Autophagy: Nuclear Events Gain More Attention. Mol. Cell 2017, 65, 781–785. [Google Scholar] [CrossRef] [Green Version]
- Trocoli, A.; Djavaheri-Mergny, M. The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am. J. Cancer Res. 2011, 1, 629. [Google Scholar]
- Barnard, R.A.; Regan, D.P.; Hansen, R.J.; Maycotte, P.; Thorburn, A.; Gustafson, D.L. Autophagy Inhibition Delays Early but Not Late-Stage Metastatic Disease. J. Pharmacol. Exp. Ther. 2016, 358, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.-B.; Shi, Y.-H.; Zhou, J.; Qiu, S.-J.; Xu, Y.; Dai, Z.; Shi, G.-M.; Wang, X.-Y.; Ke, A.-W.; Wu, B.; et al. Association of Autophagy Defect with a Malignant Phenotype and Poor Prognosis of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.R.; Kim, M.S.; Oh, J.E.; Kim, Y.R.; Song, S.Y.; Kim, S.S.; Ahn, C.H.; Yoo, N.J.; Lee, S.H. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J. Pathol. 2009, 217, 702–706. [Google Scholar] [CrossRef]
- An, C.H.; Kim, M.S.; Yoo, N.J.; Park, S.W.; Lee, S.H. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol.-Res. Pract. 2011, 207, 433–437. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS 2007, 115, 1344–1349. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B Expression Is a Common Feature of Solid Tumors and Associated with Proliferation, Metastasis, and Poor Outcome. Clin. Cancer Res. 2011, 18, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med. Oncol. 2013, 30, 1–8. [Google Scholar] [CrossRef]
- Lazova, R.; Klump, V.; Pawelek, J. Autophagy in cutaneous malignant melanoma. J. Cutan. Pathol. 2009, 37, 256–268. [Google Scholar] [CrossRef]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.L.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2012, 32, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Paul, P.; Qiao, J.; Lee, S.; Chung, D.H. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy 2013, 9, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gong, F.; Chen, Y.; Jiang, Y.; Liu, J.; Yu, M.; Zhang, S.; Wang, M.; Xiao, G.; Liao, H. Autophagy promotes paclitaxel resistance of cervical cancer cells: Involvement of Warburg effect activated hypoxia-induced factor 1-α-mediated signaling. Cell Death Dis. 2014, 5, e1367. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Li, C.; Zheng, W.; Guo, Q.; Zhang, Y.; Kang, M.; Zhang, B.; Yang, B.; Li, B.; Yang, H.; et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget 2015, 6, 39839–39854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Q.; Liang, Y.; He, Q.; You, Y.; Wu, L.; Liang, L.; Liang, J. Autophagy inhibits TLR4-mediated invasiveness of oral cancer cells via the NF-κB pathway. Oral Dis. 2020, 26, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yin, H.; Zhang, Y.; Li, X.; Tong, H.; Zeng, Y.; Wang, Q.; He, W. Hypoxia-induced autophagy promotes gemcitabine resistance in human bladder cancer cells through hypoxia-inducible factor 1α activation. Int. J. Oncol. 2018, 53, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Chen, H.; Chen, L.; Ge, M.; Yang, J.; Liu, W.; Xia, M.; Hayashi, T.; Guo, C.; Ikejima, T. Autophagy promotes apoptosis induction through repressed nitric oxide generation in the treatment of human breast cancer MCF-7 cells with L-A03, a dihydroartemisinin derivative. Med. Chem. Res. 2017, 26, 1427–1436. [Google Scholar] [CrossRef]
- Ramirez, L.V.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1–12. [Google Scholar]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial–mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Li, Y.; Dong, L.; Zong, Z.; Hua, X.; Su, D.; Li, H.; et al. Autophagy promotes metastasis and glycolysis by upregulating MCT1 expression and Wnt/β-catenin signaling pathway activation in hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2018, 37, 9. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Garner, H.; de Visser, K.E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 2020, 20, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2018, 16, 151–167. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pedro, J.M.B.-S.; Galluzzi, L.; Kroemer, G. Autophagy in natural and therapy-driven anticancer immunosurveillance. Autophagy 2017, 13, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.K.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science 2012, 339, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar]
- Talukdar, S.; Pradhan, A.K.; Bhoopathi, P.; Shen, X.-N.; August, L.A.; Windle, J.J.; Sarkar, D.; Furnari, F.B.; Cavenee, W.K.; Das, S.K.; et al. MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc. Natl. Acad. Sci. USA 2018, 115, 5768–5773. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- Buchser, W.; Laskow, T.; Pavlik, P.J.; Lin, H.-M.; Lotze, M.T. Cell-Mediated Autophagy Promotes Cancer Cell Survival. Cancer Res. 2012, 72, 2970–2979. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.-C.; Wang, T.-Y.; Chang, Y.-T.; Chu, C.-Y.; Lee, C.-L.; Hsu, H.-W.; Zhou, T.-A.; Wu, Z.; Kim, R.; Desai, S.J.; et al. Autophagy Pathway Is Required for IL-6 Induced Neuroendocrine Differentiation and Chemoresistance of Prostate Cancer LNCaP Cells. PLoS ONE 2014, 9, e88556. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tong, Y.; Ni, W.; Liu, J.; Xu, W.; Li, L.; Liu, X.; Meng, H.; Qian, W. Inhibition of autophagy induced by overexpression of mda-7/interleukin-24 strongly augments the antileukemia activity in vitro and in vivo. Cancer Gene Ther. 2009, 17, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; De Vera, M.E.; Buchser, W.; Chavez, A.R.D.V.; Loughran, P.; Stolz, D.B.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J.; et al. Inhibiting Systemic Autophagy during Interleukin 2 Immunotherapy Promotes Long-term Tumor Regression. Cancer Res. 2012, 72, 2791–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loi, M.; Müller, A.; Steinbach, K.; Niven, J.; da Silva, R.B.; Paul, P.; Ligeon, L.A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control. MHC Class. I Levels on Dendritic Cells and Shape Anti-viral CD8(+) T Cell Responses. Cell Rep. 2016, 15, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Germic, N.; Frangež, Z.; Yousefi, S.; Simon, H.-U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Germic, N.; Frangež, Z.; Yousefi, S.; Simon, H.-U. Regulation of the innate immune system by autophagy: Neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ. 2019, 26, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, Y.; Lizée, G.; Qin, H.; Liu, S.; Rabinovich, B.; Kim, G.J.; Wang, Y.H.; Ye, Y.; Sikora, A.G.; et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J. Clin. Investig. 2008, 118, 1165–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villadangos, J.A.; Schnorrer, P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat. Rev. Immunol. 2007, 7, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Liu, C.; Kim, G.J.; Liu, Y.-J.; Hwu, P.; Wang, G. Plasmacytoid Dendritic Cells Synergize with Myeloid Dendritic Cells in the Induction of Antigen-Specific Antitumor Immune Responses. J. Immunol. 2007, 178, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Labidi-Galy, S.I.; Treilleux, I.; Goddard-Leon, S.; Combes, J.-D.; Blay, J.-Y.; Ray-Coquard, I.; Caux, C.; Bendriss-Vermare, N. Plasmacytoid dendritic cells infiltrating ovarian cancer are associated with poor prognosis. OncoImmunology 2012, 1, 380–382. [Google Scholar] [CrossRef] [Green Version]
- Ghislat, G.; Lawrence, T. Autophagy in dendritic cells. Cell. Mol. Immunol. 2018, 15, 944–952. [Google Scholar] [CrossRef]
- Hubbard-Lucey, V.M.; Shono, Y.; Maurer, K.; West, M.L.; Singer, N.V.; Ziegler, C.; Lezcano, C.; Motta, A.C.; Schmid, K.; Levi, S.; et al. Autophagy Gene Atg16l1 Prevents Lethal T Cell Alloreactivity Mediated by Dendritic Cells. Immunity 2014, 41, 579–591. [Google Scholar] [CrossRef] [Green Version]
- E Wildenberg, M.; Koelink, P.J.; Diederen, K.; Velde, A.A.T.; Wolfkamp, S.C.S.; Nuij, V.J.; Peppelenbosch, M.; Nobis, M.; Sansom, O.J.; I Anderson, K.; et al. The ATG16L1 risk allele associated with Crohn’s disease results in a Rac1-dependent defect in dendritic cell migration that is corrected by thiopurines. Mucosal Immunol. 2016, 10, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Richey, L.J.; Mehta, A.J.; Shah, M.; Huber, B.T. Autophagy in Dendritic Cells and B Cells Is Critical for the Inflammatory State of TLR7-Mediated Autoimmunity. J. Immunol. 2016, 198, 1081–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical Autophagy Is Required for Type I Interferon Secretion in Response to DNA-Immune Complexes. Immunity 2012, 37, 986–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.; Swanson, M.; Lieberman, A.; Reed, M.; Yue, Z.; Lindell, D.M.; Lukacs, N.W. Autophagy-Mediated Dendritic Cell Activation Is Essential for Innate Cytokine Production and APC Function with Respiratory Syncytial Virus Responses. J. Immunol. 2011, 187, 3953–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-gamma production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect 2015, 17, 275–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Vidyarthi, A.; Pahari, S.; Negi, S.; Aqdas, M.; Nadeem, S.; Agnihotri, T.; Agrewala, J.N. Signaling through NOD-2 and TLR-4 Bolsters the T cell Priming Capability of Dendritic cells by Inducing Autophagy. Sci. Rep. 2016, 6, 19084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, A.; Duan, L.; Chen, J.; Fan, Z.; Zheng, F.; Tan, Z.; Gong, F.; Fang, M. Flt3L Combined with Rapamycin Promotes Cardiac Allograft Tolerance by Inducing Regulatory Dendritic Cells and Allograft Autophagy in Mice. PLoS ONE 2012, 7, e46230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, M.; Raffort, J.; Lareyre, F.; Tsiantoulas, D.; Newland, S.; Lu, Y.; Masters, L.; Harrison, J.; Saveljeva, S.; Ma, M.K.; et al. Impaired Autophagy in CD11b(+) Dendritic Cells Expands CD4(+) Regulatory T Cells and Limits Atherosclerosis in Mice. Circ. Res. 2019, 125, 1019–1034. [Google Scholar] [CrossRef]
- Strisciuglio, C.; Duijvestein, M.; Verhaar, A.P.; Vos, A.C.W.; Brink, G.R.V.D.; Hommes, D.W.; E Wildenberg, M. Impaired autophagy leads to abnormal dendritic cell-epithelial cell interactions. J. Crohn’s Coliti 2013, 7, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K.; et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Investig. 2017, 127, 2789–2804. [Google Scholar] [CrossRef] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.; Unanue, E.R.; Schreiber, R.D.; et al. Batf3 Deficiency Reveals a Critical Role for CD8 + Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 938. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-Stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.-G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.-R.; Corrales, L.; Gajewski, T.F. Innate Immune Recognition of Cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-Independent Essential Function for the Autophagy Protein Atg5 in Cellular Immunity to Intracellular Pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Bonilla, D.L.; Bhattacharya, A.; Sha, Y.; Xu, Y.; Xiang, Q.; Khan, A.; Jagannath, C.; Komatsu, M.; Eissa, N.T. Autophagy Regulates Phagocytosis by Modulating the Expression of Scavenger Receptors. Immunity 2013, 39, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Pu, Q.; Gan, C.; Li, R.; Li, Y.; Tan, S.; Li, X.; Wei, Y.; Lan, L.; Deng, X.; Liang, H.; et al. Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis inPseudomonasSepsis. J. Immunol. 2017, 198, 3205–3213. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.; Biswas, S.K.; Fisher, E.; Gilroy, D.; Goerdt, S.; Gordon, S.; A Hamilton, J.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.-P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Lin, E.Y.; Li, J.-F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.-N.; Pollard, J.W. Macrophages Regulate the Angiogenic Switch in a Mouse Model of Breast Cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, S.K.; Kumar, D. AKT mediated glycolytic shift regulates autophagy in classically activated macrophages. Int. J. Biochem. Cell Biol. 2015, 66, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Uchiyama, M.; Hester, J.; Wood, K.; Issa, F. Regulatory T cells for tolerance. Hum. Immunol. 2018, 79, 294–303. [Google Scholar] [CrossRef]
- Akkaya, B.; Shevach, E.M. Regulatory T cells: Master thieves of the immune system. Cell. Immunol. 2020, 355, 104160. [Google Scholar] [CrossRef] [PubMed]
- Dadey, R.E.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells in the Tumor Microenvironment. Adv. Cancer Res. 2020, 1273, 105–134. [Google Scholar]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment-The Next Generation of Immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.; Jelenčić, V.; Polić, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-Mediated Mitophagy Promotes the Generation of Natural Killer Cell Memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.; Sahai, E.; Zelenay, S.; E Sousa, C.R. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Ito, M.; Srirat, T.; Kondo, T.; Yoshimura, A. Memory T cell, exhaustion, and tumor immunity. Immunol. Med. 2020, 43, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Guerrouahen, B.S.; Maccalli, C.; Cugno, C.; Rutella, S.; Akporiaye, E.T. Reverting Immune Suppression to Enhance Cancer Immunotherapy. Front. Oncol. 2020, 9, 1554. [Google Scholar] [CrossRef] [Green Version]
- Muenst, S.; Läubli, H.; Soysal, S.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Lee, C.T.; Mace, T.; Repasky, E.A. Hypoxia-driven immunosuppression: A new reason to use thermal therapy in the treatment of cancer? Int. J. Hyperth. 2010, 26, 232–246. [Google Scholar] [CrossRef]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of Hypoxia-Mediated Immune Escape in Cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.-M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerada, C.; Ryan, K.M. Autophagy, the innate immune response and cancer. Mol. Oncol. 2020, 14, 1913–1929. [Google Scholar] [CrossRef]
- Xu, Y.; Eissa, N.T. Autophagy in Innate and Adaptive Immunity. Proc. Am. Thorac. Soc. 2010, 7, 22–28. [Google Scholar] [CrossRef]
- Chen, D.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Shravage, B.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, S.; Tortola, L.; Perlot, T.; Wirnsberger, G.H.; Novatchkova, M.; Nitsch, R.; Sykacek, P.; Frank, L.W.; Schramek, D.; Komnenovic, V.; et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014, 5, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autophagy Inhibition Synergizes with Immunotherapy in Pancreatic Cancer. Cancer Discov. 2020, 10, 760.
- Qiao, Y.; Choi, J.E.; Tien, J.C.; Simko, S.A.; Rajendiran, T.; Vo, J.N.; Delekta, A.D.; Wang, L.; Xiao, L.; Hodge, N.B.; et al. Autophagy inhibition by targeting PIKfyve potentiates response to immune checkpoint blockade in prostate cancer. Nat. Rev. Cancer 2021, 1–16. [Google Scholar]
- Duan, Y.; Tian, X.; Liu, Q.; Jin, J.; Shi, J.; Hou, Y. Role of autophagy on cancer immune escape. Cell Commun. Signal. 2021, 19, 1–12. [Google Scholar] [CrossRef]
- Dao, V.; Liu, Y.; Pandeswara, S.; Svatek, R.S.; Gelfond, J.A.; Liu, A.; Hurez, V.; Curiel, T.J. Immune-Stimulatory Effects of Rapamycin Are Mediated by Stimulation of Antitumor gammadelta T Cells. Cancer Res. 2016, 76, 5970–5982. [Google Scholar] [CrossRef] [Green Version]
- Keating, R.; Hertz, T.; Wehenkel, M.; Harris, T.L.; A Edwards, B.; McClaren, J.L.; A Brown, S.; Surman, S.; Wilson, Z.S.; Bradley, P.; et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat. Immunol. 2013, 14, 1266–1276. [Google Scholar] [CrossRef] [Green Version]
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padrón, Á.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Lin, Z.; Sun, W.; Hollinger, M.K.; Desierto, M.J.; Keyvanfar, K.; Malide, D.; Muranski, P.; Chen, J.; Young, N.S. Rapamycin is highly effective in murine models of immune-mediated bone marrow failure. Haematologica 2017, 102, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, M.R.; Fruman, D.A. Immune Regulation by Rapamycin: Moving Beyond T Cells. Sci. Signal. 2009, 2, pe25. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Infante, B.; Di Lorenzo, A.; Rascio, F.; Zaza, G.; Grandaliano, G. mTOR inhibitors effects on regulatory T cells and on dendritic cells. J. Transl. Med. 2016, 14, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrezenmeier, E.; Dörner, T. Mechanisms of action of hydroxychloroquine and chloroquine: Implications for rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef]
- Gies, V.; Bekaddour, N.; Dieudonné, Y.; Guffroy, A.; Frenger, Q.; Gros, F.; Rodero, M.P.; Herbeuval, J.-P.; Korganow, A.-S. Beyond Anti-viral Effects of Chloroquine/Hydroxychloroquine. Front. Immunol. 2020, 11, 1409. [Google Scholar] [CrossRef]
- Qin, X.; Chen, G.; Feng, Y.; Zhu, X.; Du, Y.; Pang, W.; Qi, Z.; Cao, Y. Early Treatment with Chloroquine Inhibits the Immune Response against Plasmodium yoelii Infection in Mice. Tohoku J. Exp. Med. 2014, 234, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Chandler, L.C.; Yusuf, I.H.; McClements, M.E.; Barnard, A.R.; MacLaren, R.E.; Xue, K. Immunomodulatory Effects of Hydroxychloroquine and Chloroquine in Viral Infections and Their Potential Application in Retinal Gene Therapy. Int. J. Mol. Sci. 2020, 21, 4972. [Google Scholar] [CrossRef]
- Fiegler, N.; Textor, S.; Arnold, A.; Rölle, A.; Oehme, I.; Breuhahn, K.; Moldenhauer, G.; Witzens-Harig, M.; Cerwenka, A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013, 122, 684–693. [Google Scholar] [CrossRef]
- Shen, L.; Pili, R. Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. OncoImmunology 2012, 1, 948–950. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.L.; Mirshahpanah, P.; Haas-Stapleton, E.; Asadullah, K.; Zollner, T.M.; Numerof, R.P. Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell. Immunol. 2009, 257, 97–104. [Google Scholar] [CrossRef]
- Reddy, P.; Sun, Y.; Toubai, T.; Duran-Struuck, R.; Clouthier, S.G.; Weisiger, E.; Maeda, Y.; Tawara, I.; Krijanovski, O.; Gatza, E.; et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J. Clin. Investig. 2008, 118, 2562–2573. [Google Scholar] [CrossRef]
- Laengle, J.; Kabiljo, J.; Hunter, L.; Homola, J.; Prodinger, S.; Egger, G.; Bergmann, M. Histone deacetylase inhibitors valproic acid and vorinostat enhance trastuzumab-mediated antibody-dependent cell-mediated phagocytosis. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.-T.; Tao, X.-H.; Fan, Y.-B.; Wang, S.-B. Crosstalk between oncolytic viruses and autophagy in cancer therapy. Biomed. Pharmacother. 2020, 134, 110932. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Somma, S.; Iannuzzi, C.A.; Passaro, C.; Forte, I.M.; Iannone, R.; Gigantino, V.; Indovina, P.; Botti, G.; Giordano, A.; Formisano, P.; et al. The Oncolytic Virus dl922-947 Triggers Immunogenic Cell Death in Mesothelioma and Reduces Xenograft Growth. Front. Oncol. 2019, 9, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Kang, S.; Song, J.; Kim, J.-H. The effectiveness of the oncolytic activity induced by Ad5/F35 adenoviral vector is dependent on the cumulative cellular conditions of survival and autophagy. Int. J. Oncol. 2013, 42, 1337–1348. [Google Scholar] [CrossRef] [Green Version]
- Botta, G.; Passaro, C.; Libertini, S.; Abagnale, A.; Barbato, S.; Maione, A.S.; Hallden, G.; Beguinot, F.; Formisano, P.; Portella, G. Inhibition of Autophagy Enhances the Effects of E1A-Defective Oncolytic Adenovirus dl922–947 Against Glioma Cells In Vitro and In Vivo. Hum. Gene Ther. 2012, 23, 623–634. [Google Scholar] [CrossRef]
- Tazawa, H.; Yano, S.; Yoshida, R.; Yamasaki, Y.; Sasaki, T.; Hashimoto, Y.; Kuroda, S.; Ouchi, M.; Onishi, T.; Uno, F.; et al. Genetically engineered oncolytic adenovirus induces autophagic cell death through an E2F1-microRNA-7-epidermal growth factor receptor axis. Int. J. Cancer 2012, 131, 2939–2950. [Google Scholar] [CrossRef]
- Tong, Y.; You, L.; Liu, H.; Li, L.; Meng, H.; Qian, Q.; Qian, W. Potent antitumor activity of oncolytic adenovirus expressing Beclin-1 via induction of autophagic cell death in leukemia. Oncotarget 2013, 4, 860–874. [Google Scholar] [CrossRef]
- Liikanen, I.; Ahtiainen, L.; Hirvinen, M.L.; Bramante, S.; Cerullo, V.; Nokisalmi, P.; Hemminki, O.; Diaconu, I.; Pesonen, S.; Koski-Palkén, A.; et al. Oncolytic Adenovirus With Temozolomide Induces Autophagy and Antitumor Immune Responses in Cancer Patients. Mol. Ther. 2013, 21, 1212–1223. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Baird, S.K.; Aerts, J.L.; Eddaoudi, A.; Lockley, M.; Lemoine, N.R.; A McNeish, I. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 2007, 27, 3081–3090. [Google Scholar] [CrossRef] [Green Version]
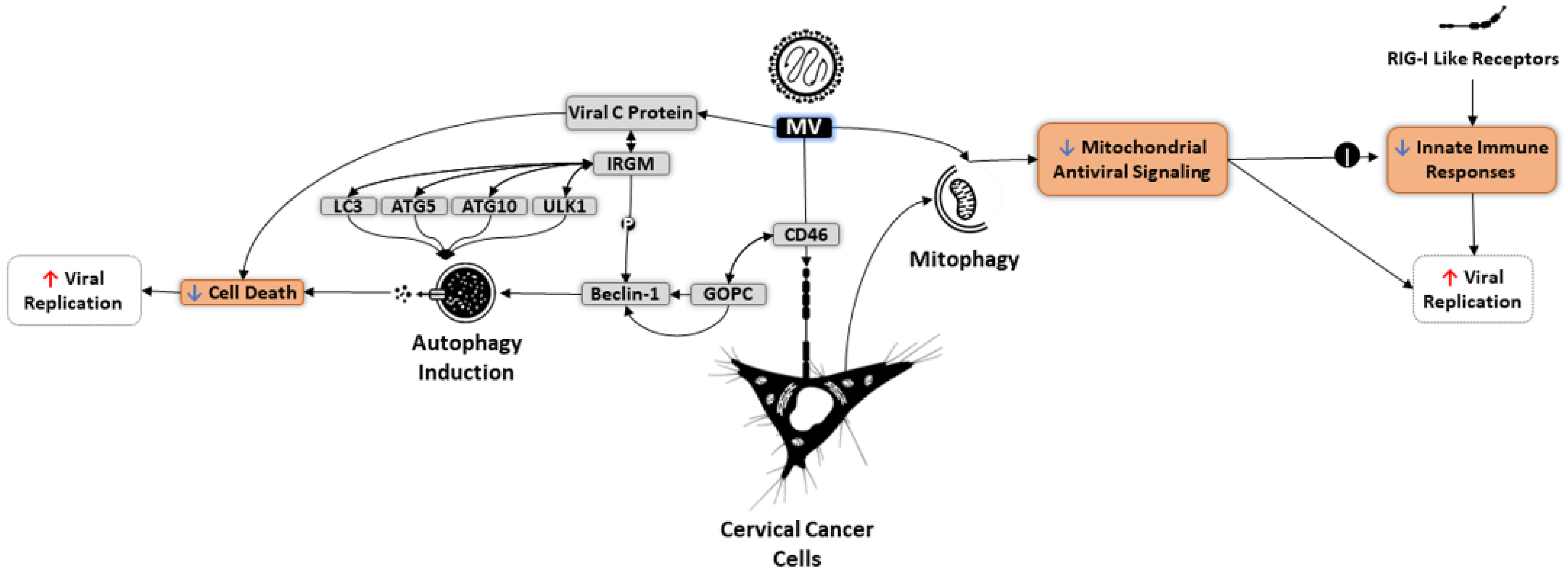
- Petkova, D.S.; Viret, C.; Faure, M. IRGM in autophagy and viral infections. Front. Immunol. 2013, 3, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Miri, S.M.; Ghaemi, A. Oncolytic paramyxoviruses-induced autophagy; a prudent weapon for cancer therapy. J. Biomed. Sci. 2019, 26, 1–11. [Google Scholar]
- Hansen, M.D.; Johnsen, I.B.; Stiberg, K.A.; Sherstova, T.; Wakita, T.; Richard, G.M.; Kandasamy, R.K.; Meurs, E.F.; Anthonsen, M.W. Hepatitis C virus triggers Golgi fragmentation and autophagy through the immunity-related GTPase M. Proc. Natl. Acad. Sci. USA 2017, 114, E3462–E3471. [Google Scholar] [CrossRef] [Green Version]
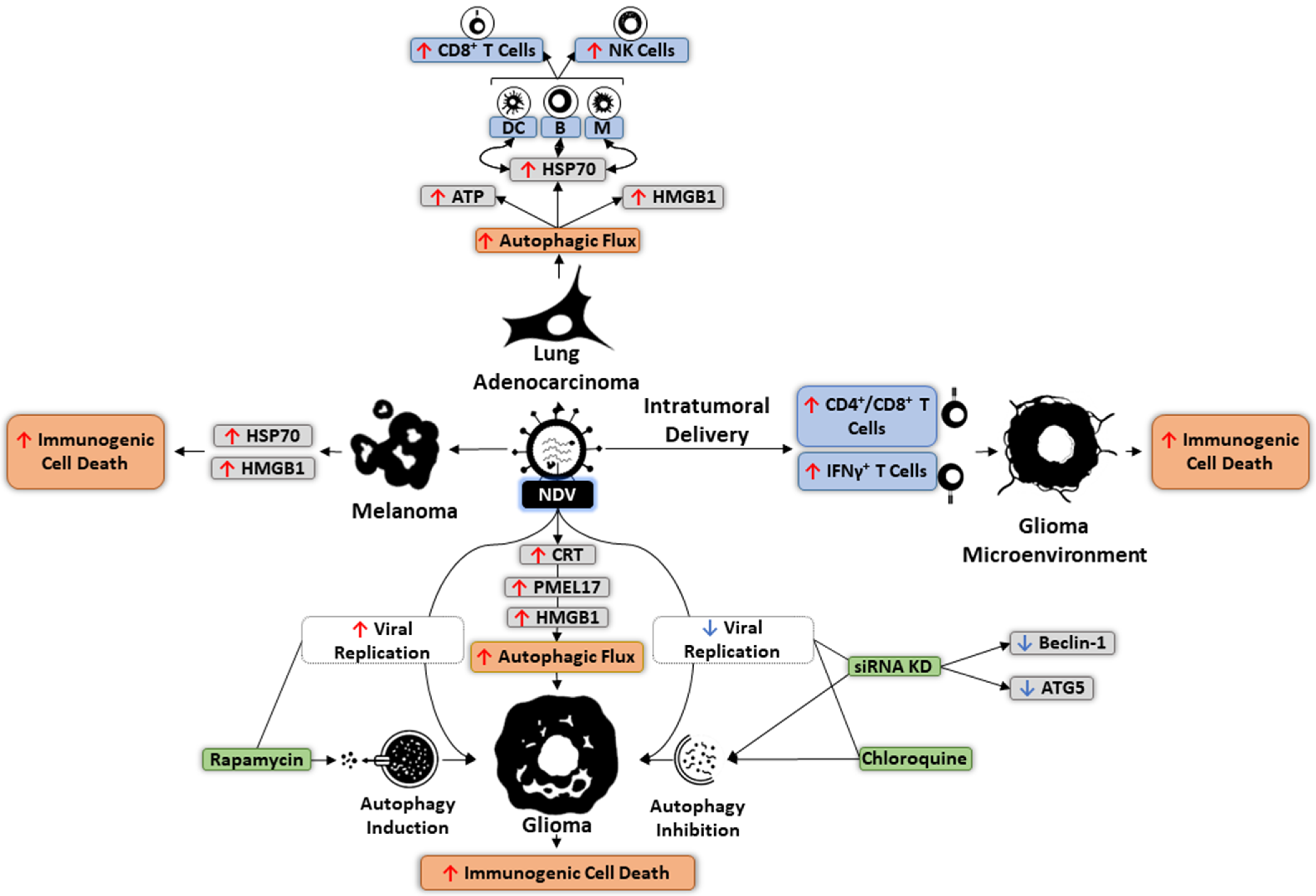
- Meng, C.; Zhou, Z.; Jiang, K.; Yu, S.; Jia, L.; Wu, Y.; Liu, Y.; Meng, S.; Ding, C. Newcastle disease virus triggers autophagy in U251 glioma cells to enhance virus replication. Arch. Virol. 2012, 157, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Koks, C.A.; Garg, A.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through the induction of immunogenic cell death. Int. J. Cancer 2014, 136, E313–E325. [Google Scholar] [CrossRef]
- Shao, X.; Wang, X.; Guo, X.; Jiang, K.; Ye, T.; Chen, J.; Fang, J.; Gu, L.; Wang, S.; Zhang, G.; et al. STAT3 Contributes To Oncolytic Newcastle Disease Virus-Induced Immunogenic Cell Death in Melanoma Cells. Front. Oncol. 2019, 9, 436. [Google Scholar] [CrossRef] [Green Version]
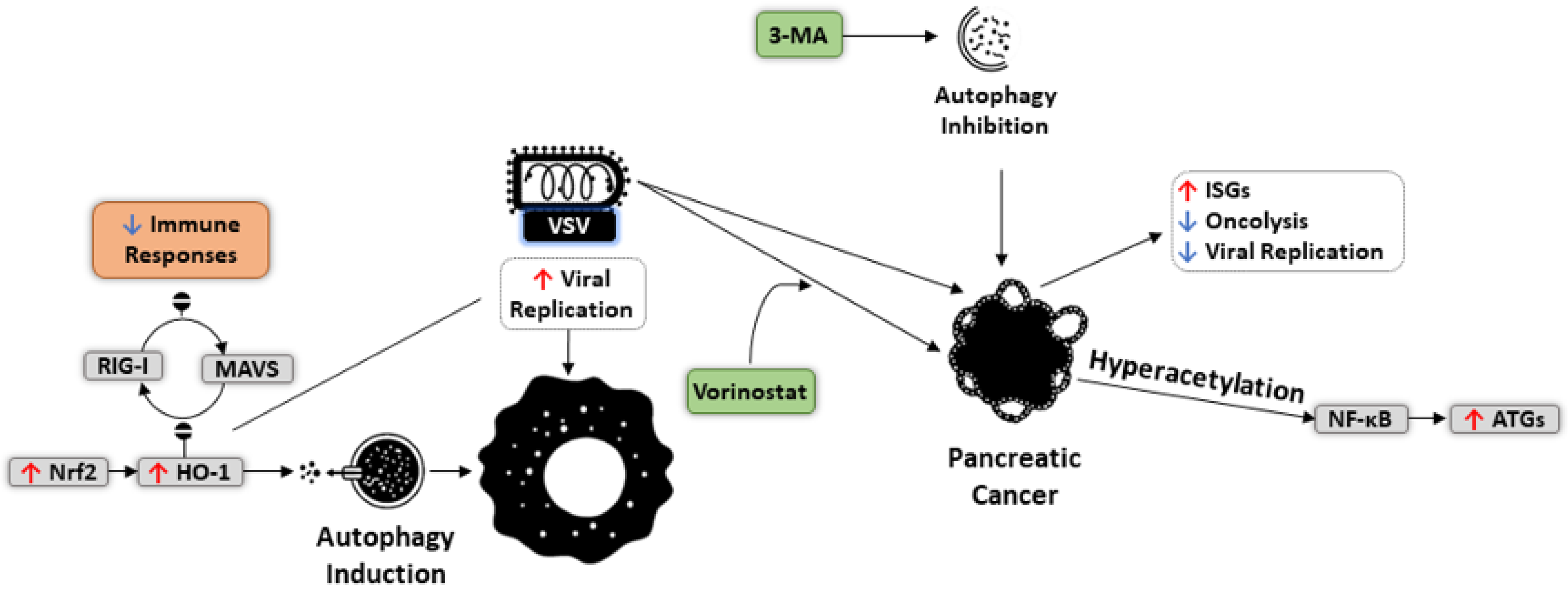
- Olagnier, D.; Lababidi, R.R.; Hadj, S.B.; Sze, A.; Liu, Y.; Naidu, S.D.; Ferrari, M.; Jiang, Y.; Chiang, C.; Beljanski, V.; et al. Activation of Nrf2 Signaling Augments Vesicular Stomatitis Virus Oncolysis via Autophagy-Driven Suppression of Antiviral Immunity. Mol. Ther. 2017, 25, 1900–1916. [Google Scholar] [CrossRef] [Green Version]
- Shulak, L. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-κB-dependent autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabizadeh, A.; Bamdad, T.; Arefian, E.; Nikoo, S.H.R. Autophagy Gene Activity May Act As a Key Factor for Sensitivity of Tumor Cells to Oncolytic Vesicular Stomatitis Virus. Iran. J. Cancer Prev. 2016, 9, e3919. [Google Scholar] [CrossRef]
- Yu, L.; Baxter, P.A.; Zhao, X.; Liu, Z.; Wadhwa, L.; Zhang, Y.; Su, J.M.F.; Tan, X.; Yang, J.; Adesina, A.; et al. A single intravenous injection of oncolytic picornavirus SVV-001 eliminates medulloblastomas in primary tumor-based orthotopic xenograft mouse models. Neuro-Oncol. 2010, 13, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Wang, S.; Xu, N.; Chen, Y.; Wu, G.; Zhang, A.; Chen, X.; Tong, Y.; Qian, W. Enhancing therapeutic efficacy of oncolytic vaccinia virus armed with Beclin-1, an autophagic Gene in leukemia and myeloma. Biomed. Pharmacother. 2020, 125, 110030. [Google Scholar] [CrossRef] [PubMed]
- Jiffry, J.; Thavornwatanayong, T.; Rao, D.; Fogel, E.J.; Saytoo, D.; Nahata, R.; Guzik, H.; Chaudhary, I.; Augustine, T.; Goel, S.; et al. Oncolytic Reovirus (pelareorep) Induces Autophagy in KRAS-mutated Colorectal Cancer. Clin. Cancer Res. 2020, 27, 865–876. [Google Scholar] [CrossRef] [PubMed]
- I Chi, P.; Huang, W.-R.; Lai, I.H.; Cheng, C.Y.; Liu, H.J. The p17 Nonstructural Protein of Avian Reovirus Triggers Autophagy Enhancing Virus Replication via Activation of Phosphatase and Tensin Deleted on Chromosome 10 (PTEN) and AMP-activated Protein Kinase (AMPK), as well as dsRNA-dependent Protein Kinase (PKR)/eIF2α Signaling Pathways. J. Biol. Chem. 2013, 288, 3571–3584. [Google Scholar] [PubMed] [Green Version]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE 1 RN ase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10, e7929. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Hu, C.; Xing, F.; Gao, M.; Liang, J.; Xiao, X.; Cai, J.; Tan, Y.; Hu, J.; Zhu, W.; et al. Deficiency of the IRE1α-Autophagy Axis Enhances the Antitumor Effects of the Oncolytic Virus M1. J. Virol. 2018, 92, e01331-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, Y.; Tan, J.; Zhang, Y.; Wong, C.W.; Lin, Z.; Liu, X.; Sander, M.; Yang, X.; Liang, L.; et al. Necroptotic virotherapy of oncolytic alphavirus M1 cooperated with Doxorubicin displays promising therapeutic efficacy in TNBC. Oncogene 2021, 1–13. [Google Scholar]
- Storniolo, A.; Alfano, V.; Carbotta, S.; Ferretti, E.; Di Renzo, L. IRE1α deficiency promotes tumor cell death and eIF2α degradation through PERK dipendent autophagy. Cell Death Discov. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tameire, F.; Verginadis, I.I.; Koumenis, C. Cell intrinsic and extrinsic activators of the unfolded protein response in cancer: Mechanisms and targets for therapy. Semin. Cancer Biol. 2015, 33, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-Tumor Interactome Screen Reveals ER Stress Response Can Reprogram Resistant Cancers for Oncolytic Virus-Triggered Caspase-2 Cell Death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V.; Suomalainen, M.; Pennauer, M.; Yakimovich, A.; Andriasyan, V.; Hemmi, S.; Greber, U.F. Chemical Induction of Unfolded Protein Response Enhances Cancer Cell Killing through Lytic Virus Infection. J. Virol. 2014, 88, 13086–13098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; HuangFu, W.-C.; Kumar, K.S.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Virus-Induced Unfolded Protein Response Attenuates Antiviral Defenses via Phosphorylation-Dependent Degradation of the Type I Interferon Receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Autophagy Role | Mechanism | Reference |
|---|---|---|---|
| Neuroblastoma (BE(2)-C and BE(2)-M17) | Inhibition | Degradation of gastrin-releasing peptide and inhibition of angiogenesis | [75] |
| Cervical Cancer (HeLa) | Promotion | Activating HIF-1α and increasing drug resistance (paclitaxel) | [76] |
| Gastric Cancer (MGC-803 and SGC-7901) | Inhibition | Downregulating HIF-1α and decreasing metastasis and glycolysis | [77] |
| Oral Cancer (SCC-9) | Inhibition | Suppressing the NF-κB pathway and inhibiting invasiveness | [78] |
| Bladder Cancer (T24) | Promotion | Increasing HIF-1α expression and counteracting gemcitabine-induced apoptosis | [79] |
| Pancreatic Cancer (PDAC) | Promotion | Degrading MHC-1 and boosting immune evasion | [80] |
| Breast Cancer (MCF-7) | Inhibition | Blocking nitric oxide generation and inducing apoptosis | [81] |
| Breast Cancer (D2A1 and MCF-7) | Promotion | Improving survival and metastasis | [82] |
| Hepatocellular Carcinoma (BEL7402 and HepG2) | Promotion | Elevating invasiveness via upregulating EMT markers (E-cadherin, CK18, and fibronectin) | [83] |
| Hepatocellular Carcinoma (SMMC-7721 and HepG2) | Promotion | Enhancing glycolysis and metastasis via upregulating MCT1 and activating Wnt/β-catenin | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahedi-Amiri, A.; Malone, K.; Beug, S.T.; Alain, T.; Yeganeh, B. Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer. Cells 2021, 10, 2672. https://doi.org/10.3390/cells10102672
Zahedi-Amiri A, Malone K, Beug ST, Alain T, Yeganeh B. Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer. Cells. 2021; 10(10):2672. https://doi.org/10.3390/cells10102672
Chicago/Turabian StyleZahedi-Amiri, Ali, Kyle Malone, Shawn T. Beug, Tommy Alain, and Behzad Yeganeh. 2021. "Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer" Cells 10, no. 10: 2672. https://doi.org/10.3390/cells10102672
APA StyleZahedi-Amiri, A., Malone, K., Beug, S. T., Alain, T., & Yeganeh, B. (2021). Autophagy in Tumor Immunity and Viral-Based Immunotherapeutic Approaches in Cancer. Cells, 10(10), 2672. https://doi.org/10.3390/cells10102672