
Impaired Bile Acid Metabolism and Gut Dysbiosis in Mice Lacking Lysosomal Acid Lipase

 , , ,
, , ,  , , and
, , and
Abstract
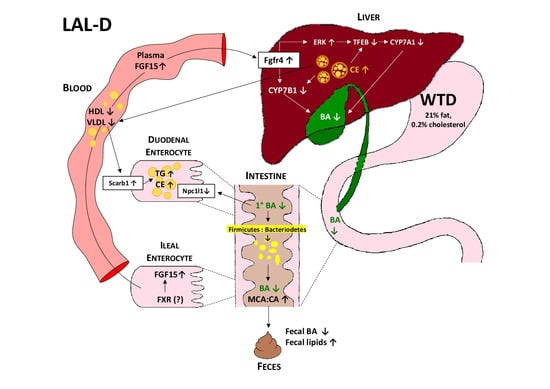
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Diets
2.2. Plasma Lipid Parameters, Lipoprotein Profiles, and 7α-hydroxy-4-cholesten-3-one (C4)
2.3. Analysis of Circulating FGF15 Concentrations
2.4. RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
2.5. Western Blotting Analysis
2.6. Electron Microscopy
2.7. Energy Metabolism In Vivo
2.8. Acute Cholesterol Absorption
2.9. Basolateral FA Uptake
2.10. Fecal Neutral Sterol Measurements
2.11. BA Measurements
2.12. Microbiota Analysis
2.13. Isolation of Primary Enterocytes
2.14. Immunohistochemical Hematoxylin and Eosin as Well as Oil-Red O (ORO) Staining
2.15. Statistics
3. Results
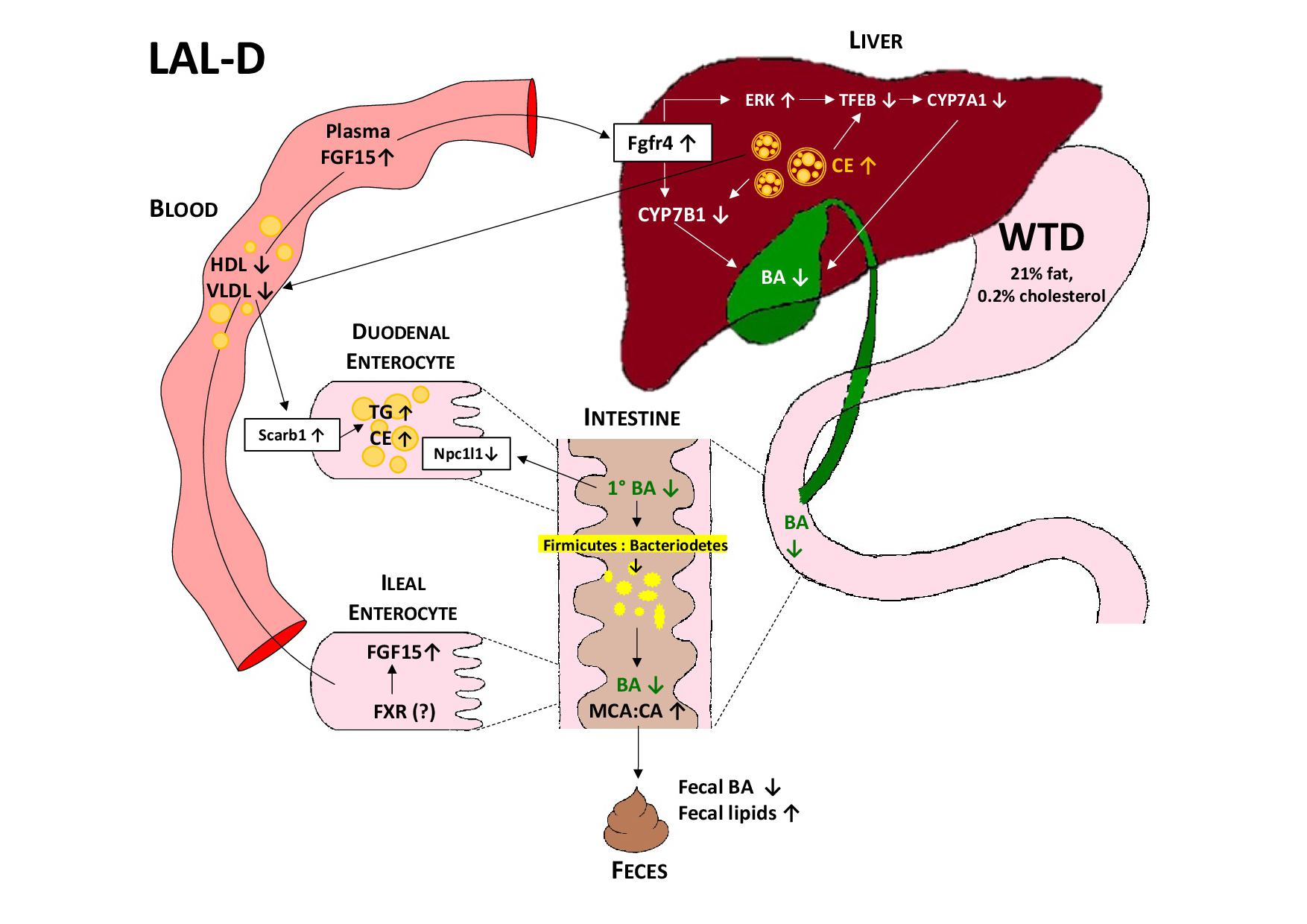
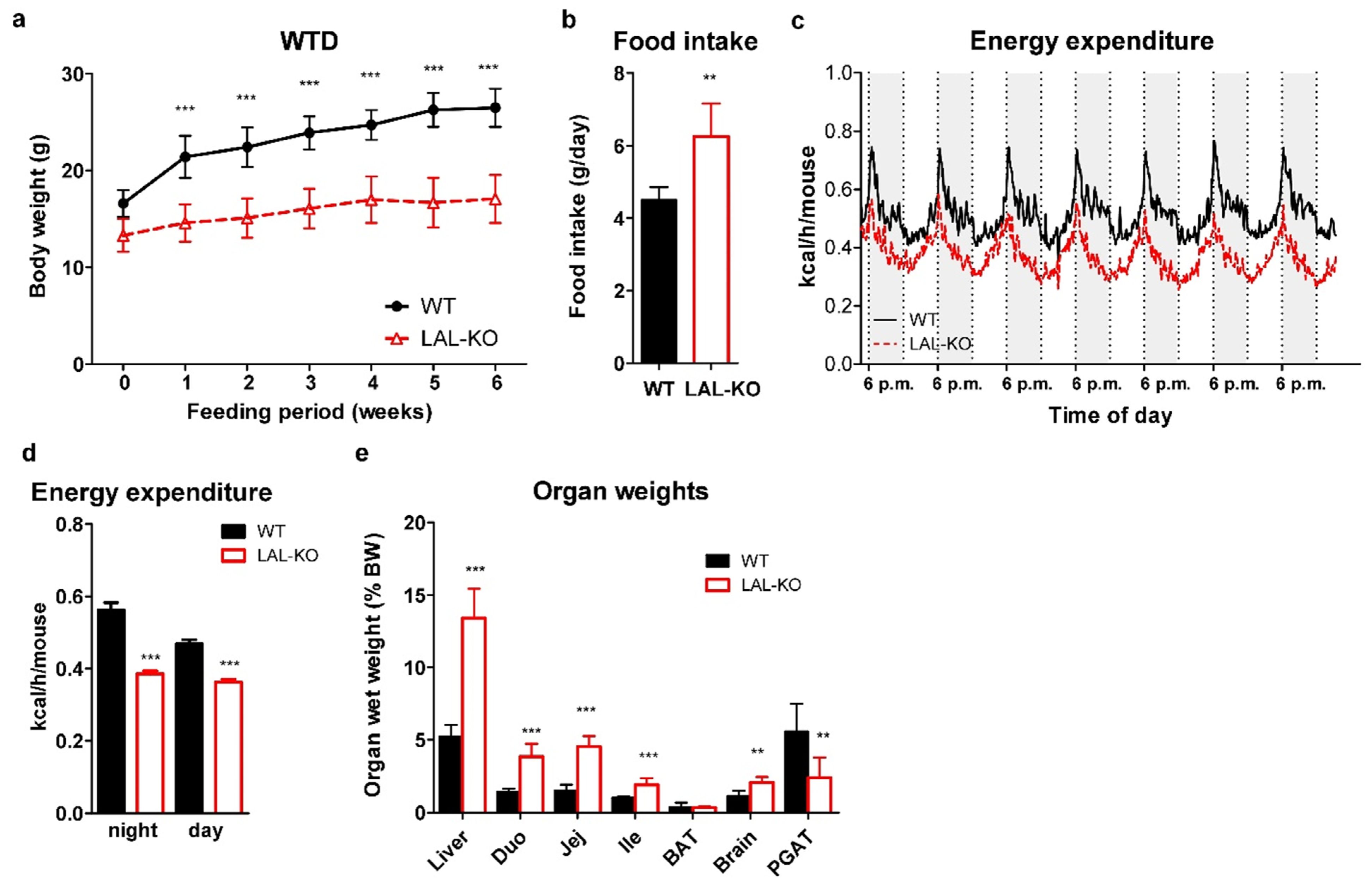
3.1. LAL-KO Mice Are Resistant to Diet-Induced Obesity
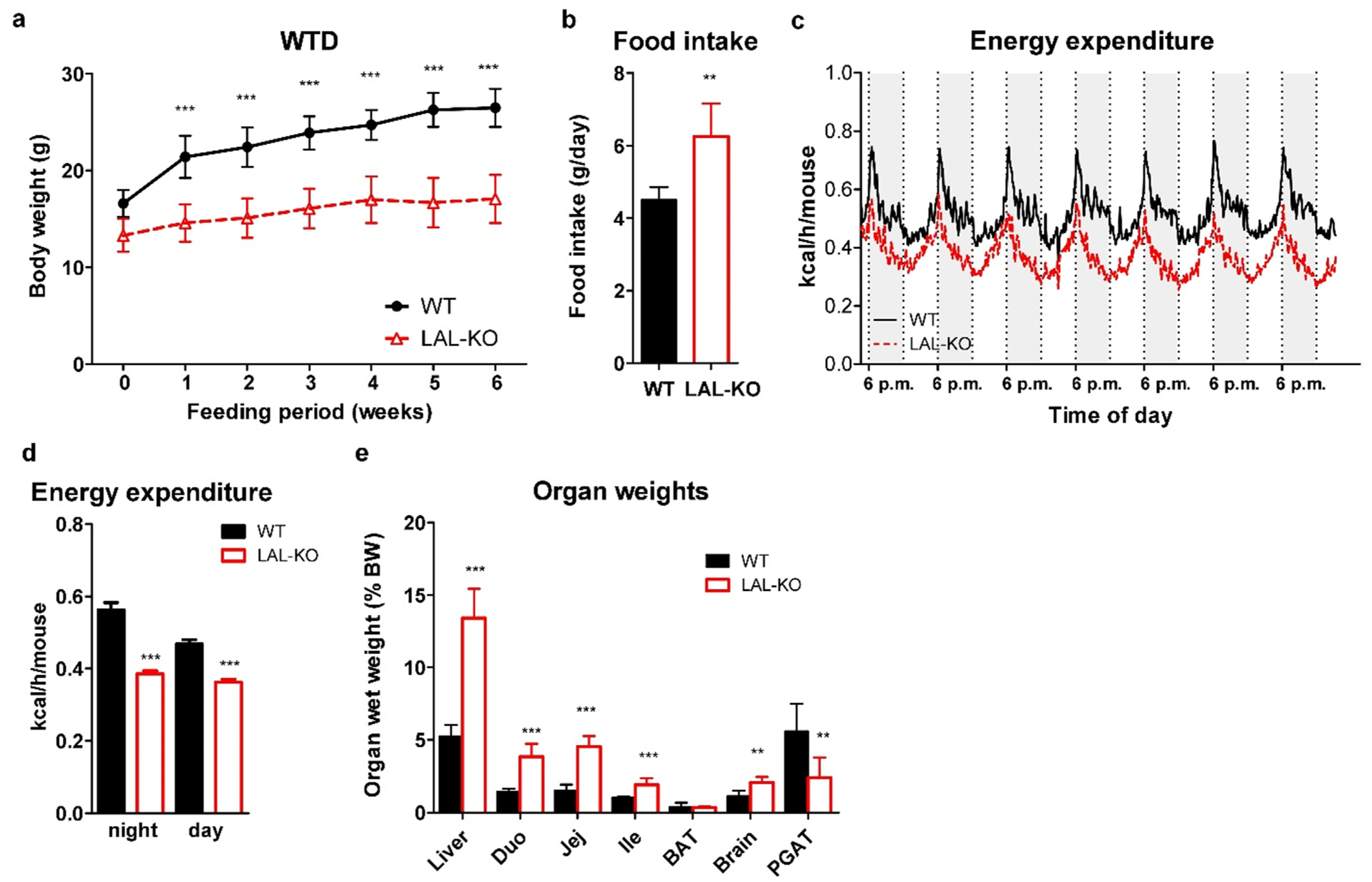
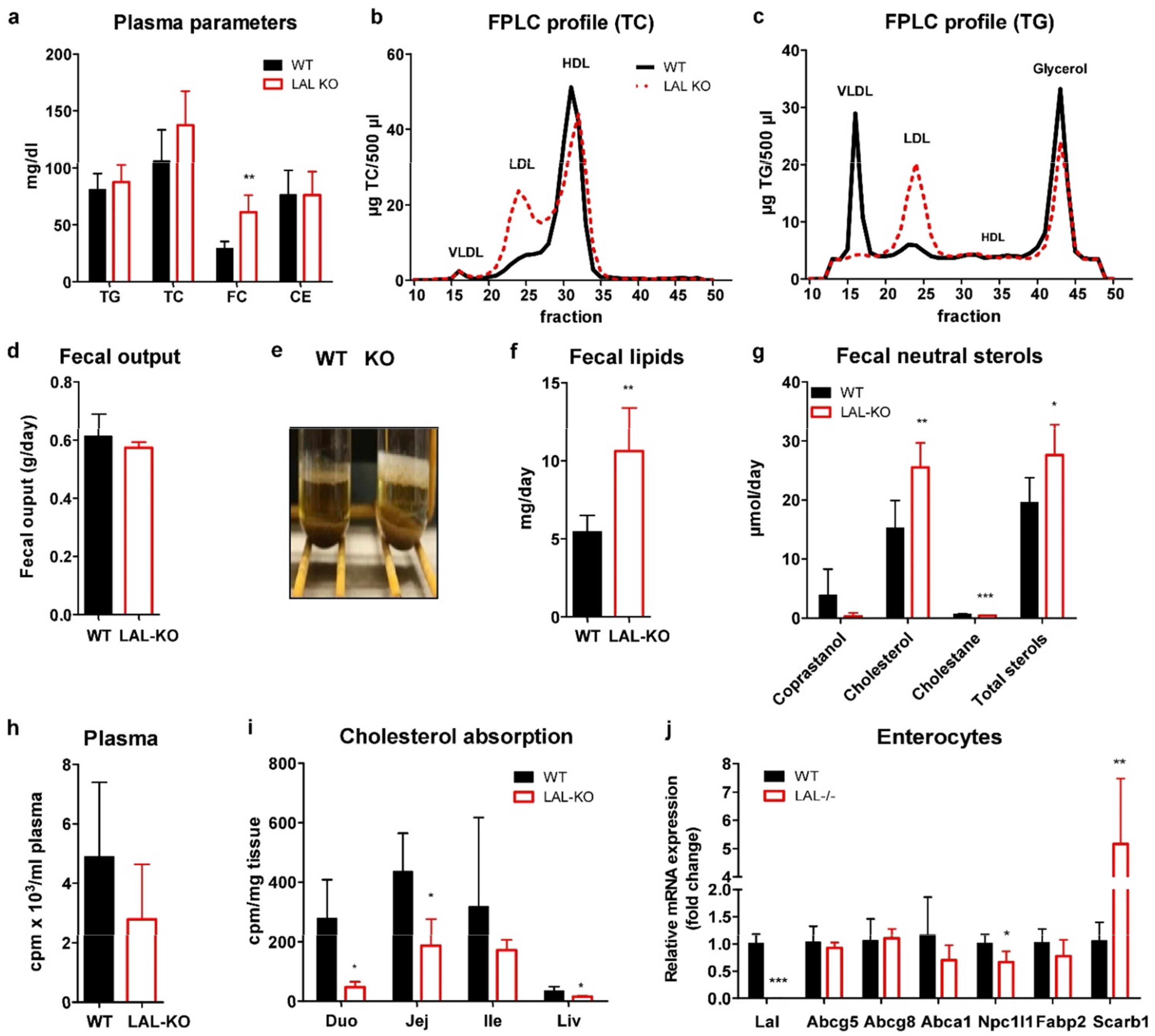
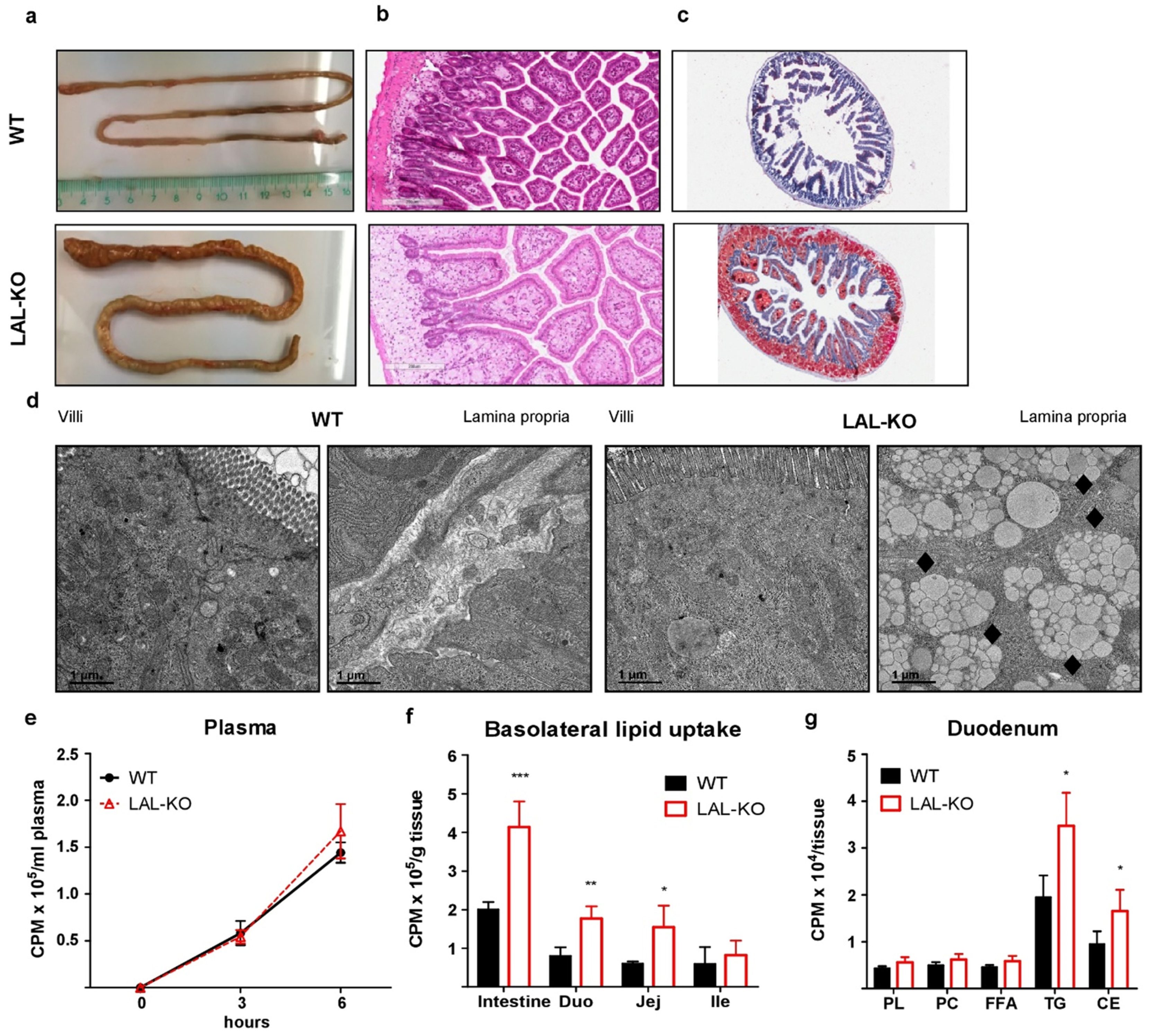
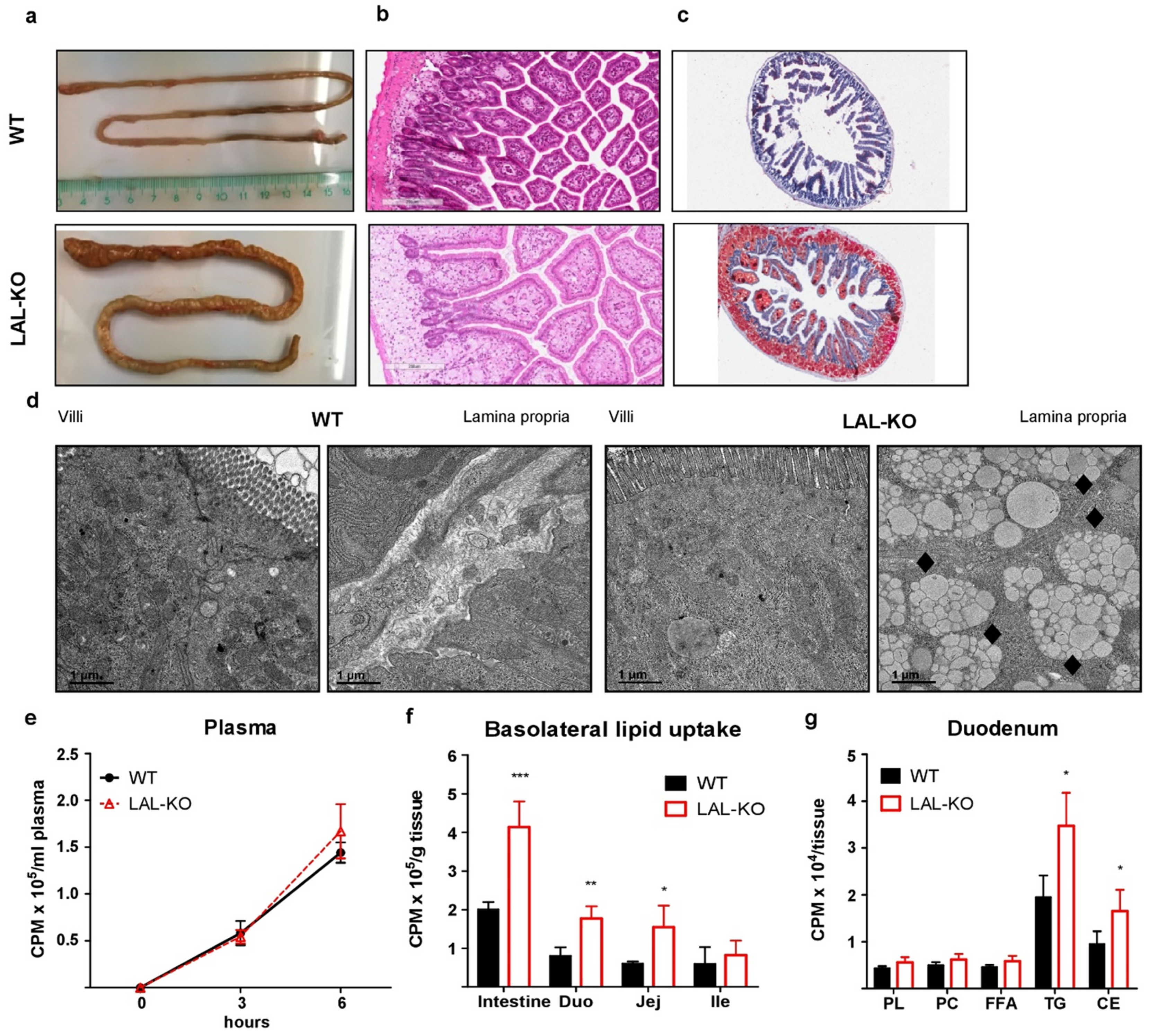
3.2. LAL-KO Mice Exhibit Impaired Cholesterol Absorption
3.3. LAL-KO Intestines Accumulate Lipids from the Systemic Circulation
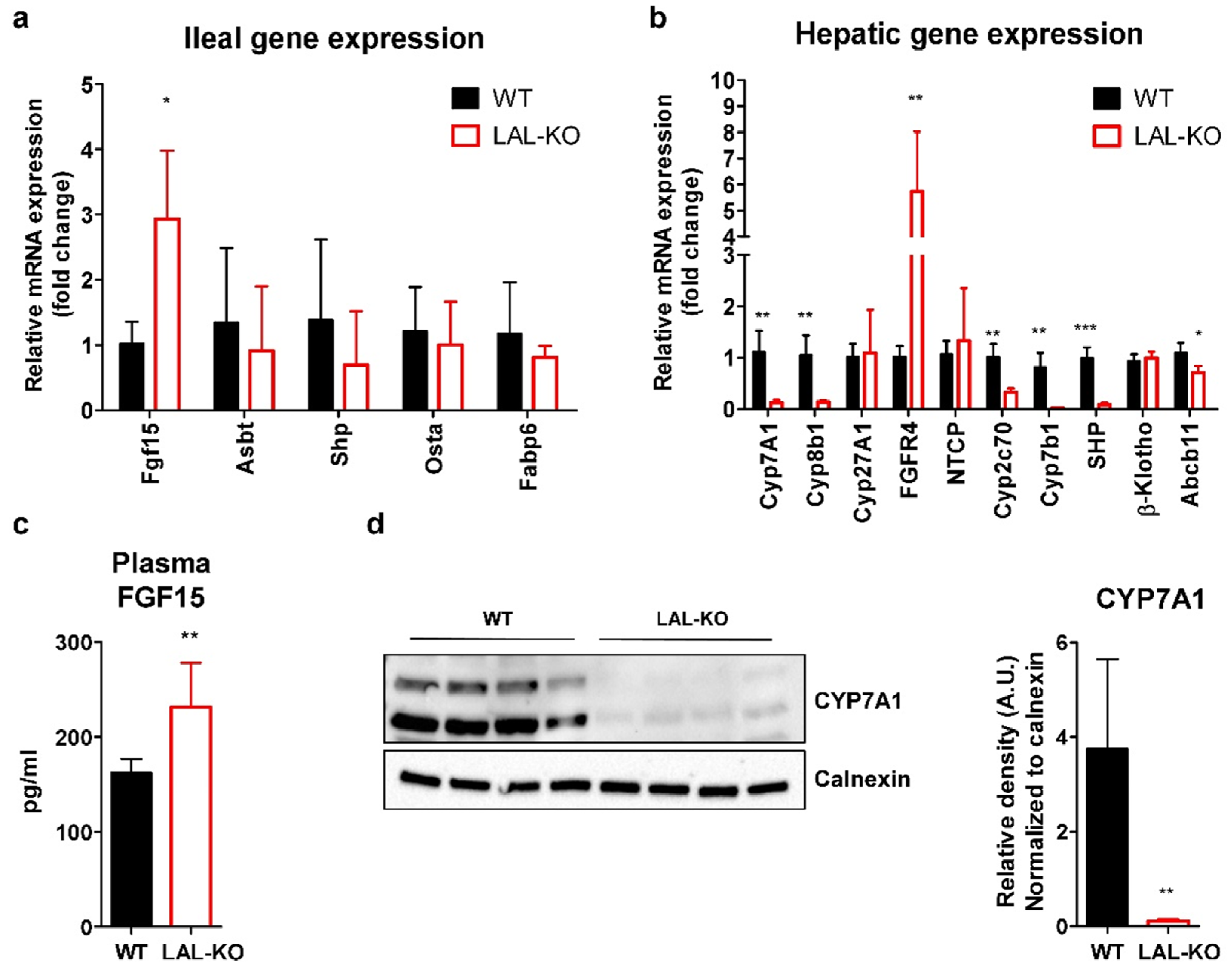
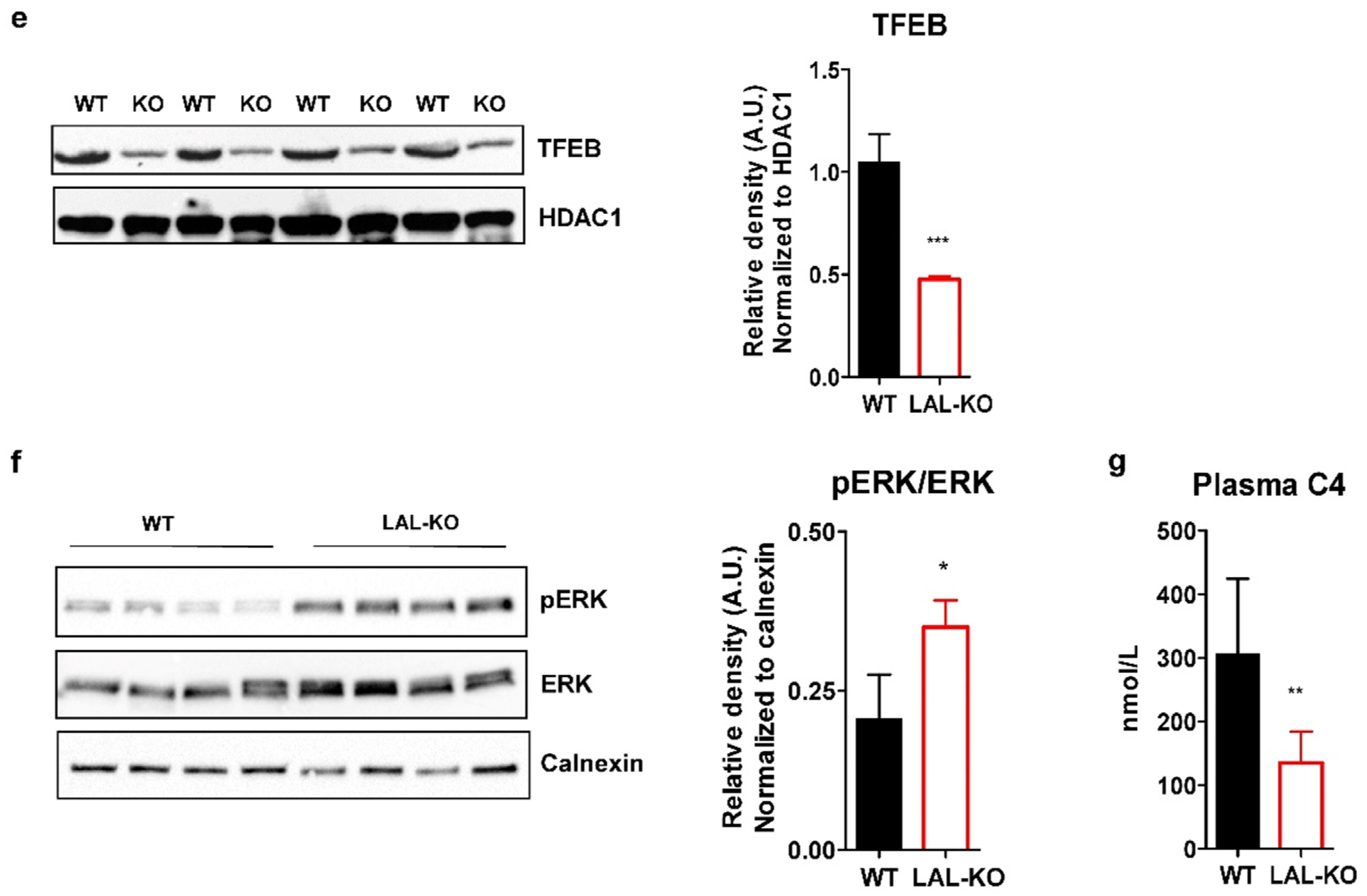
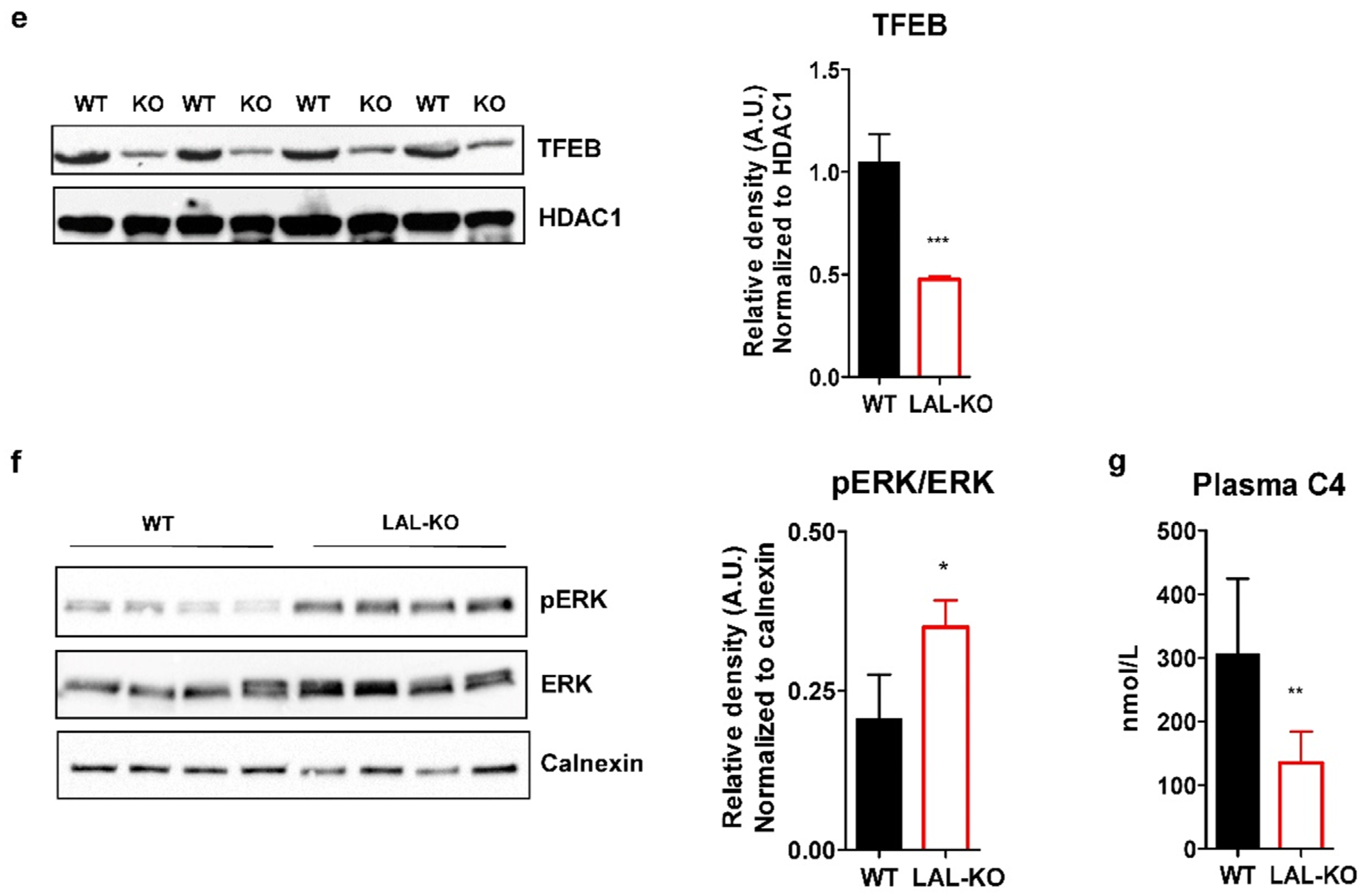
3.4. LAL-KO Mice Have Increased FGF15 Signaling
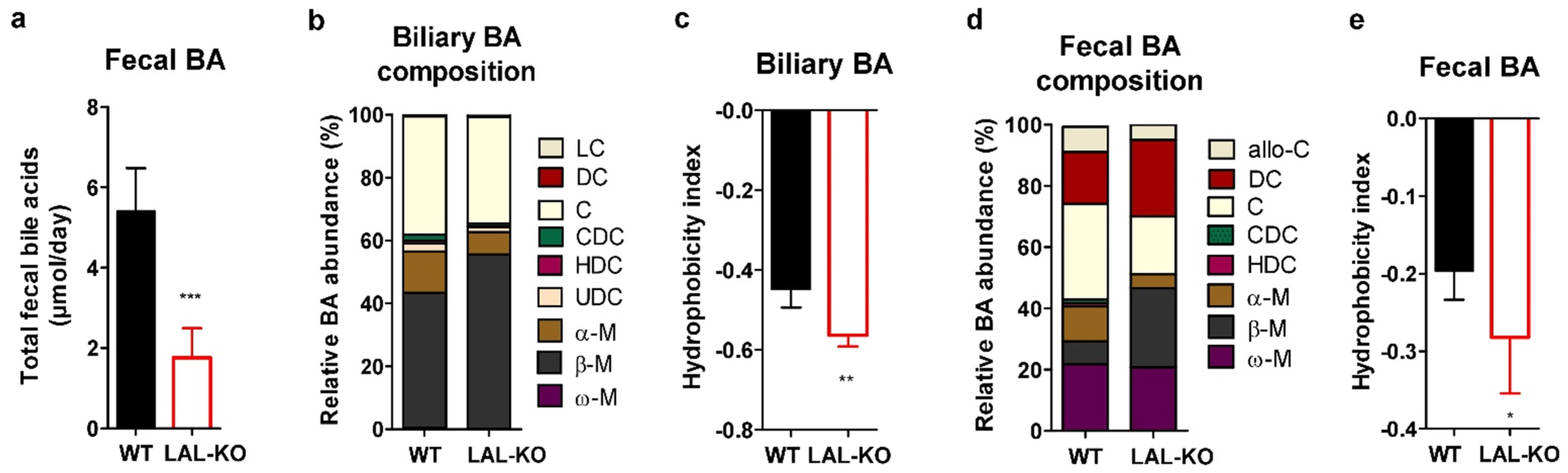
3.5. LAL-KO Mice Have Impaired BA Homeostasis
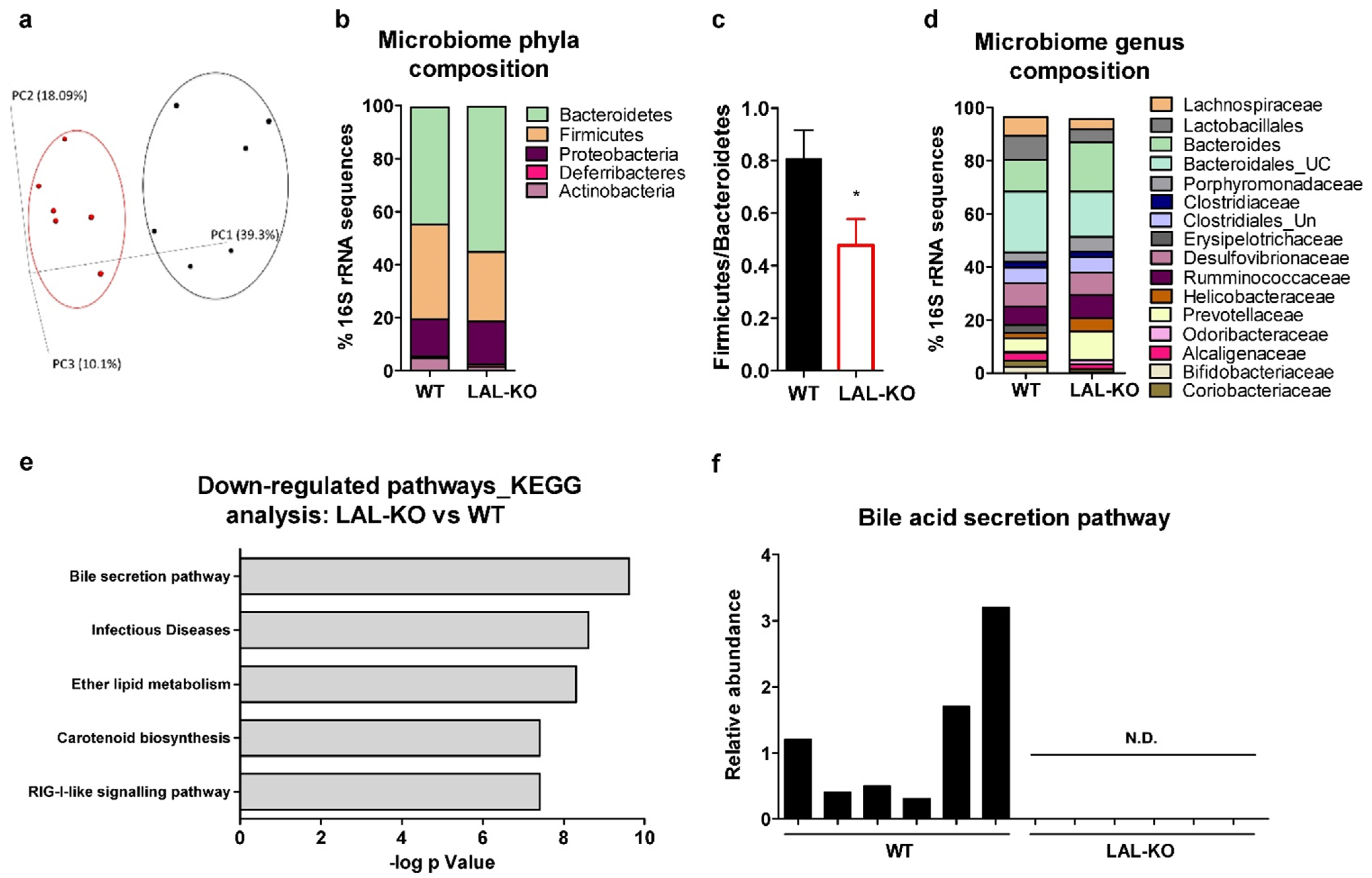
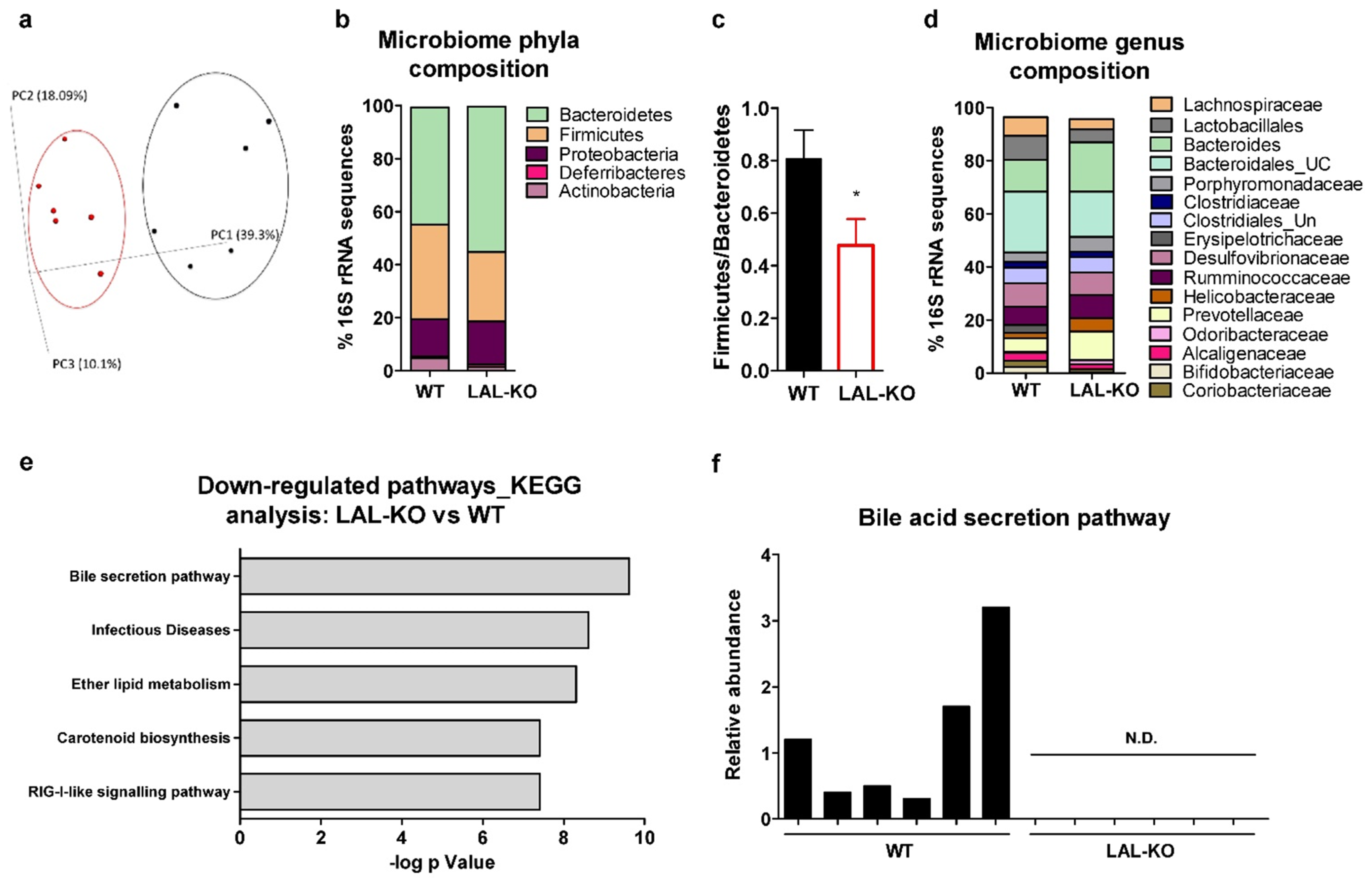
3.6. Drastic Alterations in the Gut Microbiome in LAL-KO Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nohturfft, A.; Zhang, S.C. Coordination of Lipid Metabolism in Membrane Biogenesis. Annu. Rev. Cell Dev. Biol. 2009, 25, 539–566. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, B.K.; Deegan, P.B.; Enns, G.M.; Guardamagna, O.; Horslen, S.; Hovingh, G.K.; Lobritto, S.J.; Malinova, V.; McLin, V.A.; Raiman, J.; et al. Clinical Features of Lysosomal Acid Lipase Deficiency. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Gomaraschi, M.; Bonacina, F.; Norata, G. Lysosomal Acid Lipase: From Cellular Lipid Handler to Immunometabolic Target. Trends Pharmacol. Sci. 2019, 40, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, M. Wolman’s disease and cholesteryl ester storage disorder: The phenotypic spectrum of lysosomal acid lipase deficiency. Lancet Gastroenterol. Hepatol. 2017, 2, 670–679. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Valayannopoulos, V.; Goodman, Z.D.; Balwani, M. Lysosomal Acid Lipase Deficiency: The Continuous Spectra of Disease Variants, in The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Aguisanda, F.; Thorne, N.; Zheng, W. Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development. Curr. Chem. Genom. Trans. Med. 2017, 11, 1–18. [Google Scholar] [CrossRef]
- Bernstein, D.L.; Hůlková, H.; Bialer, M.G.; Desnick, R.J. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol. 2013, 58, 1230–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Porto, A.F. Cholesteryl ester storage disease: Protean presentations of lysosomal acid lipase deficiency. J. Pediatr. Gastroenterol Nutr. 2013, 56, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.B.; Strong, A.; Ficicioglu, C. Persistent dyslipidemia in treatment of lysosomal acid lipase deficiency. Orphanet J. Rare Dis. 2020, 15, 1–7. [Google Scholar] [CrossRef]
- Du, H.; Witte, D.P.; A Grabowski, G. Tissue and cellular specific expression of murine lysosomal acid lipase mRNA and protein. J. Lipid Res. 1996, 37, 937–949. [Google Scholar] [CrossRef]
- Du, H.; Heur, M.; Duanmu, M.; Grabowski, G.A.; Hui, D.Y.; Witte, D.P.; Mishra, J. Lysosomal acid lipase-deficient mice: Depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 2001, 42, 489–500. [Google Scholar] [CrossRef]
- Schlager, S.; Vujic, N.; Korbelius, M.; Duta-Mare, M.; Dorow, J.; Leopold, C.; Rainer, S.; Wegscheider, M.; Reicher, H.; Ceglarek, U.; et al. Lysosomal lipid hydrolysis provides substrates for lipid mediator synthesis in murine macrophages. Oncotarget 2017, 8, 40037–40051. [Google Scholar] [CrossRef] [Green Version]
- Duta-Mare, M.; Sachdev, V.; Leopold, C.; Kolb, D.; Vujic, N.; Korbelius, M.; Hofer, D.C.; Xia, W.; Huber, K.; Auer, M.; et al. Lysosomal acid lipase regulates fatty acid channeling in brown adipose tissue to maintain thermogenesis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 467–478. [Google Scholar] [CrossRef]
- Gamblin, C.; Rouault, C.; Lacombe, A.; Langa-Vives, F.; Farabos, D.; Lamaziere, A.; Clément, K.; Gautier, E.L.; Yvan-Charvet, L.; Dugail, I. Lysosomal Acid Lipase Drives Adipocyte Cholesterol Homeostasis and Modulates Lipid Storage in Obesity, Independent of Autophagy. Diabetes 2020, 70, 76–90. [Google Scholar] [CrossRef]
- Radović, B.; Vujic, N.; Leopold, C.; Schlager, S.; Goeritzer, M.; Patankar, J.V.; Korbelius, M.; Kolb, D.; Reindl, J.; Wegscheider, M.; et al. Lysosomal acid lipase regulates VLDL synthesis and insulin sensitivity in mice. Diabetologia 2016, 59, 1743–1752. [Google Scholar] [CrossRef] [Green Version]
- Leopold, C.; Duta-Mare, M.; Sachdev, V.; Goeritzer, M.; Maresch, L.K.; Kolb, D.; Reicher, H.; Wagner, B.; Stojakovic, T.; Ruelicke, T.; et al. Hepatocyte-specific lysosomal acid lipase deficiency protects mice from diet-induced obesity but promotes hepatic inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 500–511. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M. Bile acid regulation of hepatic physiology: III. Regulation of bile acid synthesis: Past progress and future challenges. Am. J. Physiol. Liver Physiol. 2003, 284, G551–G557. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.F.; Hagey, L.R. Key discoveries in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [Green Version]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- de Boer, J.F.; Schonewille, M.; Boesjes, M.; Wolters, H.; Bloks, V.W.; Bos, T.; van Dijk, T.H.; Jurdzinski, A.; Boverhof, R.; Wolters, J.C.; et al. Intestinal Farnesoid X Receptor Controls Transintestinal Cholesterol Excretion in Mice. Gastroenterology 2017, 152, 1126–1138 e6. [Google Scholar] [CrossRef] [Green Version]
- Joyce, S.A.; Mac Sharry, J.; Casey, P.G.; Kinsella, M.; Murphy, E.F.; Shanahan, F.; Hill, C.; Gahan, C.G.M. Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. USA 2014, 111, 7421–7426. [Google Scholar] [CrossRef] [Green Version]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile Acid Is a Host Factor That Regulates the Composition of the Cecal Microbiota in Rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Wang, S.; Han, R.; Cao, Y.; Hua, W.; Mao, Y.; Zhang, X.; Pang, X.; Wei, C.; et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010, 4, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.F.; Cotter, P.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.; O’Toole, P.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef]
- Out, C.; Dikkers, A.; Laskewitz, A.; Boverhof, R.; Van Der Ley, C.; Kema, I.P.; Wolters, H.; Havinga, R.; Verkade, H.J.; Kuipers, F.; et al. Prednisolone increases enterohepatic cycling of bile acids by induction of Asbt and promotes reverse cholesterol transport. J. Hepatol. 2014, 61, 351–357. [Google Scholar] [CrossRef]
- Sachdev, V.; Leopold, C.; Bauer, R.; Patankar, J.V.; Iqbal, J.; Obrowsky, S.; Boverhofe, R.; Doktorovae, M.; Scheicherf, B.; Goeritzera, M.; et al. Novel role of a triglyceride-synthesizing enzyme: DGAT1 at the crossroad between triglyceride and cholesterol metabolism. Biochim. Biophys. Acta 2016, 1861, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Gupte, J.; Gong, Y.; Weiszmann, J.; Zhang, Y.; Lee, K.J. Chronic Over-expression of Fibroblast Growth Factor 21 Increases Bile Acid Biosynthesis by Opposing FGF15/19 Action. EBioMedicine 2017, 15, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Obrowsky, S.; Chandak, P.G.; Patankar, J.V.; Povoden, S.; Schlager, S.; Kershaw, E.E.; Bogner-Strauss, J.G.; Hoefler, G.; Levak-Frank, S.; Kratky, D. Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARalpha signaling. J. Lipid Res. 2013, 54, 425–435. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.F.; de Vries, H.D.; Palmiotti, A.; Li, R.; Doestzada, M.; Hoogerland, J.A.; Fu, J.; La Rose, A.M.; Westerterp, M.; Mulder, N.L.; et al. Cholangiopathy and Biliary Fibrosis in Cyp2c70-Deficient Mice Are Fully Reversed by Ursodeoxycholic Acid. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1045–1069. [Google Scholar] [CrossRef]
- Eggink, H.M.; Tambyrajah, L.L.; Berg, R.V.D.; Mol, I.M.; Heuvel, J.K.V.D.; Koehorst, M.; Groen, A.K.; Boelen, A.; Kalsbeek, A.; Romijn, J.A.; et al. Chronic infusion of taurolithocholate into the brain increases fat oxidation in mice. J. Endocrinol. 2018, 236, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Out, C.; Patankar, J.V.; Doktorova, M.; Boesjes, M.; Bos, T.; de Boer, S.; Havinga, R.; Wolters, H.; Boverhof, R.; van Dijk, T.H.; et al. Gut microbiota inhibit Asbt-dependent intestinal bile acid reabsorption via Gata4. J. Hepatol. 2015, 63, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Heuman, D.M. Quantitative estimation of the hydrophilic-hydrophobic balance of mixed bile salt solutions. J. Lipid Res. 1989, 30, 719–730. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Oresic, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-beta-muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Wurm, P.; Dörner, E.; Kremer, C.; Spranger, J.; Maddox, C.; Halwachs, B.; Harrison, U.; Blanchard, T.; Haas, R.; Högenauer, C.; et al. Qualitative and Quantitative DNA- and RNA-Based Analysis of the Bacterial Stomach Microbiota in Humans, Mice, and Gerbils. Msystems 2018, 3, e00262-18. [Google Scholar] [CrossRef] [Green Version]
- Korbelius, M.; Vujic, N.; Sachdev, V.; Obrowsky, S.; Rainer, S.; Gottschalk, B.; Graier, W.; Kratky, D. ATGL/CGI-58-Dependent Hydrolysis of a Lipid Storage Pool in Murine Enterocytes. Cell Rep. 2019, 28, 1923–1934.e4. [Google Scholar] [CrossRef] [Green Version]
- Erickson, R.P.; Bhattacharyya, A.; Hunter, R.J.; Heidenreich, R.A.; Cherrington, N.J. Liver disease with altered bile acid transport in Niemann-Pick C mice on a high-fat, 1% cholesterol diet. Am. J. Physiol. Liver Physiol. 2005, 289, G300–G307. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Lian, X.; Li, Y.; Dai, Y.; White, A.; Qin, Y.; Li, H.; Hume, D.A.; Du, H. Macrophage-specific expression of human lysosomal acid lipase corrects inflammation and pathogenic pheno-types in lal-/- mice. Am. J. Pathol. 2006, 169, 916–926. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Duanmu, M.; Witte, D.; Grabowski, G.A. Targeted disruption of the mouse lysosomal acid lipase gene: Long-term survival with massive cholesteryl ester and triglyceride storage. Hum. Mol. Genet. 1998, 7, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gunewardena, S.; Li, F.; Matye, D.J.; Chen, C.; Chao, X.; Jung, T.; Zhang, Y.; Czerwiński, M.; Ni, H.-M.; et al. An FGF15/19-TFEB regulatory loop controls hepatic cholesterol and bile acid homeostasis. Nat. Commun. 2020, 11, 3612. [Google Scholar] [CrossRef]
- Kreit, J. Microbial catabolism of sterols: Focus on the enzymes that transform the sterol 3beta-hydroxy-5-en into 3-keto-4-en. FEMS Microbiol. Lett. 2017, 364, fnx007. [Google Scholar] [CrossRef]
- Turnbaugh, P.; Ley, R.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.; Evans, T.D.; Jeong, S.-J.; Razani, B. Classical and alternative roles for autophagy in lipid metabolism. Curr. Opin. Lipidol. 2018, 29, 203–211. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Mengel, E.; Brassier, A.; Grabowski, G. Lysosomal acid lipase deficiency: Expanding differential diagnosis. Mol. Genet. Metab. 2016, 120, 62–66. [Google Scholar] [CrossRef]
- Tiwari, S.; Siddiqi, S.A. Intracellular Trafficking and Secretion of VLDL. Arter. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Bowden, K.L.; Dubland, J.A.; Chan, T.; Xu, Y.-H.; Grabowski, G.A.; Du, H.; Francis, G.A. LAL (Lysosomal Acid Lipase) Promotes Reverse Cholesterol Transport In Vitro and In Vivo. Arter. Thromb. Vasc. Biol. 2018, 38, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- Amigo, L.; Mendoza, H.; Castro, J.; Quiñones, V.; Miquel, J.F.; Zanlungo, S. Relevance of Niemann-Pick type C1 protein expression in controlling plasma cholesterol and biliary lipid secretion in mice. Hepatology 2002, 36, 819–828. [Google Scholar]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Fröhlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.; Ferguson, S.M. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Author response: Intracellular sphingosine releases calcium from lysosomes. eLife 2015. [Google Scholar] [CrossRef]
- Worthmann, A.; John, C.; Rühlemann, M.C.; Baguhl, M.; Heinsen, F.-A.; Schaltenberg, N.; Heine, M.; Schlein, C.; Evangelakos, I.; Mineo, C.; et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017, 23, 839–849. [Google Scholar] [CrossRef]
- Patankar, J.V.; Wong, C.K.; Morampudi, V.; Gibson, W.T.; Vallance, B.; Ioannou, G.N.; Hayden, M.R. Genetic ablation of Cyp8b1 preserves host metabolic function by repressing steatohepatitis and altering gut microbiota composition. Am. J. Physiol. Metab. 2018, 314, E418–E432. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Somm, E.; Henry, H.; Bruce, S.J.; Bonnet, N.; Montandon, S.A.; Niederländer, N.J.; Messina, A.; Aeby, S.; Rosikiewicz, M.; Fajas, L.; et al. beta-Klotho deficiency shifts the gut-liver bile acid axis and induces hepatic alterations in mice. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E833–E847. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.F.; Murphy, E.F.; Nilaweera, K.; Ross, P.R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity: New insights. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Li, J.; Zhou, N.-Y.; Tang, H.; Wang, Y. Gut Microbiota Composition Modifies Fecal Metabolic Profiles in Mice. J. Proteome Res. 2013, 12, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Tannock, G.W.; Dashkevicz, M.P.; Feighner, S.D. Lactobacilli and bile salt hydrolase in the murine intestinal tract. Appl. Environ. Microbiol. 1989, 55, 1848–1851. [Google Scholar] [CrossRef] [Green Version]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Di Tullio, G.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective Activation of Nuclear Bile Acid Receptor FXR in the Intestine Protects Mice Against Cholestasis. Gastroenterology 2012, 142, 355–365.e4. [Google Scholar] [CrossRef]
- de Wit, N.J.; Bosch-Vermeulen, H.; de Groot, P.J.; Hooiveld, G.J.; Bromhaar, M.M.G.; Jansen, J.; Müller, M.; van der Meer, R. The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC Med. Genom. 2008, 1, 14. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sachdev, V.; Duta-Mare, M.; Korbelius, M.; Vujić, N.; Leopold, C.; Freark de Boer, J.; Rainer, S.; Fickert, P.; Kolb, D.; Kuipers, F.; et al. Impaired Bile Acid Metabolism and Gut Dysbiosis in Mice Lacking Lysosomal Acid Lipase. Cells 2021, 10, 2619. https://doi.org/10.3390/cells10102619
Sachdev V, Duta-Mare M, Korbelius M, Vujić N, Leopold C, Freark de Boer J, Rainer S, Fickert P, Kolb D, Kuipers F, et al. Impaired Bile Acid Metabolism and Gut Dysbiosis in Mice Lacking Lysosomal Acid Lipase. Cells. 2021; 10(10):2619. https://doi.org/10.3390/cells10102619
Chicago/Turabian StyleSachdev, Vinay, Madalina Duta-Mare, Melanie Korbelius, Nemanja Vujić, Christina Leopold, Jan Freark de Boer, Silvia Rainer, Peter Fickert, Dagmar Kolb, Folkert Kuipers, and et al. 2021. "Impaired Bile Acid Metabolism and Gut Dysbiosis in Mice Lacking Lysosomal Acid Lipase" Cells 10, no. 10: 2619. https://doi.org/10.3390/cells10102619
APA StyleSachdev, V., Duta-Mare, M., Korbelius, M., Vujić, N., Leopold, C., Freark de Boer, J., Rainer, S., Fickert, P., Kolb, D., Kuipers, F., Radovic, B., Gorkiewicz, G., & Kratky, D. (2021). Impaired Bile Acid Metabolism and Gut Dysbiosis in Mice Lacking Lysosomal Acid Lipase. Cells, 10(10), 2619. https://doi.org/10.3390/cells10102619