
Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings

 , , and
, , and 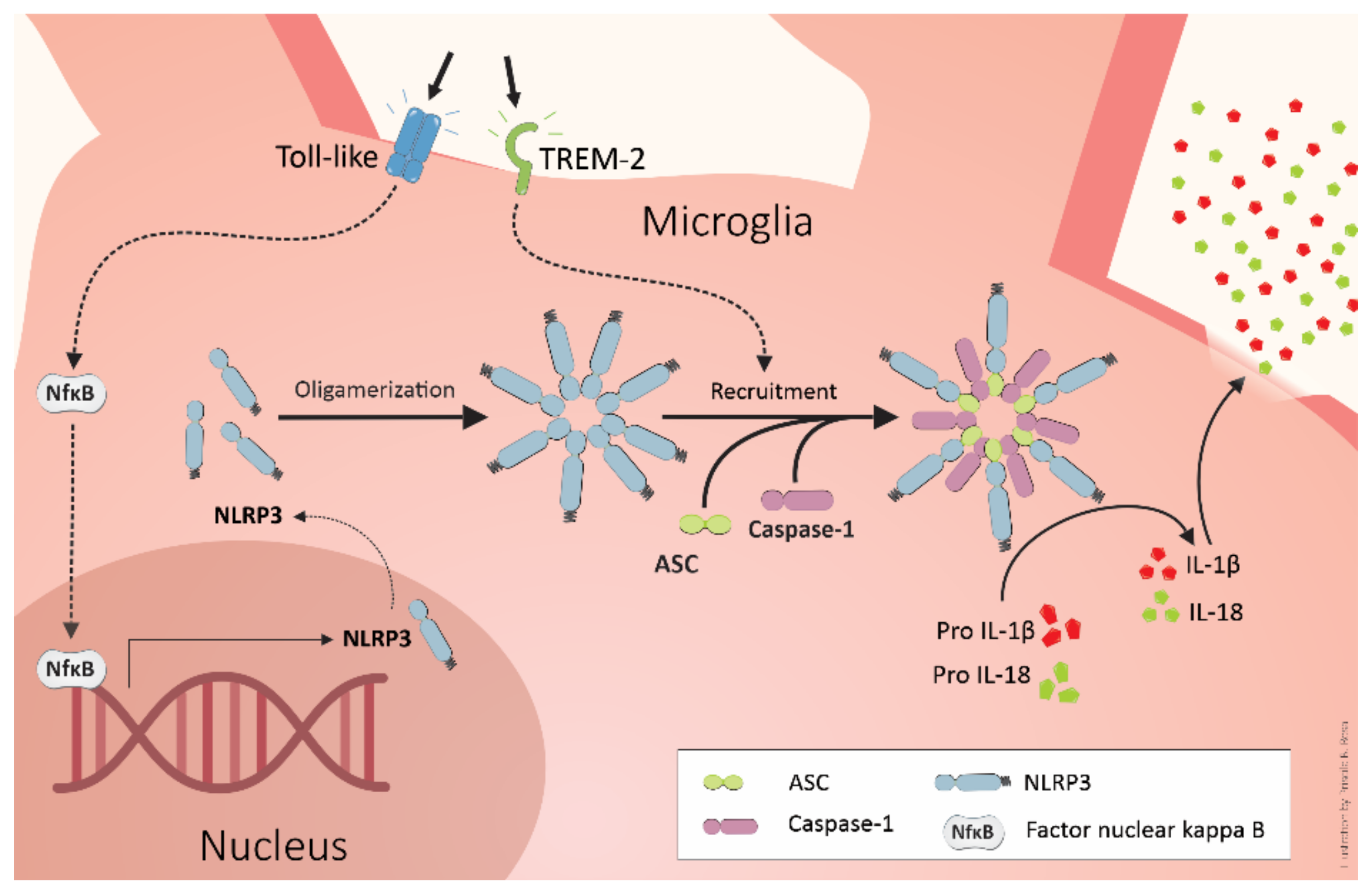{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathogenic Mechanisms of Alzheimer’s Disease
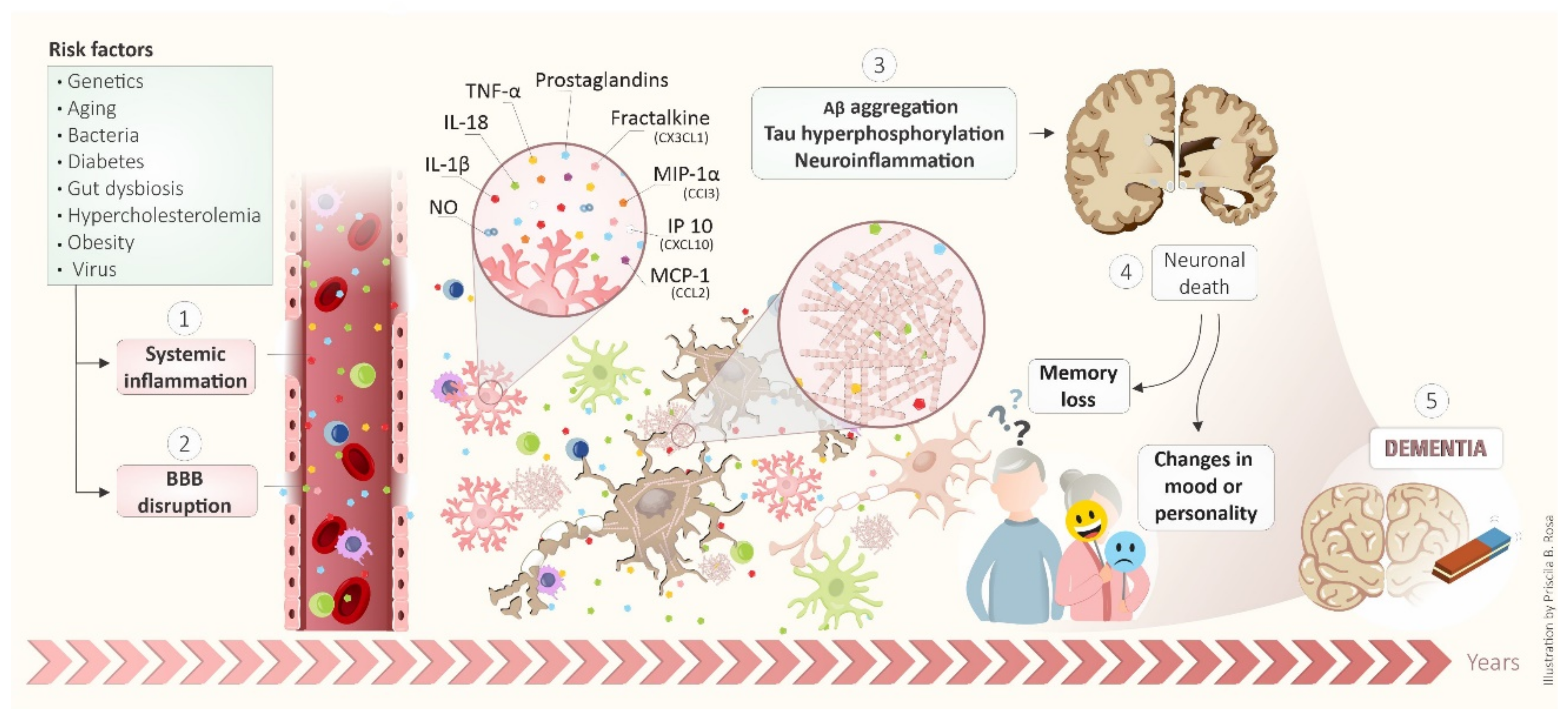
3. Inflammatory Cascade in Alzheimer’s Disease
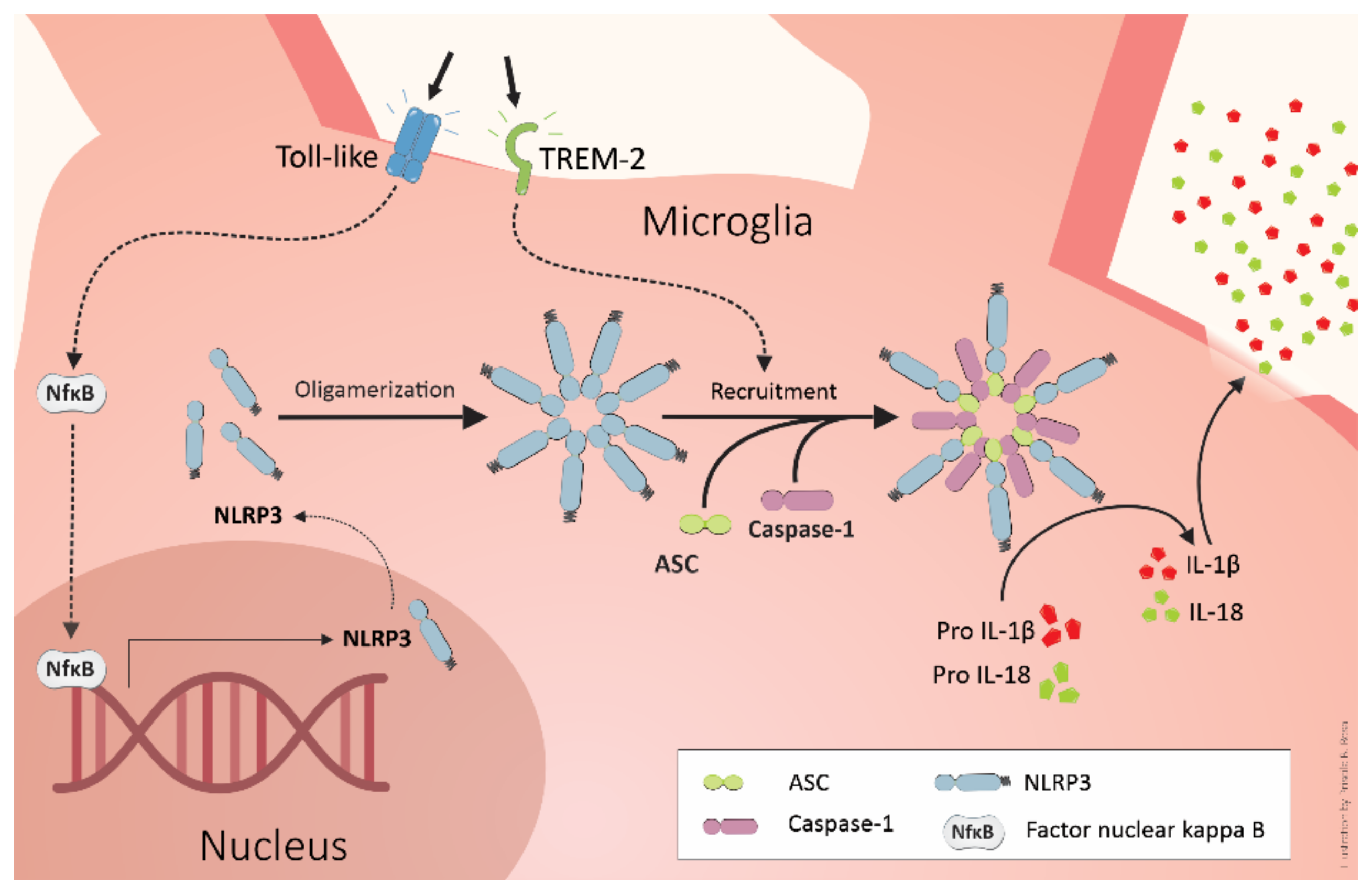
4. TREM2 and Alzheimer’s Pathogenesis
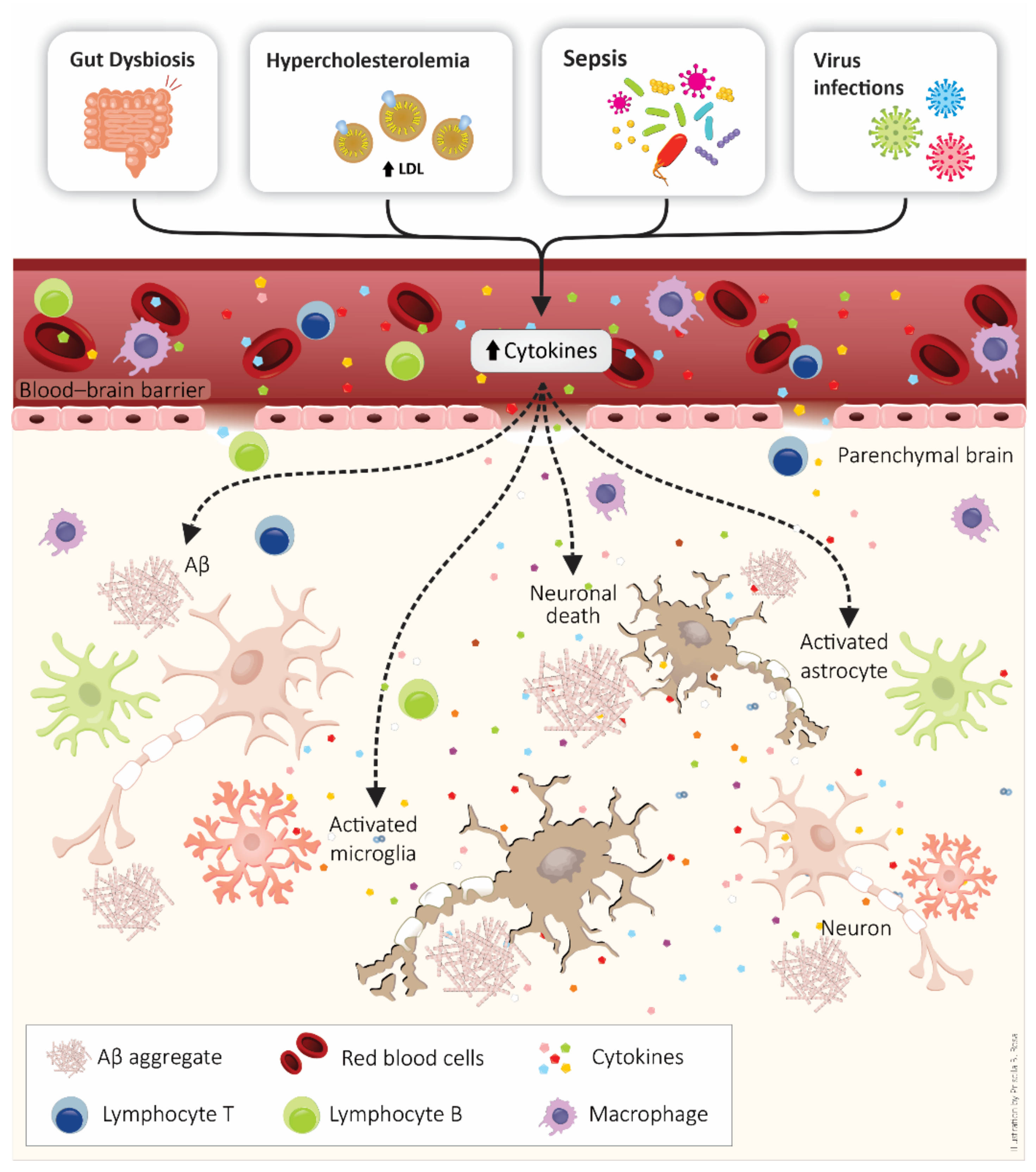
5. BBB as a Target of Systemic Inflammation: Importance to Alzheimer’s Disease Development
6. Systemic Inflammatory Diseases and the Connection with Alzheimer’s Disease Development
7. Anti-Inflammatory Approaches in the Alzheimer’s Disease
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ijaopo, E.O. Dementia-Related Agitation: A Review of Non-Pharmacological Interventions and Analysis of Risks and Benefits of Pharmacotherapy. Transl. Psychiatry 2017, 7, e1250. [Google Scholar] [CrossRef] [Green Version]
- Dementia Statistics | Alzheimer’s Disease International (ADI). Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 3 August 2021).
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., III. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimer’s Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, K. Neuroinflammation in Alzheimer’s Disease: Mechanisms, Pathologic Consequences, and Potential for Therapeutic Manipulation. J. Alzheimer’s Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Latta, C.H.; Brothers, H.M.; Wilcock, D.M. Neuroinflammation in Alzheimer’s Disease; a Source of Heterogeneity and Target for Personalized Therapy. Neuroscience 2015, 302, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Canevelli, M.; Bruno, G.; Cesari, M. The Sterile Controversy on the Amyloid Cascade Hypothesis. Neurosci. Biobehav. Rev. 2017, 83, 472–473. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Laurent, C.; Buée, L.; Blum, D. Tau and Neuroinflammation: What Impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2020, 109, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Andhey, P.S.; Ising, C.; Wang, K.; Snipes, L.L.; Boyer, K.; Lawson, S.; Yamada, K.; Qin, W.; Manis, M. Overexpressing Low-Density Lipoprotein Receptor Reduces Tau-Associated Neurodegeneration in Relation to ApoE-Linked Mechanisms. Neuron 2021, 109, 2413–2426.e7. [Google Scholar] [CrossRef] [PubMed]
- Panegyres, P.K.; Chen, H.-Y. Differences between Early and Late Onset Alzheimer’s Disease. Am. J. Neurodegener. Dis. 2013, 2, 300. [Google Scholar] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late Onset Alzheimer’s Disease Genetics Implicates Microglial Pathways in Disease Risk. Mol. Neurodegener. 2017, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s Disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—a Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A. A Path toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ma, Q.; Zhang, Y.; Xu, H. Proteolytic Processing of Alzheimer’s Β-amyloid Precursor Protein. J. Neurochem. REVIEW 2012, 120, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.-S.; Jo, S.A. Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. 2012, 20, 245. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.; Moreno-Gonzalez, I.; Taylor-Presse, K.; Edwards, G., III; Gamez, N.; Calderon, O.; Zhu, B.; Velasquez, F.C.; Soto, C.; Sevick-Muraca, E.M. Impaired Peripheral Lymphatic Function and Cerebrospinal Fluid Outflow in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 585–593. [Google Scholar] [CrossRef]
- Sun, X.; Chen, W.-D.; Wang, Y.-D. β-Amyloid: The Key Peptide in the Pathogenesis of Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 221. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of Amyloid-β Peptide across the Blood-Brain Barrier: Implication for Therapies in Alzheimer’s Disease. CNS Neurol. Disord. -Drug Targets Former. Curr. Drug Targets-CNS Neurol. Disord. 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally Secreted Oligomers of Amyloid β Protein Potently Inhibit Hippocampal Long-Term Potentiation in Vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Castellani, R.J.; Smith, M.A. Compounding Artefacts with Uncertainty, and an Amyloid Cascade Hypothesis That Is ‘Too Big to Fail’. J. Pathol. 2011, 224, 147–152. [Google Scholar] [CrossRef]
- Alexander, G.C.; Emerson, S.; Kesselheim, A.S. Evaluation of Aducanumab for Alzheimer Disease: Scientific Evidence and Regulatory Review Involving Efficacy, Safety, and Futility. JAMA 2021, 325, 1717–1718. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; de Strooper, B. The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Jones, D.T.; Greicius, M.D. Failure to Demonstrate Efficacy of Aducanumab: An Analysis of the EMERGE and ENGAGE Trials as Reported by Biogen, December 2019. Alzheimer’s Dement. 2021, 17, 696–701. [Google Scholar] [CrossRef]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory Response in the Hippocampus of PS1M146L/APP751SL Mouse Model of Alzheimer’s Disease: Age-Dependent Switch in the Microglial Phenotype from Alternative to Classic. J. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The Amyloid Cascade-Inflammatory Hypothesis of Alzheimer Disease: Implications for Therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef]
- Kidd, M. Paired Helical Filaments in Electron Microscopy of Alzheimer’s Disease. Nature 1963, 197, 192–193. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional Implications for the Microtubule-Associated Protein Tau: Localization in Oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Liu, C.-C.; Atagi, Y.; Chen, X.-F.; Jia, L.; Yang, L.; He, W.; Zhang, X.; Kang, S.S.; Rosenberry, T.L. Opposing Roles of the Triggering Receptor Expressed on Myeloid Cells 2 and Triggering Receptor Expressed on Myeloid Cells-like Transcript 2 in Microglia Activation. Neurobiol. Aging 2016, 42, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Cao, Y.; Ma, L.; Wei, Y.; Li, H. Possible Mechanisms of Tau Spread and Toxicity in Alzheimer’s Disease. Front. Cell Dev. Biol. 2021, 2064. [Google Scholar]
- Cras, P.; Kawai, M.; Lowery, D.; Gonzalez-DeWhitt, P.; Greenberg, B.; Perry, G. Senile Plaque Neurites in Alzheimer Disease Accumulate Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 1991, 88, 7552–7556. [Google Scholar] [CrossRef] [Green Version]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717F β-Amyloid Precursor Protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- McNamara, M.J.; Ruff, C.T.; Wasco, W.; Tanzi, R.E.; Thinakaran, G.; Hyman, B.T. Immunohistochemical and in Situ Analysis of Amyloid Precursor-like Protein-1 and Amyloid Precursor-like Protein-2 Expression in Alzheimer Disease and Aged Control Brains. Brain Res. 1998, 804, 45–51. [Google Scholar] [CrossRef]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M.-Y. Aβ Accelerates the Spatiotemporal Progression of Tau Pathology and Augments Tau Amyloidosis in an Alzheimer Mouse Model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef]
- Schönheit, B.; Zarski, R.; Ohm, T.G. Spatial and Temporal Relationships between Plaques and Tangles in Alzheimer-Pathology. Neurobiol. Aging 2004, 25, 697–711. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-Independent Regulators of Tau Pathology in Alzheimer Disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E.V.A. Staging of Alzheimer’s Disease-Related Neurofibrillary Changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Boche, D.; Nicoll, J.A.R. Invited Review–Understanding Cause and Effect in Alzheimer’s Pathophysiology: Implications for Clinical Trials. Neuropathol. Appl. Neurobiol. 2020, 46, 623–640. [Google Scholar] [CrossRef]
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated Microglia Mitigate Aβ-Associated Tau Seeding and Spreading. J. Exp. Med. 2021, 218, e20210542. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the Immune Response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A Meta-Analysis of Cytokines in Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D. Tracking Pathophysiological Processes in Alzheimer’s Disease: An Updated Hypothetical Model of Dynamic Biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Miller, R.G.; Madison, C.; Jin, X.; Honrada, R.; Harris, W.; Katz, J.; Forshew, D.A.; McGrath, M.S. Systemic Immune System Alterations in Early Stages of Alzheimer’s Disease. J. Neuroimmunol. 2013, 256, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Dursun, E.; Gezen-Ak, D.; Hanağası, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, İ.L.; Alaylıoğlu, M.; Araz, Ö.S.; Önal, B. The Interleukin 1 Alpha, Interleukin 1 Beta, Interleukin 6 and Alpha-2-Macroglobulin Serum Levels in Patients with Early or Late Onset Alzheimer’s Disease, Mild Cognitive Impairment or Parkinson’s Disease. J. Neuroimmunol. 2015, 283, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-C.; Han, S.-H.; Mook-Jung, I. Peripheral Inflammatory Biomarkers in Alzheimer’s Disease: A Brief Review. BMB Rep. 2020, 53, 10. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; D’Introno, A.; Colacicco, A.M.; Capurso, C.; Todarello, O.; Pellicani, V.; Capurso, S.A.; Pietrarossa, G.; Santamato, V.; Capurso, A. Circulating Biomarkers of Cognitive Decline and Dementia. Clin. Chim. Acta 2006, 364, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Harries, L.W.; Bradley-Smith, R.M.; Llewellyn, D.J.; Pilling, L.C.; Fellows, A.; Henley, W.; Hernandez, D.; Guralnik, J.M.; Bandinelli, S.; Singleton, A. Leukocyte CCR2 Expression Is Associated with Mini-Mental State Examination Score in Older Adults. Rejuvenation Res. 2012, 15, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westin, K.; Buchhave, P.; Nielsen, H.; Minthon, L.; Janciauskiene, S.; Hansson, O. CCL2 Is Associated with a Faster Rate of Cognitive Decline during Early Stages of Alzheimer’s Disease. PLoS ONE 2012, 7, e30525. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.-C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; Jun, G.; DeStefano, A.L.; Bis, J.C.; Beecham, G.W. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, P.G.; Hoyt, K.B.; Boehme, K.; Mukherjee, S.; Crane, P.K.; Haines, J.L.; Mayeux, R.; Farrer, L.A.; Pericak-Vance, M.A.; Schellenberg, G.D. Assessment of the Genetic Variance of Late-Onset Alzheimer’s Disease. Neurobiol. Aging 2016, 41, 200.e13–200.e20. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.; van der Lee, S.J.; Naj, A.C.; Bellenguez, C.; Badarinarayan, N.; Jakobsdottir, J.; Kunkle, B.W.; Boland, A.; Raybould, R.; Bis, J.C. Rare Coding Variants in PLCG2, ABI3, and TREM2 Implicate Microglial-Mediated Innate Immunity in Alzheimer’s Disease. Nat. Genet. 2017, 49, 1373–1384. [Google Scholar] [CrossRef]
- Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; Carneiro, F.G.; Luz, A.P.; Schiavo, G.L.; Andrighetti, M.S.; Scheid, M.G.; Bolfe, R.P.; Budni, J. Minocycline Reduces Inflammatory Parameters in the Brain Structures and Serum and Reverses Memory Impairment Caused by the Administration of Amyloid β (1-42) in Mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 77, 23–31. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37, 152–163. [Google Scholar] [CrossRef]
- Garcez, M.L.; Mina, F.; Bellettini-Santos, T.; da Luz, A.P.; Schiavo, G.L.; Macieski, J.M.C.; Medeiros, E.B.; Marques, A.O.; Magnus, N.Q.; Budni, J. The Involvement of NLRP3 on the Effects of Minocycline in an AD-like Pathology Induced by β-Amyloid Oligomers Administered to Mice. Mol. Neurobiol. 2019, 56, 2606–2617. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s Disease Brain-Derived Amyloid-β-Mediated Inhibition of LTP in Vivo Is Prevented by Immunotargeting Cellular Prion Protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef]
- Borlikova, G.G.; Trejo, M.; Mably, A.J.; Mc Donald, J.M.; Frigerio, C.S.; Regan, C.M.; Murphy, K.J.; Masliah, E.; Walsh, D.M. Alzheimer Brain-Derived Amyloid β-Protein Impairs Synaptic Remodeling and Memory Consolidation. Neurobiol. Aging 2013, 34, 1315–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 Induces Alzheimer-Type Phosphorylation of Tau Protein by Deregulating the Cdk5/P35 Pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern Recognition Receptors and Central Nervous System Repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Huang, Q.-X.; Yang, S.-S.; Chu, J.; Wang, J.-Z.; Tian, Q. Melatonin in Alzheimer’s Disease. Int. J. Mol. Sci. 2013, 14, 14575–14593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Pozo, A.; Muzikansky, A.; Gómez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential Relationships of Reactive Astrocytes and Microglia to Fibrillar Amyloid Deposits in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Plaque-Associated Local Toxicity Increases over the Clinical Course of Alzheimer Disease. Am. J. Pathol. 2016, 186, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Zolezzi, J.M.; Inestrosa, N.C. Wnt/TLR Dialog in Neuroinflammation, Relevance in Alzheimer’s Disease. Front. Immunol. 2017, 8, 187. [Google Scholar] [CrossRef] [Green Version]
- el Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 Mediates the Innate Host Response to β-Amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [Green Version]
- Lukose, B.; Rani, P. G82S RAGE Polymorphism Influences Amyloid-RAGE Interactions Relevant in Alzheimer’s Disease Pathology. PLoS ONE 2020, 15, e0225487. [Google Scholar]
- Udan, M.L.D.; Ajit, D.; Crouse, N.R.; Nichols, M.R. Toll-like Receptors 2 and 4 Mediate Aβ (1–42) Activation of the Innate Immune Response in a Human Monocytic Cell Line. J. Neurochem. 2008, 104, 524–533. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T. TLR2 Is a Primary Receptor for Alzheimer’s Amyloid β Peptide to Trigger Neuroinflammatory Activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balducci, C.; Frasca, A.; Zotti, M.; la Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L. Toll-like Receptor 4-Dependent Glial Cell Activation Mediates the Impairment in Memory Establishment Induced by β-Amyloid Oligomers in an Acute Mouse Model of Alzheimer’s Disease. Brain Behav. Immun. 2017, 60, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Babić Leko, M.; Nikolac Perković, M.; Klepac, N.; Štrac, D.Š.; Borovečki, F.; Pivac, N.; Hof, P.R.; Šimić, G. IL-1β, IL-6, IL-10, and TNF α Single Nucleotide Polymorphisms in Human Influence the Susceptibility to Alzheimer’s Disease Pathology. J. Alzheimer’s Dis. 2020, 75, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate Immune Activation in Neurodegenerative Disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric Amyloid β Induces IL-1 β Processing via Production of ROS: Implication in Alzheimer’s Disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 Inflammasome Is Involved in the Innate Immune Response to Amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q. NLRP3 Deficiency Ameliorates Neurovascular Damage in Experimental Ischemic Stroke. J. Cereb. Blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.-S.; Yu, J.-T.; Jiang, T.; Zhu, X.-C.; Tan, L. The NLRP3 Inflammasome in Alzheimer’s Disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S. Fenamate NSAIDs Inhibit the NLRP3 Inflammasome and Protect against Alzheimer’s Disease in Rodent Models. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M. AIM2 and NLRP3 Inflammasomes Activate Both Apoptotic and Pyroptotic Death Pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s Disease: Chemokines Produced by Astrocytes and Chemokine Receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342. [Google Scholar]
- Gold, M.; el Khoury, J. β-Amyloid, Microglia, and the Inflammasome in Alzheimer’s Disease. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 37, pp. 607–611. [Google Scholar]
- Merino, J.J.; Muñetón-Gómez, V.; Alvárez, M.-I.; Toledano-Díaz, A. Effects of Cx3cr1 and Fractalkine Chemokines in Amyloid Beta Clearance and P-Tau Accumulation in Alzheimer, s Disease (Ad) Rodent Models: Is Fractalkine a Systemic Biomarker for Ad? Curr. Alzheimer Res. 2016, 13, 403–412. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2–DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Shen, Y.; Chuang, H.; Chiu, C.; Ye, Y.; Zhao, L. Neuroinflammation in Alzheimer’s Disease: Microglia, Molecular Participants and Therapeutic Choices. Curr. Alzheimer Res. 2019, 16, 659–674. [Google Scholar] [CrossRef]
- Butler, A.W.; Ng, M.Y.M.; Hamshere, M.L.; Forabosco, P.; Wroe, R.; Al-Chalabi, A.; Lewis, C.M.; Powell, J.F. Meta-Analysis of Linkage Studies for Alzheimer’s Disease—a Web Resource. Neurobiol. Aging 2009, 30, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roussos, P.; Katsel, P.; Fam, P.; Tan, W.; Purohit, D.P.; Haroutunian, V. The Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) Is Associated with Enhanced Inflammation, Neuropathological Lesions and Increased Risk for Alzheimer’s Dementia. Alzheimer’s Dement. 2015, 11, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kober, D.L.; Stuchell-Brereton, M.D.; Kluender, C.E.; Dean, H.B.; Strickland, M.R.; Steinberg, D.F.; Nelson, S.S.; Baban, B.; Holtzman, D.M.; Frieden, C. Functional Insights from Biophysical Study of TREM2 Interactions with ApoE and Aβ1-42. Alzheimer’s Dement. 2021, 17, 475–488. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New Insights into the Role of TREM2 in Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kober, D.L.; Brett, T.J. TREM2-Ligand Interactions in Health and Disease. J. Mol. Biol. 2017, 429, 1607–1629. [Google Scholar] [CrossRef]
- Kleinberger, G.; Brendel, M.; Mracsko, E.; Wefers, B.; Groeneweg, L.; Xiang, X.; Focke, C.; Deußing, M.; Suárez-Calvet, M.; Mazaheri, F. The FTD-like Syndrome Causing TREM 2 T66M Mutation Impairs Microglia Function, Brain Perfusion, and Glucose Metabolism. EMBO J. 2017, 36, 1837–1853. [Google Scholar] [CrossRef]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential Proteolytic Processing of the Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) Protein by Ectodomain Shedding and γ-Secretase-Dependent Intramembranous Cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R. Amyloid-Beta Modulates Microglial Responses by Binding to the Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 1–12. [Google Scholar] [CrossRef]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Fernandes, A.; Brites, D. Microglia Change from a Reactive to an Age-like Phenotype with the Time in Culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia Show Altered Morphology and Reduced Arborization in Human Brain during Aging and A Lzheimer’s Disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.-L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [Green Version]
- van Acker, Z.P.; Perdok, A.; Bretou, M.; Annaert, W. The Microglial Lysosomal System in Alzheimer’s Disease: Guardian against Proteinopathy. Ageing Res. Rev. 2021, 101444. [Google Scholar] [CrossRef]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Casali, B.T.; Reed-Geaghan, E.G. Microglial Function and Regulation during Development, Homeostasis and Alzheimer’s Disease. Cells 2021, 10, 957. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, N.; Miller, F.; Cazaubon, S.; Couraud, P.-O. The Blood-Brain Barrier in Brain Homeostasis and Neurological Diseases. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood–Brain Barrier Impairment in Alzheimer Disease: Stability and Functional Significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [Green Version]
- Dickstein, D.L.; Walsh, J.; Brautigam, H.; Stockton, S.D., Jr.; Gandy, S.; Hof, P.R. Role of Vascular Risk Factors and Vascular Dysfunction in Alzheimer’s Disease. Mt. Sinai J. Med. J. Transl. Pers. Med. 2010, 77, 82–102. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujiie, M.; Dickstein, D.L.; Carlow, D.A.; Jefferies, W.A. Blood–Brain Barrier Permeability Precedes Senile Plaque Formation in an Alzheimer Disease Model. Microcirculation 2003, 10, 463–470. [Google Scholar]
- van de Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; van Osch, M.J.P.; van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Kozlowski, P.B. Evidence for Blood-brain Barrier Changes in Senile Dementia of the Alzheimer Type (SDAT). Ann. N. Y. Acad. Sci. 1982, 396, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, H.M.; Vorbrodt, A.W.; Wegiel, J. Amyloid Angiopathy and Blood–Brain Barrier Changes in Alzheimer’s Disease a, b. Ann. N. Y. Acad. Sci. 1997, 826, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Blood–Brain Barrier Dysfunction as a Cause and Consequence of Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Reuck, J.L. The Significance of Small Cerebral Bleeds in Neurodegenerative Dementia Syndromes. Aging Dis. 2012, 3, 307. [Google Scholar]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship Between Amyloid-β Deposition and Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Front. Cell. Neurosci. 2021, 15, 271. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J. Clearance of Alzheimer’s Amyloid-β 1-40 Peptide from Brain by LDL Receptor–Related Protein-1 at the Blood-Brain Barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A.; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E. RAGE, LRP-1, and Amyloid-Beta Protein in Alzheimer’s Disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- Gali, C.C.; Fanaee-Danesh, E.; Zandl-Lang, M.; Albrecher, N.M.; Tam-Amersdorfer, C.; Stracke, A.; Sachdev, V.; Reichmann, F.; Sun, Y.; Avdili, A. Amyloid-Beta Impairs Insulin Signaling by Accelerating Autophagy-Lysosomal Degradation of LRP-1 and IR-β in Blood-Brain Barrier Endothelial Cells in Vitro and in 3XTg-AD Mice. Mol. Cell. Neurosci. 2019, 99, 103390. [Google Scholar] [CrossRef]
- Huang, Z.; Wong, L.-W.; Su, Y.; Huang, X.; Wang, N.; Chen, H.; Yi, C. Blood-Brain Barrier Integrity in the Pathogenesis of Alzheimer’s Disease. Front. Neuroendocrinol. 2020, 59, 100857. [Google Scholar] [CrossRef]
- Kovac, A.; Zilkova, M.; Deli, M.A.; Zilka, N.; Novak, M. Human Truncated Tau Is Using a Different Mechanism from Amyloid-β to Damage the Blood-Brain Barrier. J. Alzheimer’s Dis. 2009, 18, 897–906. [Google Scholar] [CrossRef]
- Michalicova, A.; Majerova, P.; Kovac, A. Tau Protein and Its Role in Blood–Brain Barrier Dysfunction. Front. Mol. Neurosci. 2020, 13, 178. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Lee, S.C.; Mattiace, L.A.; Yen, S.C.; Brosnan, C. Microglia and Cytokines in Neurological Disease, with Special Reference to AIDS and Alzheimer’s Disease. Glia 1993, 7, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, F.L.; Ferri, C.; Licastro, F.; Veglia, F.; Biunno, I.; Gavazzi, A.; Calabrese, E.; Boneschi, F.M.; Sorbi, S.; Mariani, C. Interleukin-1B Polymorphism Is Associated with Age at Onset of Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 927–931. [Google Scholar] [CrossRef]
- Letiembre, M.; Liu, Y.; Walter, S.; Hao, W.; Pfander, T.; Wrede, A.; Schulz-Schaeffer, W.; Fassbender, K. Screening of Innate Immune Receptors in Neurodegenerative Diseases: A Similar Pattern. Neurobiol. Aging 2009, 30, 759–768. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 Signaling Rescues Cognition, Attenuates Tau Pathology, and Restores Neuronal β-Catenin Pathway Function in an Alzheimer’s Disease Model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eikelenboom, P.; van Exel, E.; Veerhuis, R.; Rozemuller, A.J.M.; van Gool, W.A.; Hoozemans, J.J.M. Innate Immunity and the Etiology of Late-Onset Alzheimer’s Disease. Neurodegener. Dis. 2012, 10, 271–273. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome Signalling in Brain Function and Neurodegenerative Disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Comparative Overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licastro, F.; Pedrini, S.; Caputo, L.; Annoni, G.; Davis, L.J.; Ferri, C.; Casadei, V.; Grimaldi, L.M.E. Increased Plasma Levels of Interleukin-1, Interleukin-6 and α-1-Antichymotrypsin in Patients with Alzheimer’s Disease: Peripheral Inflammation or Signals from the Brain? J. Neuroimmunol. 2000, 103, 97–102. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; van Swieten, J.C.; Stijnen, T.; Hofman, A.; Witteman, J.C.M.; Breteler, M.M.B. Inflammatory Proteins in Plasma and the Risk of Dementia: The Rotterdam Study. Arch. Neurol. 2004, 61, 668–672. [Google Scholar] [CrossRef] [Green Version]
- Tilvis, R.S.; Kähönen-Väre, M.H.; Jolkkonen, J.; Valvanne, J.; Pitkala, K.H.; Strandberg, T.E. Predictors of Cognitive Decline and Mortality of Aged People over a 10-Year Period. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2004, 59, M268–M274. [Google Scholar] [CrossRef] [Green Version]
- MERAZ RIOS, M.A.; Toral-Rios, D.; Franco-Bocanegra, D.; Villeda-Hernández, J.; Campos-Peña, V. Inflammatory Process in Alzheimer’s Disease. Front. Integr. Neurosci. 2013, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Takeda, S.; Sato, N.; Morishita, R. Systemic Inflammation, Blood-Brain Barrier Vulnerability and Cognitive/Non-Cognitive Symptoms in Alzheimer Disease: Relevance to Pathogenesis and Therapy. Front. Aging Neurosci. 2014, 6, 171. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The Blood-Brain Barrier in Systemic Inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The Effect of Systemic Inflammation on Human Brain Barrier Function. Brain Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef]
- Wichmann, M.A.; Cruickshanks, K.J.; Carlsson, C.M.; Chappell, R.; Fischer, M.E.; Klein, B.E.K.; Klein, R.; Tsai, M.Y.; Schubert, C.R. Long-term Systemic Inflammation and Cognitive Impairment in a Population-based Cohort. J. Am. Geriatr. Soc. 2014, 62, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic Risk Factors of Sporadic Alzheimer’s Disease: Implications in the Pathology, Pathogenesis and Treatment. Aging Dis. 2015, 6, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Gonzalez, I.; Edwards, G., III; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular Interaction between Type 2 Diabetes and Alzheimer’s Disease through Cross-Seeding of Protein Misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G.A., III; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, S.; Peferoen, L.A.N.; Vogel, D.Y.S.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in Neurodegenerative Diseases–an Update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Clarke, J.R.; Bomfim, T.R.; de Felice, F.G. Inflammation, Defective Insulin Signaling, and Neuronal Dysfunction in Alzheimer’s Disease. Alzheimer’s Dement. 2014, 10, S76–S83. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Chawla, A. Pleiotropic Actions of Insulin Resistance and Inflammation in Metabolic Homeostasis. Science 2013, 339, 172–177. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Parimisetty, A.; Dorsemans, A.-C.; Awada, R.; Ravanan, P.; Diotel, N.; d’Hellencourt, C.L. Secret Talk between Adipose Tissue and Central Nervous System via Secreted Factors—an Emerging Frontier in the Neurodegenerative Research. J. Neuroinflamm. 2016, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Martini, S.R.; Kent, T.A. Hyperglycemia in Acute Ischemic Stroke: A Vascular Perspective. J. Cereb. Blood Flow Metab. 2007, 27, 435–451. [Google Scholar] [CrossRef] [Green Version]
- Lohmann, C.; Schäfer, N.; von Lukowicz, T.; Stein, M.A.S.; Borén, J.; Rütti, S.; Wahli, W.; Donath, M.Y.; Lüscher, T.F.; Matter, C.M. Atherosclerotic Mice Exhibit Systemic Inflammation in Periadventitial and Visceral Adipose Tissue, Liver, and Pancreatic Islets. Atherosclerosis 2009, 207, 360–367. [Google Scholar] [CrossRef]
- Gustafson, D.R.; Karlsson, C.; Skoog, I.; Rosengren, L.; Lissner, L.; Blennow, K. Mid-life Adiposity Factors Relate to Blood–Brain Barrier Integrity in Late Life. J. Intern. Med. 2007, 262, 643–650. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes Mellitus and Blood-Brain Barrier Dysfunction: An Overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar]
- de Oliveira, J.; Moreira, E.L.G.; dos Santos, D.B.; Piermartiri, T.C.; Dutra, R.C.; Pinton, S.; Tasca, C.I.; Farina, M.; Prediger, R.D.S.; de Bem, A.F. Increased Susceptibility to Amyloid-β-Induced Neurotoxicity in Mice Lacking the Low-Density Lipoprotein Receptor. J. Alzheimer’s Dis. 2014, 41, 43–60. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, J.; Engel, D.F.; de Paula, G.C.; dos Santos, D.B.; Lopes, J.B.; Farina, M.; Moreira, E.L.G.; de Bem, A.F. High Cholesterol Diet Exacerbates Blood-Brain Barrier Disruption in LDLr–/–Mice: Impact on Cognitive Function. J. Alzheimer’s Dis. 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sparks, D.L. Activation of Microglia in the Brains of Humans with Heart Disease and Hypercholesterolemic Rabbits. J. Mol. Med. 1997, 75, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Sparks, D.L.; KUO, Y.; Roher, A.; Martin, T.; Lukas, R.J. Alterations of Alzheimer’s Disease in the Cholesterol-fed Rabbit, Including Vascular Inflammation: Preliminary Observations. Ann. N. Y. Acad. Sci. 2000, 903, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, R.P.J.; Schommer, E.; Thomasson, S.; Thompson, A.; Feist, G.; Ghribi, O. Regulation of β-Amyloid Levels in the Brain of Cholesterol-Fed Rabbit, a Model System for Sporadic Alzheimer’s Disease. Mech. Ageing Dev. 2008, 129, 649–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High Cholesterol-induced Neuroinflammation and Amyloid Precursor Protein Processing Correlate with Loss of Working Memory in Mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlokovic, B.V. Clearing Amyloid through the Blood–Brain Barrier. J. Neurochem. 2004, 89, 807–811. [Google Scholar] [CrossRef]
- Desale, S.E.; Chinnathambi, S. Role of Dietary Fatty Acids in Microglial Polarization in Alzheimer’s Disease. J. Neuroinflamm. 2020, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.; Brorson, J.R.; Alexander, J.J. Septic Encephalopathy: Inflammation in Man and Mouse. Neurochem. Int. 2011, 58, 472–476. [Google Scholar] [CrossRef]
- Dal-Pizzol, F.; Tomasi, C.D.; Ritter, C. Septic Encephalopathy: Does Inflammation Drive the Brain Crazy? Braz. J. Psychiatry 2014, 36, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Barichello, T.; Generoso, J.S.; Goularte, J.A.; Collodel, A.; Pitcher, M.R.; Simões, L.R.; Quevedo, J.; Dal-Pizzol, F. Does Infection-Induced Immune Activation Contribute to Dementia? Aging Dis. 2015, 6, 342. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, V.V.; Masud, F.; Petronilho, F.; Dal-Pizzol, F.; Barichello, T. Infection-Induced Systemic Inflammation Is a Potential Driver of Alzheimer’s Disease Progression. Front. Aging Neurosci. 2019, 11, 122. [Google Scholar] [CrossRef]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.C.F.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T. Receptor for Advanced Glycation End Products Mediates Sepsis-Triggered Amyloid-β Accumulation, Tau Phosphorylation, and Cognitive Impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef] [Green Version]
- de Felice, F.G.; Tovar-Moll, F.; Moll, J.; Munoz, D.P.; Ferreira, S.T. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and the Central Nervous System. Trends Neurosci. 2020, 43, 355–357. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bai, W.; Hashikawa, T. The Neuroinvasive Potential of SARS-CoV2 May Play a Role in the Respiratory Failure of COVID-19 Patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological Associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and Long-Term Consequences of COVID-19 Infections for the Development of Neurological Disease. Alzheimer’s Res. Ther. 2020, 12, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Fotuhi, M.; Mian, A.; Meysami, S.; Raji, C.A. Neurobiology of COVID-19. J. Alzheimer’s Dis. 2020, 76, 3–19. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, L.; Zhu, Q.; Shu, M.; Cai, Q. Common Infections May Lead to Alzheimer’s Disease. Virol. Sin. 2018, 33, 456–458. [Google Scholar] [CrossRef]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Van Giau, V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.; Berk, M.; Maes, M.; Puri, B.K. Could Alzheimer’s Disease Originate in the Periphery and If so How so? Mol. Neurobiol. 2019, 56, 406–434. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Duffy, A. Factors Influencing the Gut Microbiota, Inflammation, and Type 2 Diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.-C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutiérrez-Vázquez, C.; Hewson, P.; Staszewski, O. Microglial Control of Astrocytes in Response to Microbial Metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef]
- Sochocka, M.; Donskow-Łysoniewska, K.; Diniz, B.S.; Kurpas, D.; Brzozowska, E.; Leszek, J. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease—A Critical Review. Mol. Neurobiol. 2019, 56, 1841–1851. [Google Scholar] [CrossRef] [Green Version]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C. Porphyromonas Gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [Green Version]
- Beydoun, M.A.; Beydoun, H.A.; Hossain, S.; El-Hajj, Z.W.; Weiss, J.; Zonderman, A.B. Clinical and Bacterial Markers of Periodontitis and Their Association with Incident All-Cause and Alzheimer’s Disease Dementia in a Large National Survey. J. Alzheimer’s Dis. 2020, 75, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kolltveit, K.M.; Tronstad, L.; Olsen, I. Systemic Diseases Caused by Oral Infection. Clin. Microbiol. Rev. 2000, 13, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, X.; Yan, X.; Mizutani, S.; Kashiwazaki, H.; Ni, J.; Wu, Z. GSK3β Is Involved in Promoting Alzheimer’s Disease Pathologies Following Chronic Systemic Exposure to Porphyromonas Gingivalis Lipopolysaccharide in Amyloid Precursor ProteinNL-F/NL-F Knock-in Mice. Brain Behav. Immun. 2021, 98, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s Disease: Are They Effective? Pharm. Ther. 2010, 35, 208. [Google Scholar]
- Martyn, C. Anti-Inflammatory Drugs and Alzheimer’s Disease. BMJ 2003, 327, 353. [Google Scholar] [CrossRef]
- Breitner, J.C.S.; Gau, B.A.; Welsh, K.A.; Plassman, B.L.; McDonald, W.M.; Helms, M.J.; Anthony, J.C. Inverse Association of Anti-inflammatory Treatments and Alzheimer’s Disease: Initial Results of a Co-twin Control Study. Neurology 1994, 44, 227. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.; Baker, L.D.; Montine, T.J.; Meinert, C.L.; Lyketsos, C.G.; Ashe, K.H.; Brandt, J.; Craft, S.; Evans, D.E.; Green, R.C. Extended Results of the Alzheimer’s Disease Anti-Inflammatory Prevention Trial. Alzheimer’s Dement. 2011, 7, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.; Launer, L.J.; Ott, A.; Hoes, A.W.; Breteler, M.M.B.; Hofman, A. Do Nonsteroidal Anti-Inflammatory Drugs Decrease the Risk for Alzheimer’s Disease?: The Rotterdam Study. Neurology 1995, 45, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Ongini, E.; Wenk, G. Non-steroidal Anti-inflammatory Drugs (NSAIDs) in Alzheimer’s Disease: Old and New Mechanisms of Action. J. Neurochem. 2004, 91, 521–536. [Google Scholar] [CrossRef]
- Shadfar, S.; Hwang, C.J.; Lim, M.-S.; Choi, D.-Y.; Hong, J.T. Involvement of Inflammation in Alzheimer’s Disease Pathogenesis and Therapeutic Potential of Anti-Inflammatory Agents. Arch. Pharmacal Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef]
- McGeer, P.L.; Guo, J.P.; Lee, M.; Kennedy, K.; McGeer, E.G. Alzheimer’s Disease Can Be Spared by Nonsteroidal Anti-Inflammatory Drugs. J. Alzheimer’s Dis. 2018, 62, 1219–1222. [Google Scholar] [CrossRef] [Green Version]
- Sastre, M.; Dewachter, I.; Landreth, G.E.; Willson, T.M.; Klockgether, T.; van Leuven, F.; Heneka, M.T. Nonsteroidal Anti-Inflammatory Drugs and Peroxisome Proliferator-Activated Receptor-γ Agonists Modulate Immunostimulated Processing of Amyloid Precursor Protein through Regulation of β-Secretase. J. Neurosci. 2003, 23, 9796–9804. [Google Scholar] [CrossRef] [Green Version]
- Sastre, M.; Dewachter, I.; Rossner, S.; Bogdanovic, N.; Rosen, E.; Borghgraef, P.; Evert, B.O.; Dumitrescu-Ozimek, L.; Thal, D.R.; Landreth, G. Nonsteroidal Anti-Inflammatory Drugs Repress β-Secretase Gene Promoter Activity by the Activation of PPARγ. Proc. Natl. Acad. Sci. USA 2006, 103, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, J.L.; Sagi, S.A.; Smith, T.E.; Weggen, S.; Das, P.; McLendon, D.C.; Ozols, V.V.; Jessing, K.W.; Zavitz, K.H.; Koo, E.H. NSAIDs and Enantiomers of Flurbiprofen Target γ-Secretase and Lower Aβ42 in Vivo. J. Clin. Investig. 2003, 112, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, S.; Yang, H.; Uryu, K.; Lee, E.B.; Zhao, L.; Shineman, D.; Trojanowski, J.Q.; Lee, V.M.-Y.; Praticò, D. Modulation of Nuclear Factor-ΚB Activity by Indomethacin Influences Aβ Levels but Not Aβ Precursor Protein Metabolism in a Model of Alzheimer’s Disease. Am. J. Pathol. 2004, 165, 2197–2206. [Google Scholar] [CrossRef]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; van Leuven, F.; Landreth, G.E. Acute Treatment with the PPARγ Agonist Pioglitazone and Ibuprofen Reduces Glial Inflammation and Aβ1–42 Levels in APPV717I Transgenic Mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, R.; Kitazawa, M.; Passos, G.F.; Baglietto-Vargas, D.; Cheng, D.; Cribbs, D.H.; LaFerla, F.M. Aspirin-Triggered Lipoxin A4 Stimulates Alternative Activation of Microglia and Reduces Alzheimer Disease–Like Pathology in Mice. Am. J. Pathol. 2013, 182, 1780–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; McCleery, J.; Tabet, N. Aspirin, Steroidal and Non-steroidal Anti-inflammatory Drugs for the Treatment of Alzheimer’s Disease. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Lichtenstein, M.P.; Carriba, P.; Masgrau, R.; Pujol, A.; Galea, E. Staging Anti-Inflammatory Therapy in Alzheimer’s Disease. Front. Aging Neurosci. 2010, 2, 142. [Google Scholar] [CrossRef] [Green Version]
- Dhouafli, Z.; Cuanalo-Contreras, K.; Hayouni, E.A.; Mays, C.E.; Soto, C.; Moreno-Gonzalez, I. Inhibition of Protein Misfolding and Aggregation by Natural Phenolic Compounds. Cell. Mol. Life Sci. 2018, 75, 3521–3538. [Google Scholar] [CrossRef] [PubMed]
- Habtemariam, S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules 2019, 24, 1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales-Corral, S.; Tan, D.; Reiter, R.J.; Valdivia-Velázquez, M.; Martínez-Barboza, G.; Pablo Acosta-Martínez, J.; Ortiz, G.G. Orally Administered Melatonin Reduces Oxidative Stress and Proinflammatory Cytokines Induced by Amyloid-β Peptide in Rat Brain: A Comparative, in Vivo Study versus Vitamin C and E. J. Pineal Res. 2003, 35, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Gattoni-Celli, M.; Zhu, H.; Bhat, N.R.; Sambamurti, K.; Gattoni-Celli, S.; Kindy, M.S. Vitamin D 3-Enriched Diet Correlates with a Decrease of Amyloid Plaques in the Brain of AβPP Transgenic Mice. J. Alzheimer’s Dis. 2011, 25, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.F.; Li, N.; Wang, Q.; Cheng, X.J.; Li, X.M.; Liu, T.T. Resveratrol Decreases the Insoluble Aβ1–42 Level in Hippocampus and Protects the Integrity of the Blood–Brain Barrier in AD Rats. Neuroscience 2015, 310, 641–649. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Thiel, A.; Lauer, A.A.; Winkler, J.; Lehmann, J.; Regner, L.; Nelke, C.; Janitschke, D.; Benoist, C.; Streidenberger, O. Vitamin D and Its Analogues Decrease Amyloid-β (Aβ) Formation and Increase Aβ-Degradation. Int. J. Mol. Sci. 2017, 18, 2764. [Google Scholar] [CrossRef] [Green Version]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M. do C. Natural Compounds for Alzheimer’s Disease Therapy: A Systematic Review of Preclinical and Clinical Studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef] [Green Version]
- Cederholm, T.; Salem, N., Jr.; Palmblad, J. ω-3 Fatty Acids in the Prevention of Cognitive Decline in Humans. Adv. Nutr. 2013, 4, 672–676. [Google Scholar] [CrossRef] [Green Version]
- Hjorth, E.; Zhu, M.; Toro, V.C.; Vedin, I.; Palmblad, J.; Cederholm, T.; Freund-Levi, Y.; Faxen-Irving, G.; Wahlund, L.-O.; Basun, H. Omega-3 Fatty Acids Enhance Phagocytosis of Alzheimer’s Disease-Related Amyloid-β 42 by Human Microglia and Decrease Inflammatory Markers. J. Alzheimer’s Dis. 2013, 35, 697–713. [Google Scholar] [CrossRef] [Green Version]
- della Giustina, A.; de Souza Goldim, M.P.; Danielski, L.G.; Garbossa, L.; Junior, A.N.O.; Cidreira, T.; Denicol, T.; Bonfante, S.; da Rosa, N.; Fortunato, J.J. Lipoic Acid and Fish Oil Combination Potentiates Neuroinflammation and Oxidative Stress Regulation and Prevents Cognitive Decline of Rats After Sepsis. Mol. Neurobiol. 2020, 57, 4451–4466. [Google Scholar] [CrossRef]
- Lawlor, B.; Segurado, R.; Kennelly, S.; Olde Rikkert, M.G.M.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W. Nilvadipine in Mild to Moderate Alzheimer Disease: A Randomised Controlled Trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef]
- Meyer, P.-F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.-É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J. INTREPAD: A Randomized Trial of Naproxen to Slow Progress of Presymptomatic Alzheimer Disease. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P. Minocycline at 2 Different Dosages vs Placebo for Patients with Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Ryan, J.; Storey, E.; Murray, A.M.; Woods, R.L.; Wolfe, R.; Reid, C.M.; Nelson, M.R.; Chong, T.T.J.; Williamson, J.D.; Ward, S.A. Randomized Placebo-Controlled Trial of the Effects of Aspirin on Dementia and Cognitive Decline. Neurology 2020, 95, e320–e331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.P.; Ferreira, G.K.; Pires, A.J.; de Bem Silveira, G.; de Souza, D.L.; de Abreu Brandolfi, J.; de Souza, C.T.; Paula, M.M.S.; Silveira, P.C.L. Gold Nanoparticles Prevent Cognitive Deficits, Oxidative Stress and Inflammation in a Rat Model of Sporadic Dementia of Alzheimer’s Type. Mater. Sci. Eng. C 2017, 77, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Sanati, M.; Khodagholi, F.; Aminyavari, S.; Ghasemi, F.; Gholami, M.; Kebriaeezadeh, A.; Sabzevari, O.; Hajipour, M.J.; Imani, M.; Mahmoudi, M. Impact of Gold Nanoparticles on Amyloid β-Induced Alzheimer’s Disease in a Rat Animal Model: Involvement of STIM Proteins. ACS Chem. Neurosci. 2019, 10, 2299–2309. [Google Scholar] [CrossRef]
- Rodrigues, M.S.; de Paula, G.C.; Duarte, M.B.; de Rezende, V.L.; Possato, J.C.; Farias, H.R.; Medeiros, E.B.; Feuser, P.E.; Streck, E.L.; de Ávila, R.A.M. Nanotechnology as a Therapeutic Strategy to Prevent Neuropsychomotor Alterations Associated with Hypercholesterolemia. Colloids Surf. B Biointerfaces 2021, 201, 111608. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings. Cells 2021, 10, 2581. https://doi.org/10.3390/cells10102581
de Oliveira J, Kucharska E, Garcez ML, Rodrigues MS, Quevedo J, Moreno-Gonzalez I, Budni J. Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings. Cells. 2021; 10(10):2581. https://doi.org/10.3390/cells10102581
Chicago/Turabian Stylede Oliveira, Jade, Ewa Kucharska, Michelle Lima Garcez, Matheus Scarpatto Rodrigues, João Quevedo, Ines Moreno-Gonzalez, and Josiane Budni. 2021. "Inflammatory Cascade in Alzheimer’s Disease Pathogenesis: A Review of Experimental Findings" Cells 10, no. 10: 2581. https://doi.org/10.3390/cells10102581