
NanoHIV: A Bioinformatics Pipeline for Producing Accurate, Near Full-Length HIV Proviral Genomes Sequenced Using the Oxford Nanopore Technology

 , , and
, , and
Abstract
:1. Introduction
2. Data Generation
2.1. Inclusion Criteria & Data Collection
2.2. Near Full-Length Amplicon Generation
2.3. MiSeq™ Library Preparation
2.4. Bioinformatic Analysis of MiSeq Data
2.5. Oxford Nanopore Technologies GridION Sequencing
2.6. ONT Library Preparation
2.7. ONT Sequencing Conditions
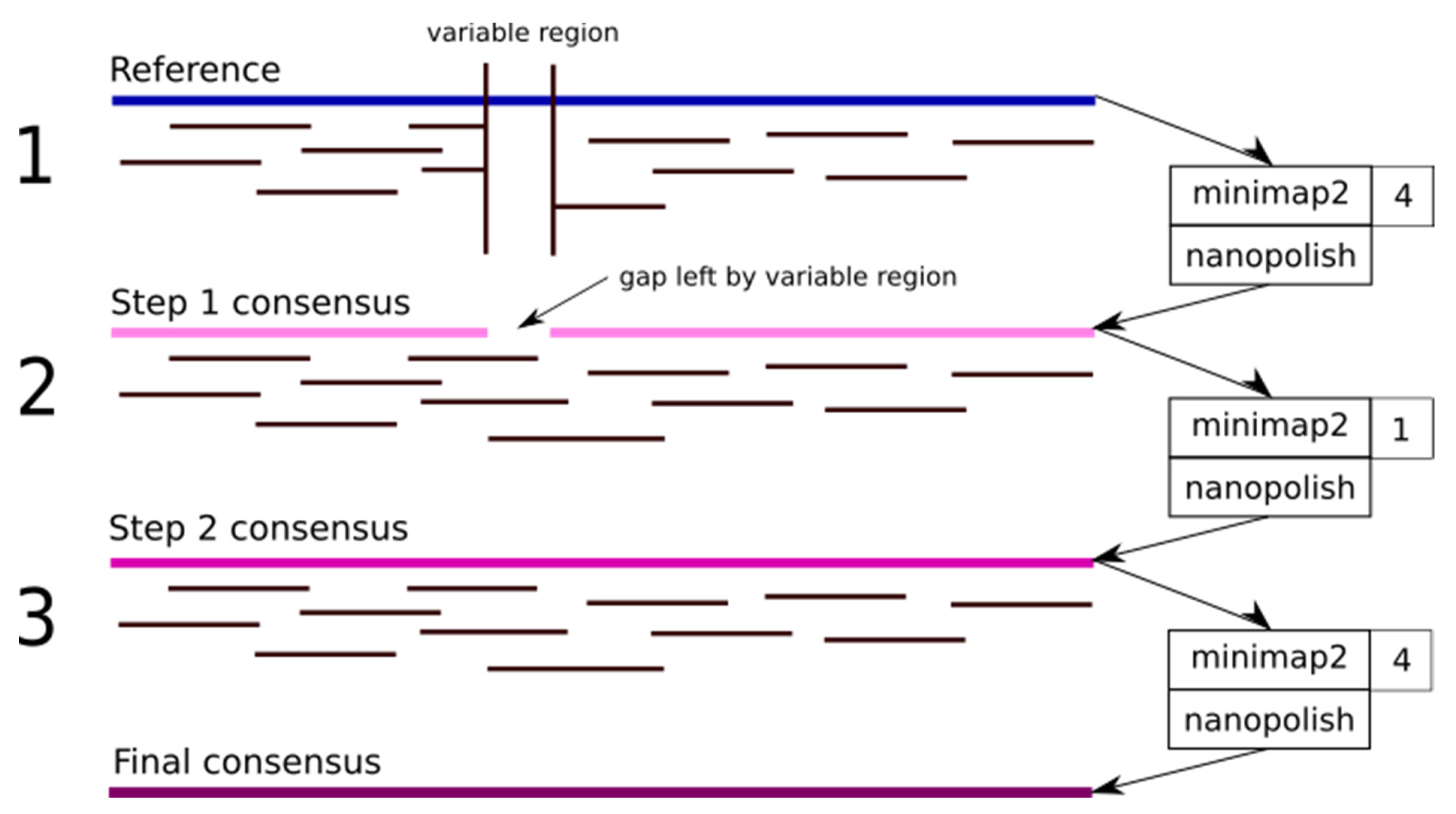
3. Pipeline Description
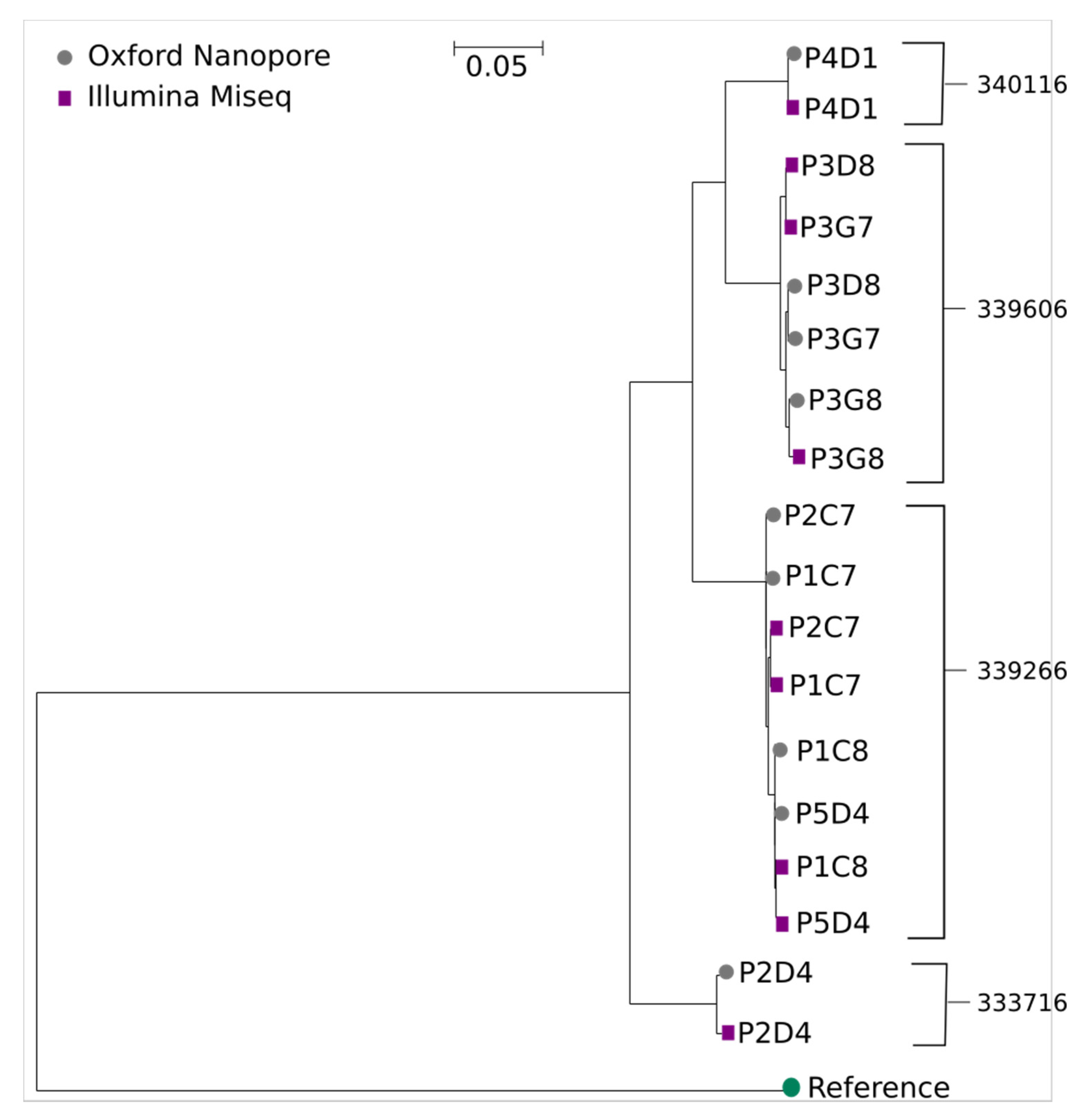
4. Results
5. Pipeline and Data Availability
6. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS. Global HIV & AIDS Statistics—2020 Fact Sheet; UNAIDS: Geneve, Switzerland, 2020. [Google Scholar]
- The INSIGHT START Study Group; INSIGHT START Study Group; Lundgren, J.D.; Babiker, A.G.; Gordin, F.; Emery, S.; Grund, B.; Sharma, S.; Avihingsanon, A.; Cooper, D.A.; et al. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N. Engl. J. Med. 2015, 373, 795–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisinger, R.W.; Dieffenbach, C.W.; Fauci, A.S. HIV Viral Load and Transmissibility of HIV Infection: Undetectable Equals Untransmittable. JAMA 2019, 321, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments. Int. J. Environ. Res. Public Health 2018, 15, 1697. [Google Scholar] [CrossRef] [Green Version]
- Ávila-Ríos, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12, 617. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.M.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, T.-W.; Engel, D.; Berrey, M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiener, B.; Horsburgh, B.A.; Eden, J.S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Pinzone, M.R.; VanBelzen, D.J.; Weissman, S.; Bertuccio, M.P.; Cannon, L.; Venanzi-Rullo, E.; Migueles, S.; Jones, R.B.; Mota, T.; Joseph, S.B.; et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat. Commun. 2019, 10, 728. [Google Scholar] [CrossRef]
- Katusiime, M.G.; Halvas, E.K.; Wright, I.; Joseph, K.; Bale, M.J.; Kirby-McCullough, B.; Engelbrecht, S.; Shao, W.; Hu, W.-S.; Cotton, M.F.; et al. Intact HIV Proviruses Persist in Children Seven to Nine Years after Initiation of Antiretroviral Therapy in the First Year of Life. J. Virol. 2020, 94, 1519–1538. [Google Scholar] [CrossRef]
- Halvas, E.K.; Joseph, K.W.; Brandt, L.D.; Guo, S.; Sobolewski, M.D.; Jacobs, J.L.; Tumiotto, C.; Bui, J.K.; Cyktor, J.C.; Keele, B.F.; et al. HIV-1 viremia not suppressible by antiretroviral therapy can originate from large T cell clones producing infectious virus. J. Clin. Investig. 2020, 130, 5847–5857. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.A.; Zhou, S.; Xu, Y.; Moeser, M.J.; MacMillan, D.R.; Council, O.; Kirchherr, J.; Sung, J.M.; Roan, N.R.; Adimora, A.A.; et al. The HIV-1 latent reservoir is largely sensitive to circulating T cells. eLife 2020, 9, e57246. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.C.; Wagner, G.A.; Hightower, G.K.; Caballero, G.; Phung, P.; Richman, D.D.; Pond, S.L.K.; Smith, D.M. HIV-1 neutralizing antibody response and viral genetic diversity characterized with next generation sequencing. Virology 2015, 474, 34–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrens, N.; Love, M.; Bandlamuri, M.; Bernhardt, D.; Wertheimer, A.; Klotz, S.; Ahmad, N. Characterization of HIV-1 Envelope V3 Region Sequences from Virologically Controlled HIV-Infected Older Patients on Long Term Antiretroviral Therapy. AIDS Res. Hum. Retroviruses 2021, 37, 233–245. [Google Scholar] [CrossRef]
- Tovanabutra, S.; Sirijatuphat, R.; Pham, P.T.; Bonar, L.; Harbolick, E.A.; Bose, M.; Song, H.; Chang, D.; Oropeza, C.; O’Sullivan, A.M.; et al. Deep Sequencing Reveals Central Nervous System Compartmentalization in Multiple Transmitted/Founder Virus Acute HIV-1 Infection. Cells 2019, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Brese, R.L.; Gonzalez-Perez, M.P.; Koch, M.; O’Connell, O.; Luzuriaga, K.; Somasundaran, M.; Clapham, P.R.; Dollar, J.J.; Nolan, D.J.; Rose, R.; et al. Ultradeep single-molecule real-time sequencing of HIV envelope reveals complete compartmentalization of highly macrophage-tropic R5 proviral variants in brain and CXCR4-using variants in immune and peripheral tissues. J. Neurovirol. 2018, 24, 439–453. [Google Scholar] [CrossRef]
- Kariuki, S.M.; Selhorst, P.; Anthony, C.; Matten, D.; Abrahams, M.-R.; Martin, D.P.; Ariën, K.K.; Rebe, K.; Williamson, C.; Dorfman, J.R. Compartmentalization and Clonal Amplification of HIV-1 in the Male Genital Tract Characterized Using Next-Generation Sequencing. J. Virol. 2020, 94, e00229-20. [Google Scholar] [CrossRef]
- Malet, I.; Subra, F.F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.-G.A.-G.; Delelis, O. Mutations Located outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. mBio 2017, 8, e00922-17. [Google Scholar] [CrossRef] [Green Version]
- Hikichi, Y.; Van Duyne, R.; Pham, P.; Groebner, J.L.; Wiegand, A.; Mellors, J.W.; Kearney, M.F.; Freed, E.O. Mechanistic analysis of the broad antiretroviral resistance conferred by hiv-1 envelope glycoprotein mutations. mBio 2021, 12, e03134-20. [Google Scholar] [CrossRef]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.; Au, K.F. PacBio Sequencing and Its Applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Quainoo, S.; Coolen, J.P.; van Hijum, S.A.; Huynen, M.A.; Melchers, W.J.; van Schaik, W.; Wertheim, H.F. Whole-Genome Sequencing of Bacterial Pathogens : The Future of Nosocomial. Clin. Microbiol. Rev. 2017, 30, 1015–1064. [Google Scholar] [CrossRef] [Green Version]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Schloissnig, S.; Franchini, P.; Du, K.; Woltering, J.; Irisarri, I.; Wong, W.Y.; Nowoshilow, S.; Kneitz, S.; Kawaguchi, A.; et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 2021, 590, 284–289. [Google Scholar] [CrossRef]
- Brown, C. Nanopore Community Meeting 2019 Technology Update. In Proceedings of the Nanopore Community Meeting 2019 Technology Update; Oxford Nanopore Technologies, Resource Centre, 2019. Available online: https://nanoporetech.com/resource-centre/nanopore-community-meeting-2019-technology-update (accessed on 15 July 2020).
- Pfeiffer, F.; Gröber, C.; Blank, M.; Händler, K.; Beyer, M.; Schultze, J.L.; Mayer, G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018, 8, 10950. [Google Scholar] [CrossRef] [Green Version]
- Fujita, S.; Masago, K.; Okuda, C.; Hata, A.; Kaji, R.; Katakami, N.; Hirata, Y. Single nucleotide variant sequencing errors in whole exome sequencing using the ion proton system. Biomed. Rep. 2017, 7, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bhatia, P. Comparative sequencing data analysis of Ion Torrent and MinION sequencing platforms using a clinical diagnostic haematology panel. Int. J. Lab. Hematol. 2020, 42, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Koopmans, M. Validating whole genome nanopore sequencing, using USUTU virus as an example. J. Vis. Exp. 2020, 2020, 60906. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, R.; Sinha, A.; Bird, S.W.; Jayamohan, H.; Edwards, H.S.; Schoeniger, J.S.; Patel, K.D.; Branda, S.S.; Bartsch, M.S. Systematic and stochastic influences on the performance of the MinION nanopore sequencer across a range of nucleotide bias. Sci. Rep. 2018, 8, 3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, R.M.; Shen, T.; Gnanakaran, S.; Derdeyn, C.A. Appreciating HIV type 1 diversity: Subtype differences in env. AIDS Res. Hum. Retroviruses 2009, 25, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.B.; Hoffman, N.G.; Swanstrom, R. Subtype-Specific Conformational Differences within the V3 Region of Subtype B and Subtype C Human Immunodeficiency Virus Type 1 Env Proteins. J. Virol. 2008, 82, 903–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chen, Y.; Mu, D.; Yuan, J.; Shi, Y.; Zhang, H.; Gan, J.; Li, N.; Hu, X.; Liu, B.; et al. Comparison of the two major classes of assembly algorithms: Overlap-layout-consensus and de-bruijn-graph. Brief. Funct. Genom. 2012, 11, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Aga, E.; Cillo, A.R.; Yates, A.L.; Besson, G.; Fyne, E.; Koontz, D.L.; Jennings, C.; Zheng, L.; Mellors, J.W. Novel Assays for Measurement of Total Cell-Associated HIV-1 DNA and RNA. J. Clin. Microbiol. 2016, 54, 902–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zyl, G.U.; Bedison, M.A.; Rensburg, A.J. Van Early Antiretroviral Therapy in South African Children Reduces HIV-1—Infected Cells and Cell-Associated HIV-1 RNA in Blood Mononuclear Cells. J. Infect. Dis. 2015, 212, 39–43. [Google Scholar] [CrossRef] [Green Version]
- Salminen, M.O.; Koch, C.; Sanders-Buell, E.; Ehrenberg, P.K.; Michael, N.L.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Recovery of Virtually Full-Length HIV-1 Provirus of Diverse Subtypes from Primary Virus Cultures Using the Polymerase Chain Reaction. Virology 1995, 213, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.T.; Pasternak, A.O.; Berkhout, B. On the generation of the MSD class of defective HIV proviruses. Retrovirology 2019, 16, 19. [Google Scholar] [CrossRef]
- Wright, I.A.; Bale, M.J.; Shao, W.; Hu, W.-S.; Coffin, J.M.; Van Zyl, G.U.; Kearney, M.F. HIVIntact: A python-based tool for HIV-1 genome intactness inference. Retrovirology 2021, 18, 16. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP. 2009. Available online: https://evolution.genetics.washington.edu/phylip.html (accessed on 2 September 2021).
- Piñar, G.; Poyntner, C.; Lopandic, K.; Tafer, H.; Sterflinger, K. Rapid diagnosis of biological colonization in cultural artefacts using the MinION nanopore sequencing technology. Int. Biodeterior. Biodegrad. 2020, 148, 104908. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routh, A.L.; Torbett, B.E. MrHAMER yields highly accurate single molecule viral sequences enabling analysis of intra-host evolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sarkozy, P.; Jobbágy; Antal, P. Calling homopolymer stretches from raw nanopore reads by analyzing k-mer dwell times. In EMBEC & NBC 2017; Springer: Singapore, 2017; Volume 65, pp. 241–244. [Google Scholar]



{kind=link}
{kind=link}
| PID | Age ART Initiated (Months) | ART Regimen | Time on ART (Years) | No. of Proviral Genomes Sequenced with ONT | |
|---|---|---|---|---|---|
| Identified as Intact | Identified as Defective | ||||
| 333716 | 2.3 | AZT/3TC/LPV/r | 8.55 | 0 | 1 |
| 339606 | 8.5 | AZT/3TC/LPV/r | 7.93 | 2 | 1 |
| 339266 | 9.23 | AZT/3TC/LPV/r | 8.2 | 4 | 0 |
| 340116 | 9.32 | AZT/3TC/LPV/r | 8.83 | 1 | 0 |
| Pre-Nested Primers | |||
| Primer Name | Primer Direction | Nucleotide Position in HXB2 (bp) | 5′-3′ Sequence |
| Li_OuterF +,* | Forward | 623–649 | AAATCTCTAGCAGTGGCGCCCGAACAG |
| Li_OuterR | Reverse | 9662–9686 | TGAGGGATCTCTAGTTACCAGAGTC |
| Nested Primers | |||
| Primer Name | Primer Direction | Nucleotide Position in HXB2 (bp) | 5′-3′ Sequence |
| Li_InnerF * | Forward | 769–793 | GCGGAGGCTAGAAGGAGAGAGATGG |
| Li_InnerR + | Reverse | 9604–9632 | GCACTCAAGGCAAGCTTTATTGAGGCTTA |
| NFL-alt_in_F # | Forward | 642–664 | CCG AAC AGG GAC BHG AAA GCG AA |
| Patient Identifier | Sample Identifier | Proviral Genome Status | Total Aligned Similarity Percentage (%) |
|---|---|---|---|
| 340116 | P4D1 | Intact | 99.3 |
| 339606 | P3D8 | Intact | 98.6 |
| P3G7 | Intact | 99.4 | |
| P3G8 | Defective | 99.6 | |
| 333716 | P2D4 | Defective | 99.1 |
| 339266 | P2C7 | Intact | 99.6 |
| P1C7 | Intact | 99.6 | |
| P1C8 | Intact | 99.7 | |
| P5D4 | Intact | 99.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, I.A.; Delaney, K.E.; Katusiime, M.G.K.; Botha, J.C.; Engelbrecht, S.; Kearney, M.F.; van Zyl, G.U. NanoHIV: A Bioinformatics Pipeline for Producing Accurate, Near Full-Length HIV Proviral Genomes Sequenced Using the Oxford Nanopore Technology. Cells 2021, 10, 2577. https://doi.org/10.3390/cells10102577
Wright IA, Delaney KE, Katusiime MGK, Botha JC, Engelbrecht S, Kearney MF, van Zyl GU. NanoHIV: A Bioinformatics Pipeline for Producing Accurate, Near Full-Length HIV Proviral Genomes Sequenced Using the Oxford Nanopore Technology. Cells. 2021; 10(10):2577. https://doi.org/10.3390/cells10102577
Chicago/Turabian StyleWright, Imogen A., Kayla E. Delaney, Mary Grace K. Katusiime, Johannes C. Botha, Susan Engelbrecht, Mary F. Kearney, and Gert U. van Zyl. 2021. "NanoHIV: A Bioinformatics Pipeline for Producing Accurate, Near Full-Length HIV Proviral Genomes Sequenced Using the Oxford Nanopore Technology" Cells 10, no. 10: 2577. https://doi.org/10.3390/cells10102577
APA StyleWright, I. A., Delaney, K. E., Katusiime, M. G. K., Botha, J. C., Engelbrecht, S., Kearney, M. F., & van Zyl, G. U. (2021). NanoHIV: A Bioinformatics Pipeline for Producing Accurate, Near Full-Length HIV Proviral Genomes Sequenced Using the Oxford Nanopore Technology. Cells, 10(10), 2577. https://doi.org/10.3390/cells10102577