
Formins in Human Disease


Abstract
1. Introduction

2. Monogenic Disorders Caused by Formin Mutation
2.1. Nephrotic Syndrome and Charcot-Marie-Tooth Disease
2.2. Hearing Loss
2.3. Thrombocytopenia
2.4. Microcephaly and Intellectual Disability
2.5. Primary Ovarian Insufficiency
2.6. Cardiomyopathy
3. Disorders Associated with Formin Mutation and Dysregulation
3.1. Developmental Cardiovascular Disorders
3.2. Neurodevelopmental Disorders and Mental Diseases
3.3. Other Developmental Defects
3.4. Aging-Related Diseases
3.5. Other Disorders
4. Formins in Cancer
4.1. Expression of Formins in Cancer
4.2. The Role of Formins in Cancer
4.3. Formins as Prognostic Biomarkers and Therapeutical Targets in Cancer
5. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schönichen, A.; Geyer, M. Fifteen Formins for an Actin Filament: A Molecular View on the Regulation of Human Formins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 152–163. [Google Scholar] [CrossRef]
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing Formins to Remodel the Actin and Microtubule Cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Bartolini, F.; Gundersen, G.G. Formins and Microtubules. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 164–173. [Google Scholar] [CrossRef]
- Fernandez-Barrera, J.; Alonso, M.A. Coordination of Microtubule Acetylation and the Actin Cytoskeleton by Formins. Cell. Mol. Life Sci. 2018, 75, 3181–3191. [Google Scholar] [CrossRef]
- Lynch, E.D.; Lee, M.K.; Morrow, J.E.; Welcsh, P.L.; León, P.E.; King, M.-C. Nonsyndromic Deafness DFNA1 Associated with Mutation of a Human Homolog of the Drosophila Gene Diaphanous. Science 1997, 278, 1315–1318. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- Hamosh, A.; Amberger, J.S.; Bocchini, C.; Scott, A.F.; Rasmussen, S.A. Online Mendelian Inheritance in Man (OMIM®): Victor McKusick’s Magnum Opus. Am. J. Med. Genet. Part A 2021. [Google Scholar] [CrossRef]
- Iwasa, Y.; Nishio, S.; Usami, S. Comprehensive Genetic Analysis of Japanese Autosomal Dominant Sensorineural Hearing Loss Patients. PLoS ONE 2016, 11, e0166781. [Google Scholar] [CrossRef]
- Kaustio, M.; Nayebzadeh, N.; Hinttala, R.; Tapiainen, T.; Åström, P.; Mamia, K.; Pernaa, N.; Lehtonen, J.; Glumoff, V.; Rahikkala, E.; et al. Loss of DIAPH1 Causes SCBMS, Combined Immunodeficiency, and Mitochondrial Dysfunction. J. Allergy Clin. Immunol. 2021, 148, 599–611. [Google Scholar] [CrossRef]
- Brozkova, D.S.; Marková, S.P.; Mészárosová, A.U.; Jenčík, J.; Čejnová, V.; Čada, Z.; Laštůvková, J.; Rašková, D.; Seeman, P. Spectrum and Frequencies of Non GJB2 Gene Mutations in Czech Patients with Early Non-Syndromic Hearing Loss Detected by Gene Panel NGS and Whole-Exome Sequencing. Clin. Genet. 2020, 98, 548–554. [Google Scholar] [CrossRef]
- Kim, B.J.; Ueyama, T.; Miyoshi, T.; Lee, S.; Han, J.H.; Park, H.-R.; Kim, A.R.; Oh, J.; Kim, M.Y.; Kang, Y.S.; et al. Differential Disruption of Autoinhibition and Defect in Assembly of Cytoskeleton during Cell Division Decide the Fate of Human DIAPH1-Related Cytoskeletopathy. J. Med. Genet. 2019, 56, 818–827. [Google Scholar] [CrossRef]
- Kang, T.-H.; Baek, J.-I.; Sagong, B.; Park, H.-J.; Park, C.I.; Lee, K.-Y.; Kim, U.-K. A Novel Missense Variant in the DIAPH1 Gene in a Korean Family with Autosomal Dominant Nonsyndromic Hearing Loss. Genes Genet. Syst. 2016, 91, 289–292. [Google Scholar] [CrossRef]
- Ercan-Sencicek, A.G.; Jambi, S.; Franjic, D.; Nishimura, S.; Li, M.; El-Fishawy, P.; Morgan, T.M.; Sanders, S.J.; Bilguvar, K.; Suri, M.; et al. Homozygous Loss of DIAPH1 Is a Novel Cause of Microcephaly in Humans. Eur. J. Hum. Genet. 2015, 23, 165–172. [Google Scholar] [CrossRef]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef]
- Al-Maawali, A.; Barry, B.J.; Rajab, A.; El-Quessny, M.; Seman, A.; Coury, S.N.; Barkovich, A.J.; Yang, E.; Walsh, C.A.; Mochida, G.H.; et al. Novel Loss-of-Function Variants in DIAPH1 Associated with Syndromic Microcephaly, Blindness, and Early Onset Seizures. Am. J. Med. Genet. Part A 2016, 170, 435–440. [Google Scholar] [CrossRef]
- Yavarna, T.; Al-Dewik, N.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; Lakhani, S.; AlMulla, M.; Nawaz, Z.; et al. High Diagnostic Yield of Clinical Exome Sequencing in Middle Eastern Patients with Mendelian Disorders. Hum. Genet. 2015, 134, 967–980. [Google Scholar] [CrossRef]
- Wu, K.; Wang, H.; Guan, J.; Lan, L.; Zhao, C.; Zhang, M.; Wang, D.; Wang, Q. A Novel Variant in Diaphanous Homolog 1 (DIAPH1) as the Cause of Auditory Neuropathy in a Chinese Family. Int. J. Pediatr. Otorhinolaryngol. 2020, 133, 109947. [Google Scholar] [CrossRef]
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype Description and Response to Thrombopoietin Receptor Agonist in DIAPH1-Related Disorder. Blood Adv. 2018, 2, 2341–2346. [Google Scholar] [CrossRef]
- Shearer, A.E.; Black-Ziegelbein, E.A.; Hildebrand, M.S.; Eppsteiner, R.W.; Ravi, H.; Joshi, S.; Guiffre, A.C.; Sloan, C.M.; Happe, S.; Howard, S.D.; et al. Advancing Genetic Testing for Deafness with Genomic Technology. J. Med. Genet. 2013, 50, 627–634. [Google Scholar] [CrossRef]
- Ueyama, T.; Ninoyu, Y.; Nishio, S.; Miyoshi, T.; Torii, H.; Nishimura, K.; Sugahara, K.; Sakata, H.; Thumkeo, D.; Sakaguchi, H.; et al. Constitutive Activation of DIA1 (DIAPH1) via C-terminal Truncation Causes Human Sensorineural Hearing Loss. EMBO Mol. Med. 2016, 8, 1310–1324. [Google Scholar] [CrossRef]
- Neuhaus, C.; Lang-Roth, R.; Zimmermann, U.; Heller, R.; Eisenberger, T.; Weikert, M.; Markus, S.; Knipper, M.; Bolz, H.J. Extension of the Clinical and Molecular Phenotype of DIAPH1-Associated Autosomal Dominant Hearing Loss (DFNA1). Clin. Genet. 2017, 91, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing High-Throughput Sequencing into Mainstream Genetic Diagnosis Practice in Inherited Platelet Disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Ganaha, A.; Kaname, T.; Shinjou, A.; Chinen, Y.; Yanagi, K.; Higa, T.; Kondo, S.; Suzuki, M. Progressive Macrothrombocytopenia and Hearing Loss in a Large Family with DIAPH1 Related Disease. Am. J. Med. Genet. 2017, 173, 2826–2830. [Google Scholar] [CrossRef] [PubMed]
- Karki, N.R.; Ajebo, G.; Savage, N.; Kutlar, A. DIAPH1 Mutation as a Novel Cause of Autosomal Dominant Macrothrombocytopenia and Hearing Loss. Acta Haematol. 2021, 144, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Rabbolini, D.; Connor, D.; Morel-Kopp, M.-C.; Donikian, D.; Kondo, M.; Chen, W.; Alessi, M.-C.; Stevenson, W.; Chen, V.; Joseph, J.; et al. An Integrated Approach to Inherited Platelet Disorders: Results from a Research Collaborative, the Sydney Platelet Group. Pathology 2020, 52, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Stritt, S.; Nurden, P.; Turro, E.; Greene, D.; Jansen, S.B.; Westbury, S.K.; Petersen, R.; Astle, W.J.; Marlin, S.; Bariana, T.K.; et al. A Gain-of-Function Variant in DIAPH1 Causes Dominant Macrothrombocytopenia and Hearing Loss. Blood 2016, 127, 2903–2914. [Google Scholar] [CrossRef]
- Ji, H.; Lu, J.; Wang, J.; Li, H.; Lin, X. Combined Examination of Sequence and Copy Number Variations in Human Deafness Genes Improves Diagnosis for Cases of Genetic Deafness. BMC Ear Nose Throat Disord. 2014, 14, 9. [Google Scholar] [CrossRef]
- Bione, S.; Sala, C.; Manzini, C.; Arrigo, G.; Zuffardi, O.; Banfi, S.; Borsani, G.; Jonveaux, P.; Philippe, C.; Zuccotti, M.; et al. A Human Homologue of the Drosophila Melanogaster Diaphanous Gene Is Disrupted in a Patient with Premature Ovarian Failure: Evidence for Conserved Function in Oogenesis and Implications for Human Sterility. Am. J. Hum. Genet. 1998, 62, 533–541. [Google Scholar] [CrossRef]
- Sala, C.; Arrigo, G.; Torri, G.; Martinazzi, F.; Riva, P.; Larizza, L.; Philippe, C.; Jonveaux, P.; Sloan, F.; Labella, T.; et al. Eleven X Chromosome Breakpoints Associated with Premature Ovarian Failure (POF) Map to a 15-Mb YAC Contig Spanning Xq21. Genomics 1997, 40, 123–131. [Google Scholar] [CrossRef]
- Marozzi, A.; Manfredini, E.; Tibiletti, M.; Furlan, D.; Villa, N.; Vegetti, W.; Crosignani, P.; Ginelli, E.; Meneveri, R.; Dalprà, L. Molecular Definition of Xq Common-Deleted Region in Patients Affected by Premature Ovarian Failure. Hum. Genet. 2000, 107, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Rødningen, O.K.; Barøy, T.; Sorte, H.; Mellembakken, J.R.; Strømme, P.; Fannemel, M.; Frengen, E. A Translocation between Xq21.33 and 22q13.33 Causes an Intragenic SHANK3 Deletion in a Woman with Phelan-McDermid Syndrome and Hypergonadotropic Hypogonadism. Am. J. Med. Genet. 2011, 155, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Genesio, R.; Mormile, A.; Licenziati, M.R.; De Brasi, D.; Leone, G.; Balzano, S.; Izzo, A.; Bonfiglio, F.; Conti, A.; Fioretti, G.; et al. Short Stature and Primary Ovarian Insufficiency Possibly Due to Chromosomal Position Effect in a Balanced X;1 Translocation. Mol. Cytogenet. 2015, 8, 50. [Google Scholar] [CrossRef][Green Version]
- Bestetti, I.; Castronovo, C.; Sironi, A.; Caslini, C.; Sala, C.; Rossetti, R.; Crippa, M.; Ferrari, I.; Pistocchi, A.; Toniolo, D.; et al. High-Resolution Array-CGH Analysis on 46, XX Patients Affected by Early Onset Primary Ovarian Insufficiency Discloses New Genes Involved in Ovarian Function. Hum. Reprod. 2019, 34, 574–583. [Google Scholar] [CrossRef]
- Sánchez-Martínez, A.; Benito-Orejas, J.I.; Tellería-Orriols, J.J.; Alonso-Ramos, M.J. Autosomal Dominant Auditory Neuropathy and Variant DIAPH3 (c.-173C>T). Acta Otorrinolaringol. Esp. 2017, 68, 183–185. [Google Scholar] [CrossRef]
- Schoen, C.J.; Emery, S.B.; Thorne, M.C.; Ammana, H.R.; Śliwerska, E.; Arnett, J.; Hortsch, M.; Hannan, F.; Burmeister, M.; Lesperance, M.M. Increased Activity of Diaphanous Homolog 3 (DIAPH3)/Diaphanous Causes Hearing Defects in Humans with Auditory Neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 13396–13401. [Google Scholar] [CrossRef]
- Schneider, R.; Deutsch, K.; Hoeprich, G.J.; Marquez, J.; Hermle, T.; Braun, D.A.; Seltzsam, S.; Kitzler, T.M.; Mao, Y.; Buerger, F.; et al. DAAM2 Variants Cause Nephrotic Syndrome via Actin Dysregulation. Am. J. Hum. Genet. 2020, 107, 1113–1128. [Google Scholar] [CrossRef]
- Law, R.; Dixon-Salazar, T.; Jerber, J.; Cai, N.; Abbasi, A.A.; Zaki, M.S.; Mittal, K.; Gabriel, S.B.; Rafiq, M.A.; Khan, V.; et al. Biallelic Truncating Mutations in FMN2, Encoding the Actin-Regulatory Protein Formin 2, Cause Nonsyndromic Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 2014, 95, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Gorukmez, O.; Gorukmez, O.; Ekici, A. A Novel Nonsense FMN2 Mutation in Nonsyndromic Autosomal Recessive Intellectual Disability Syndrome. Fetal Pediatr. Pathol. 2020, 39, 1–5. [Google Scholar] [CrossRef]
- Marco, E.J.; Aitken, A.B.; Nair, V.P.; da Gente, G.; Gerdes, M.R.; Bologlu, L.; Thomas, S.; Sherr, E.H. Burden of de Novo Mutations and Inherited Rare Single Nucleotide Variants in Children with Sensory Processing Dysfunction. BMC Med. Genom. 2018, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.D.; Rocca, M.S.; Bruno, I.; Faletra, F.; Pecile, V.; Gasparini, P. De Novo 911 kb Interstitial Deletion on Chromosome 1q43 in a Boy with Mental Retardation and Short Stature. Eur. J. Med. Genet. 2012, 55, 117–119. [Google Scholar] [CrossRef]
- Almuqbil, M.; Hamdan, F.F.; Mathonnet, G.; Rosenblatt, B.; Srour, M. De Novo Deletion of FMN2 in a Girl with Mild Non-Syndromic Intellectual Disability. Eur. J. Med. Genet. 2013, 56, 686–688. [Google Scholar] [CrossRef]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the Formin Protein INF2 Cause Focal Segmental Glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Nagano, C.; Yamamura, T.; Horinouchi, T.; Aoto, Y.; Ishiko, S.; Sakakibara, N.; Shima, Y.; Nakanishi, K.; Nagase, H.; Iijima, K.; et al. Comprehensive Genetic Diagnosis of Japanese Patients with Severe Proteinuria. Sci. Rep. 2020, 10, 270. [Google Scholar] [CrossRef]
- Varner, J.D.; Chryst-Stangl, M.; Esezobor, C.I.; Solarin, A.; Wu, G.; Lane, B.; Hall, G.; Abeyagunawardena, A.; Matory, A.; Hunley, T.E.; et al. Genetic Testing for Steroid-Resistant-Nephrotic Syndrome in an Outbred Population. Front. Pediatr. 2018, 6, 307. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Pinto, E.; Vairo, F.; Hogan, M.C.; Erickson, S.B.; El Ters, M.; Bentall, A.J.; Kukla, A.; Greene, E.L.; Hernandez, L.H.; et al. Identification of Genetic Causes of Focal Segmental Glomerulosclerosis Increases with Proper Patient Selection. Mayo Clin. Proc. 2021, 96, 2342–2353. [Google Scholar] [CrossRef]
- Laššuthová, P.; Šafka Brožková, D.; Krůtová, M.; Neupauerová, J.; Haberlová, J.; Mazanec, R.; Dřímal, P.; Seeman, P. Improving Diagnosis of Inherited Peripheral Neuropathies through Gene Panel Analysis. Orphanet J. Rare Dis. 2016, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Huynh Cong, E.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot-Marie-Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef]
- Roos, A.; Weis, J.; Korinthenberg, R.; Fehrenbach, H.; Häusler, M.; Züchner, S.; Mache, C.; Hubmann, H.; Auer-Grumbach, M.; Senderek, J. Inverted Formin 2-Related Charcot-Marie-Tooth Disease: Extension of the Mutational Spectrum and Pathological Findings in Schwann Cells and Axons. J. Peripher. Nerv. Syst. 2015, 20, 52–59. [Google Scholar] [CrossRef]
- Toyota, K.; Ogino, D.; Hayashi, M.; Taki, M.; Saito, K.; Abe, A.; Hashimoto, T.; Umetsu, K.; Tsukaguchi, H.; Hayasaka, K. INF2 Mutations in Charcot-Marie-Tooth Disease Complicated with Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2013, 18, 97–98. [Google Scholar] [CrossRef]
- Mademan, I.; Deconinck, T.; Dinopoulos, A.; Voit, T.; Schara, U.; Devriendt, K.; Meijers, B.; Lerut, E.; De Jonghe, P.; Baets, J. De Novo INF2 Mutations Expand the Genetic Spectrum of Hereditary Neuropathy with Glomerulopathy. Neurology 2013, 81, 1953–1958. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 Gene Account for a Significant Proportion of Familial but Not Sporadic Focal and Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef]
- Snoek, R.; Nguyen, T.Q.; van der Zwaag, B.; van Zuilen, A.D.; Kruis, H.M.E.; van Gils-Verrij, L.A.; Goldschmeding, R.; Knoers, N.V.A.M.; Rookmaaker, M.B.; van Eerde, A.M. Importance of Genetic Diagnostics in Adult-Onset Focal Segmental Glomerulosclerosis. Nephron 2019, 142, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome Sequencing and Identification of Phenocopies in Patients with Clinically Presumed Hereditary Nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Benoit, G.; Gribouval, O.; Nevo, F.; Tête, M.-J.; Dantal, J.; Gilbert-Dussardier, B.; Touchard, G.; Karras, A.; Presne, C.; et al. Mutations in INF2 Are a Major Cause of Autosomal Dominant Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Lee, C.; Kim, N.K.D.; Ahn, Y.H.; Park, Y.S.; Lee, J.H.; Kim, S.H.; Cho, M.H.; Cho, H.; Yoo, K.H.; et al. Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. J. Clin. Med. 2020, 9, 2013. [Google Scholar] [CrossRef]
- Rodriguez, P.Q.; Lohkamp, B.; Celsi, G.; Mache, C.J.; Auer-Grumbach, M.; Wernerson, A.; Hamajima, N.; Tryggvason, K.; Patrakka, J. Novel INF2 Mutation p. L77P in a Family with Glomerulopathy and Charcot-Marie-Tooth Neuropathy. Pediatr. Nephrol. 2013, 28, 339–343. [Google Scholar] [CrossRef]
- Xie, J.; Hao, X.; Azeloglu, E.U.; Ren, H.; Wang, Z.; Ma, J.; Liu, J.; Ma, X.; Wang, W.; Pan, X.; et al. Novel Mutations in the Inverted Formin 2 Gene of Chinese Families Contribute to Focal Segmental Glomerulosclerosis. Kidney Int. 2015, 88, 593–604. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Latour, P. A Cryptic Splicing Mutation in the INF2 Gene Causing Charcot-Marie-Tooth Disease with Minimal Glomerular Dysfunction. J. Peripher. Nerv. Syst. 2019, 24, 120–124. [Google Scholar] [CrossRef]
- Challis, R.C.; Ring, T.; Xu, Y.; Wong, E.K.S.; Flossmann, O.; Roberts, I.S.D.; Ahmed, S.; Wetherall, M.; Salkus, G.; Brocklebank, V.; et al. Thrombotic Microangiopathy in Inverted Formin 2-Mediated Renal Disease. J. Am. Soc. Nephrol. 2017, 28, 1084–1091. [Google Scholar] [CrossRef]
- Bacquet, J.; Stojkovic, T.; Boyer, A.; Martini, N.; Audic, F.; Chabrol, B.; Salort-Campana, E.; Delmont, E.; Desvignes, J.-P.; Verschueren, A.; et al. Molecular Diagnosis of Inherited Peripheral Neuropathies by Targeted Next-Generation Sequencing: Molecular Spectrum Delineation. BMJ Open 2018, 8, e021632. [Google Scholar] [CrossRef]
- Lemieux, G.; Neemeh, J.A. Charcot-Marie-Tooth Disease and Nephritis. Can. Med. Assoc. J. 1967, 97, 1193–1198. [Google Scholar]
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Caridi, G.; Lugani, F.; Dagnino, M.; Gigante, M.; Iolascon, A.; Falco, M.; Graziano, C.; Benetti, E.; Dugo, M.; Del Prete, D.; et al. Novel INF2 Mutations in an Italian Cohort of Patients with Focal Segmental Glomerulosclerosis, Renal Failure and Charcot-Marie-Tooth Neuropathy. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), iv80–iv86. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Ma, M.; Pang, M.; Yang, L.; Li, G.; Song, J.; Zhang, J. Analysis of a pedigree with autosomal dominant intermediate Charcot-Marie-Tooth disease type E and nephropathy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2019, 36, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wang, W.; Wang, R.; Lv, H.; Zhang, W.; Wang, Z.; Jiao, J.; Yuan, Y. INF2 Mutations Associated with Dominant Inherited Intermediate Charcot-Marie-Tooth Neuropathy with Focal Segmental Glomerulosclerosis in Two Chinese Patients. Clin. Neuropathol. 2015, 34, 275–281. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, H.J.; Hong, Y.B.; Nam, S.H.; Chung, K.W.; Choi, B.-O. A Novel INF2 Mutation in a Korean Family with Autosomal Dominant Intermediate Charcot-Marie-Tooth Disease and Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2014, 19, 175–179. [Google Scholar] [CrossRef]
- Laurin, L.-P.; Lu, M.; Mottl, A.K.; Blyth, E.R.; Poulton, C.J.; Weck, K.E. Podocyte-Associated Gene Mutation Screening in a Heterogeneous Cohort of Patients with Sporadic Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2014, 29, 2062–2069. [Google Scholar] [CrossRef]
- Wang, M.; Chun, J.; Genovese, G.; Knob, A.U.; Benjamin, A.; Wilkins, M.S.; Friedman, D.J.; Appel, G.B.; Lifton, R.P.; Mane, S.; et al. Contributions of Rare Gene Variants to Familial and Sporadic FSGS. J. Am. Soc. Nephrol. 2019, 30, 1625–1640. [Google Scholar] [CrossRef]
- Münch, J.; Grohmann, M.; Lindner, T.H.; Bergmann, C.; Halbritter, J. Diagnosing FSGS without Kidney Biopsy—A Novel INF2-Mutation in a Family with ESRD of Unknown Origin. BMC Med. Genet. 2016, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Büscher, A.K.; Celebi, N.; Hoyer, P.F.; Klein, H.-G.; Weber, S.; Hoefele, J. Mutations in INF2 May Be Associated with Renal Histology other than Focal Segmental Glomerulosclerosis. Pediatr. Nephrol. 2018, 33, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.-M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of Disease-Causing Variants by Comprehensive Genetic Testing with Exome Sequencing in Adults with Suspicion of Hereditary FSGS. Eur. J. Hum. Genet. 2021, 29, 262–270. [Google Scholar] [CrossRef]
- Rood, I.M.; Bongers, E.M.H.F.; Lugtenberg, D.; Klein, I.H.H.T.; Steenbergen, E.J.; Wetzels, J.F.M.; Deegens, J.K.J. Familial Focal Segmental Glomerulosclerosis: Mutation in Inverted Formin 2 Mimicking Alport Syndrome. Neth. J. Med. 2016, 74, 82–85. [Google Scholar] [PubMed]
- Gbadegesin, R.A.; Lavin, P.J.; Hall, G.; Bartkowiak, B.; Homstad, A.; Jiang, R.; Wu, G.; Byrd, A.; Lynn, K.; Wolfish, N.; et al. Inverted Formin 2 Mutations with Variable Expression in Patients with Sporadic and Hereditary Focal and Segmental Glomerulosclerosis. Kidney Int. 2012, 81, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Tan, W.; Lovric, S.; Ashraf, S.; Rao, J.; Schapiro, D.; Airik, M.; Shril, S.; Gee, H.Y.; Baum, M.; Daouk, G.; et al. Analysis of 24 Genes Reveals a Monogenic Cause in 11.1% of Cases with Steroid-Resistant Nephrotic Syndrome at a Single Center. Pediatr. Nephrol. 2018, 33, 305–314. [Google Scholar] [CrossRef]
- Ogino, D.; Hashimoto, T.; Hattori, M.; Sugawara, N.; Akioka, Y.; Tamiya, G.; Makino, S.; Toyota, K.; Mitsui, T.; Hayasaka, K. Analysis of the Genes Responsible for Steroid-Resistant Nephrotic Syndrome and/or Focal Segmental Glomerulosclerosis in Japanese Patients by Whole-Exome Sequencing Analysis. J. Hum. Genet. 2016, 61, 137–141. [Google Scholar] [CrossRef]
- Shang, S.; Peng, F.; Wang, T.; Wu, X.; Li, P.; Li, Q.; Chen, X.M. Genotype-Phenotype Correlation and Prognostic Impact in Chinese Patients with Alport Syndrome. Mol. Genet. Genom. Med. 2019, 7, e00741. [Google Scholar] [CrossRef]
- Gribouval, O.; Boyer, O.; Hummel, A.; Dantal, J.; Martinez, F.; Sberro-Soussan, R.; Etienne, I.; Chauveau, D.; Delahousse, M.; Lionet, A.; et al. Identification of Genetic Causes for Sporadic Steroid-Resistant Nephrotic Syndrome in Adults. Kidney Int. 2018, 94, 1013–1022. [Google Scholar] [CrossRef]
- Singh, A.; Singh, A.; Mishra, O.P.; Prasad, R.; Narayan, G.; Batra, V.V.; Tabatabaeifar, M.; Schaefer, F. Molecular Study of Childhood Steroid-Resistant Nephrotic Syndrome: A Hospital-Based Study. J. Pediatr. Genet. 2021. [Google Scholar] [CrossRef]
- Santín, S.; Bullich, G.; Tazón-Vega, B.; García-Maset, R.; Giménez, I.; Silva, I.; Ruíz, P.; Ballarín, J.; Torra, R.; Ars, E. Clinical Utility of Genetic Testing in Children and Adults with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2011, 6, 1139–1148. [Google Scholar] [CrossRef]
- Larsen, C.P.; Durfee, T.; Wilson, J.D.; Beggs, M.L. A Custom Targeted Next-Generation Sequencing Gene Panel for the Diagnosis of Genetic Nephropathies. Am. J. Kidney Dis. 2016, 67, 992–993. [Google Scholar] [CrossRef]
- Zhao, W.; Ma, X.; Zhang, X.; Luo, D.; Zhang, J.; Li, M.; Ye, Z.; Peng, H. INF2 p.Arg214Cys Mutation in a Chinese Family with Rapidly Progressive Renal Failure and Follow-up of Renal Transplantation: Case Report and Literature Review. BMC Nephrol. 2021, 22, 51. [Google Scholar] [CrossRef]
- Safarikova, M.; Stekrova, J.; Honsova, E.; Horinova, V.; Tesar, V.; Reiterova, J. Mutational Screening of Inverted Formin 2 in Adult-Onset Focal Segmental Glomerulosclerosis or Minimal Change Patients from the Czech Republic. BMC Med. Genet. 2018, 19, 147. [Google Scholar] [CrossRef]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) Mutations Are the Most Frequent Mutations Underlying Adult Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Lipska, B.S.; Iatropoulos, P.; Maranta, R.; Caridi, G.; Ozaltin, F.; Anarat, A.; Balat, A.; Gellermann, J.; Trautmann, A.; Erdogan, O.; et al. Genetic Screening in Adolescents with Steroid-Resistant Nephrotic Syndrome. Kidney Int. 2013, 84, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Bullich, G.; Trujillano, D.; Santín, S.; Ossowski, S.; Mendizábal, S.; Fraga, G.; Madrid, Á.; Ariceta, G.; Ballarín, J.; Torra, R.; et al. Targeted Next-Generation Sequencing in Steroid-Resistant Nephrotic Syndrome: Mutations in Multiple Glomerular Genes May Influence Disease Severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Han, K.H.; Jung, Y.H.; Kang, H.G.; Moon, K.C.; Ha, I.S.; Choi, Y.; Cheong, H.I. Variable Renal Phenotype in a Family with an INF2 Mutation. Pediatr. Nephrol. 2011, 26, 73–76. [Google Scholar] [CrossRef]
- Sanchez-Ares, M.; Garcia-Vidal, M.; Antucho, E.-E.; Julio, P.; Eduardo, V.-M.; Lens, X.M.; Garcia-Gonzalez, M.A. A Novel Mutation, Outside of the Candidate Region for Diagnosis, in the Inverted Formin 2 Gene Can Cause Focal Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Büscher, A.K.; Beck, B.B.; Melk, A.; Hoefele, J.; Kranz, B.; Bamborschke, D.; Baig, S.; Lange-Sperandio, B.; Jungraithmayr, T.; Weber, L.T.; et al. Rapid Response to Cyclosporin A and Favorable Renal Outcome in Nongenetic Versus Genetic Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 245–253. [Google Scholar] [CrossRef]
- Weber, S.; Büscher, A.K.; Hagmann, H.; Liebau, M.C.; Heberle, C.; Ludwig, M.; Rath, S.; Alberer, M.; Beissert, A.; Zenker, M.; et al. Dealing with the Incidental Finding of Secondary Variants by the Example of SRNS Patients Undergoing Targeted Next-Generation Sequencing. Pediatr. Nephrol. 2016, 31, 73–81. [Google Scholar] [CrossRef]
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Sinha, R.; Maiti, R.; Das, D.; Mandal, K. Steroid Resistant Nephrotic Syndrome with Clumsy Gait Associated with INF2 Mutation. Indian Pediatr. 2020, 57, 764. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Ruan, J.; Liu, J.; Zhang, C.; Kang, L.; Wang, J.; Zou, Y.; Song, L. Variant Spectrum of Formin Homology 2 Domain-Containing 3 Gene in Chinese Patients with Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e018236. [Google Scholar] [CrossRef]
- Huang, S.; Pu, T.; Wei, W.; Xu, R.; Wu, Y. Exome Sequencing Identifies a FHOD3 p.S527del Mutation in a Chinese Family with Hypertrophic Cardiomyopathy. J. Gene Med. 2020, 22, e3146. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Bagnall, R.D. Revisiting Genome Sequencing Data in Light of Novel Disease Gene Associations. J. Am. Coll. Cardiol. 2019, 73, 1365–1366. [Google Scholar] [CrossRef]
- Ochoa, J.P.; Lopes, L.R.; Perez-Barbeito, M.; Cazón-Varela, L.; de la Torre-Carpente, M.M.; Sonicheva-Paterson, N.; Uña-Iglesias, D.D.; Quinn, E.; Kuzmina-Krutetskaya, S.; Garrote, J.A.; et al. Deletions of Specific Exons of FHOD3 Detected by Next-Generation Sequencing Are Associated with Hypertrophic Cardiomyopathy. Clin. Genet. 2020, 98, 86–90. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanimoto, K.; Hirayama-Yamada, K.; Tsuda, E.; Ayusawa, M.; Nunoda, S.; Hosaki, A.; Kimura, A. Genetic Background of Japanese Patients with Pediatric Hypertrophic and Restrictive Cardiomyopathy. J. Hum. Genet. 2018, 63, 989–996. [Google Scholar] [CrossRef]
- DeWard, A.D.; Eisenmann, K.M.; Matheson, S.F.; Alberts, A.S. The Role of Formins in Human Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Randall, T.S.; Ehler, E. A Formin-g Role during Development and Disease. Eur. J. Cell Biol. 2014, 93, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Scott, R.P.; Quaggin, S.E. The Cell Biology of Renal Filtration. J. Cell Biol. 2015, 209, 199–210. [Google Scholar] [CrossRef]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Labat-de-Hoz, L.; Alonso, M.A. The Formin INF2 in Disease: Progress from 10 Years of Research. Cell. Mol. Life Sci. 2020, 77, 4581–4600. [Google Scholar] [CrossRef]
- Bose, B.; Cattran, D. Glomerular Diseases: FSGS. Clin. J. Am. Soc. Nephrol. 2014, 9, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Fogo, A.B. Causes and Pathogenesis of Focal Segmental Glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical Implications of Genetic Advances in Charcot-Marie-Tooth Disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef]
- Bayraktar, S.; Nehrig, J.; Menis, E.; Karli, K.; Janning, A.; Struk, T.; Halbritter, J.; Michgehl, U.; Krahn, M.P.; Schuberth, C.E.; et al. A Deregulated Stress Response Underlies Distinct INF2-Associated Disease Profiles. J. Am. Soc. Nephrol. 2020, 31, 1296–1313. [Google Scholar] [CrossRef]
- Mu, A.; Fung, T.S.; Kettenbach, A.N.; Chakrabarti, R.; Higgs, H.N. A Complex Containing Lysine-Acetylated Actin Inhibits the Formin INF2. Nat. Cell Biol. 2019, 21, 592–602. [Google Scholar] [CrossRef]
- Subramanian, B.; Sun, H.; Yan, P.; Charoonratana, V.T.; Higgs, H.N.; Wang, F.; Lai, K.-M.V.; Valenzuela, D.M.; Brown, E.J.; Schlöndorff, J.S.; et al. Mice with Mutant Inf2 Show Impaired Podocyte and Slit Diaphragm Integrity in Response to Protamine-Induced Kidney Injury. Kidney Int. 2016, 90, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Mathis, S.; Funalot, B.; Boyer, O.; Lacroix, C.; Marcorelles, P.; Magy, L.; Richard, L.; Antignac, C.; Vallat, J.-M. Neuropathologic Characterization of INF2-Related Charcot-Marie-Tooth Disease: Evidence for a Schwann Cell Actinopathy. J. Neuropathol. Exp. Neurol. 2014, 73, 223–233. [Google Scholar] [CrossRef]
- Tricaud, N. Myelinating Schwann Cell Polarity and Mechanically-Driven Myelin Sheath Elongation. Front. Cell. Neurosci. 2018, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Takemon, Y.; Wright, V.; Davenport, B.; Gatti, D.M.; Sheehan, S.M.; Letson, K.; Savage, H.S.; Lennon, R.; Korstanje, R. Uncovering Modifier Genes of X-Linked Alport Syndrome Using a Novel Multiparent Mouse Model. J. Am. Soc. Nephrol. 2021, 32, 1961–1973. [Google Scholar] [CrossRef]
- Schwander, M.; Kachar, B.; Müller, U. Review Series: The Cell Biology of Hearing. J. Cell Biol. 2010, 190, 9–20. [Google Scholar] [CrossRef]
- Drummond, M.C.; Belyantseva, I.A.; Friderici, K.H.; Friedman, T.B. Actin in Hair Cells and Hearing Loss. Hear Res. 2012, 288, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Wallar, B.J.; Stropich, B.N.; Schoenherr, J.A.; Holman, H.A.; Kitchen, S.M.; Alberts, A.S. The Basic Region of the Diaphanous-Autoregulatory Domain (DAD) Is Required for Autoregulatory Interactions with the Diaphanous-Related Formin Inhibitory Domain. J. Biol. Chem. 2006, 281, 4300–4307. [Google Scholar] [CrossRef]
- Miyoshi, T.; Belyantseva, I.A.; Kitajiri, S.-I.; Miyajima, H.; Nishio, S.-Y.; Usami, S.-I.; Kim, B.J.; Choi, B.Y.; Omori, K.; Shroff, H.; et al. Human Deafness-Associated Variants Alter the Dynamics of Key Molecules in Hair Cell Stereocilia F-Actin Cores. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Lakha, R.; Montero, A.M.; Jabeen, T.; Costeas, C.C.; Ma, J.; Vizcarra, C.L. Variable Autoinhibition among Deafness-Associated Variants of Diaphanous 1 (DIAPH1). Biochemistry 2021, 60, 2320–2329. [Google Scholar] [CrossRef]
- Ninoyu, Y.; Sakaguchi, H.; Lin, C.; Suzuki, T.; Hirano, S.; Hisa, Y.; Saito, N.; Ueyama, T. The Integrity of Cochlear Hair Cells Is Established and Maintained through the Localization of Dia1 at Apical Junctional Complexes and Stereocilia. Cell Death Dis. 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Schoen, C.J.; Burmeister, M.; Lesperance, M.M. Diaphanous Homolog 3 (Diap3) Overexpression Causes Progressive Hearing Loss and Inner Hair Cell Defects in a Transgenic Mouse Model of Human Deafness. PLoS ONE 2013, 8, e56520. [Google Scholar] [CrossRef]
- Surel, C.; Guillet, M.; Lenoir, M.; Bourien, J.; Sendin, G.; Joly, W.; Delprat, B.; Lesperance, M.M.; Puel, J.-L.; Nouvian, R. Remodeling of the Inner Hair Cell Microtubule Meshwork in a Mouse Model of Auditory Neuropathy AUNA1. eNeuro 2016, 3, 0295-16.2016. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.-H.; Baek, J.-I.; Lee, J.D.; Song, M.H.; Kwon, T.-J.; Oh, S.-K.; Jeong, J.Y.; Choi, J.Y.; Lee, K.-Y.; Kim, U.-K. Genetic Analysis of Auditory Neuropathy Spectrum Disorder in the Korean Population. Gene 2013, 522, 65–69. [Google Scholar] [CrossRef]
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants which May Be Associated with Bleeding. Front. Cardiovasc. Med. 2019, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Becker, I.C.; Scheller, I.; Wackerbarth, L.M.; Beck, S.; Heib, T.; Aurbach, K.; Manukjan, G.; Gross, C.; Spindler, M.; Nagy, Z.; et al. Actin/Microtubule Crosstalk during Platelet Biogenesis in Mice Is Critically Regulated by Twinfilin1 and Cofilin1. Blood Adv. 2020, 4, 2124–2134. [Google Scholar] [CrossRef]
- Italiano, J.E.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood Platelets Are Assembled Principally at the Ends of Proplatelet Processes Produced by Differentiated Megakaryocytes. J. Cell Biol. 1999, 147, 1299–1312. [Google Scholar] [CrossRef]
- Patel, S.R. The Biogenesis of Platelets from Megakaryocyte Proplatelets. J. Clin. Investig. 2005, 115, 3348–3354. [Google Scholar] [CrossRef]
- Potts, K.S.; Farley, A.; Dawson, C.A.; Rimes, J.; Biben, C.; de Graaf, C.; Potts, M.A.; Stonehouse, O.J.; Carmagnac, A.; Gangatirkar, P.; et al. Membrane Budding Is a Major Mechanism of in Vivo Platelet Biogenesis. J. Exp. Med. 2020, 217, e20191206. [Google Scholar] [CrossRef]
- Pan, J.; Lordier, L.; Meyran, D.; Rameau, P.; Lecluse, Y.; Kitchen-Goosen, S.; Badirou, I.; Mokrani, H.; Narumiya, S.; Alberts, A.S.; et al. The Formin DIAPH1 (MDia1) Regulates Megakaryocyte Proplatelet Formation by Remodeling the Actin and Microtubule Cytoskeletons. Blood 2014, 124, 3967–3977. [Google Scholar] [CrossRef]
- Zuidscherwoude, M.; Green, H.L.H.; Thomas, S.G. Formin Proteins in Megakaryocytes and Platelets: Regulation of Actin and Microtubule Dynamics. Platelets 2019, 30, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of Microtubules and the Actin Cytoskeleton by the Rho Effector MDia1. Nat. Cell Biol. 2001, 3, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Cook, T.A.; Alberts, A.S.; Gundersen, G.G. mDia Mediates Rho-Regulated Formation and Orientation of Stable Microtubules. Nat. Cell Biol. 2001, 3, 723–729. [Google Scholar] [CrossRef]
- Heremans, J.; Garcia-Perez, J.E.; Turro, E.; Schlenner, S.M.; Casteels, I.; Collin, R.; de Zegher, F.; Greene, D.; Humblet-Baron, S.; Lesage, S.; et al. Abnormal Differentiation of B Cells and Megakaryocytes in Patients with Roifman Syndrome. J. Allergy Clin. Immunol. 2018, 142, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Roifman, M.; Braunschweig, U.; Yuen, R.K.C.; Alexandrova, R.; Bates, A.; Reid, B.; Nalpathamkalam, T.; Wang, Z.; Thiruvahindrapuram, B.; et al. Compound Heterozygous Mutations in the Noncoding RNU4ATAC Cause Roifman Syndrome by Disrupting Minor Intron Splicing. Nat. Commun. 2015, 6, 8718. [Google Scholar] [CrossRef]
- Dutta, P.; Maiti, S. Expression of Multiple Formins in Adult Tissues and during Developmental Stages of Mouse Brain. Gene Expr. Patterns 2015, 19, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kawabata Galbraith, K.; Kengaku, M. Multiple Roles of the Actin and Microtubule-Regulating Formins in the Developing Brain. Neurosci. Res. 2019, 138, 59–69. [Google Scholar] [CrossRef]
- Damiani, D.; Goffinet, A.M.; Alberts, A.; Tissir, F. Lack of Diaph3 Relaxes the Spindle Checkpoint Causing the Loss of Neural Progenitors. Nat. Commun. 2016, 7, 13509. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Shinohara, R.; Watanabe, K.; Takebayashi, H.; Toyoda, Y.; Tohyama, K.; Ishizaki, T.; Furuyashiki, T.; Narumiya, S. Deficiency of mDia, an Actin Nucleator, Disrupts Integrity of Neuroepithelium and Causes Periventricular Dysplasia. PLoS ONE 2011, 6, e25465. [Google Scholar] [CrossRef]
- Eisenmann, K.M.; West, R.A.; Hildebrand, D.; Kitchen, S.M.; Peng, J.; Sigler, R.; Zhang, J.; Siminovitch, K.A.; Alberts, A.S. T Cell Responses in Mammalian Diaphanous-Related Formin MDia1 Knock-out Mice. J. Biol. Chem. 2007, 282, 25152–25158. [Google Scholar] [CrossRef]
- Sakata, D.; Taniguchi, H.; Yasuda, S.; Adachi-Morishima, A.; Hamazaki, Y.; Nakayama, R.; Miki, T.; Minato, N.; Narumiya, S. Impaired T Lymphocyte Trafficking in Mice Deficient in an Actin-Nucleating Protein, MDia1. J. Exp. Med. 2007, 204, 2031–2038. [Google Scholar] [CrossRef]
- Kundu, T.; Dutta, P.; Nagar, D.; Maiti, S.; Ghose, A. Coupling of Dynamic Microtubules to F-Actin by Fmn2 Regulates Chemotaxis of Neuronal Growth Cones. J. Cell Sci. 2021, 134, jcs252916. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, A.; Ghate, K.; Mutalik, S.; Jacob, A.; Ghose, A. Formin 2 Regulates the Stabilization of Filopodial Tip Adhesions in Growth Cones and Affects Neuronal Outgrowth and Pathfinding in Vivo. Development 2016, 143, 449–460. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leader, B.; Lim, H.; Carabatsos, M.J.; Harrington, A.; Ecsedy, J.; Pellman, D.; Maas, R.; Leder, P. Formin-2, Polyploidy, Hypofertility and Positioning of the Meiotic Spindle in Mouse Oocytes. Nat. Cell Biol. 2002, 4, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Dettenhofer, M.; Lu, J.; Downing, M.; Chenn, A.; Wong, T.; Sheen, V. Filamin A- and Formin 2-Dependent Endocytosis Regulates Proliferation via the Canonical Wnt Pathway. Development 2016, 143, 4509–4520. [Google Scholar] [CrossRef] [PubMed]
- Lybaek, H.; Ørstavik, K.H.; Prescott, T.; Hovland, R.; Breilid, H.; Stansberg, C.; Steen, V.M.; Houge, G. An 8.9 Mb 19p13 Duplication Associated with Precocious Puberty and a Sporadic 3.9 Mb 2q23.3q24.1 Deletion Containing NR4A2 in Mentally Retarded Members of a Family with an Intrachromosomal 19p-into-19q between-Arm Insertion. Eur. J. Hum. Genet. 2009, 17, 904–910. [Google Scholar] [CrossRef]
- Castrillon, D.H.; Wasserman, S.A. Diaphanous Is Required for Cytokinesis in Drosophila and Shares Domains of Similarity with the Products of the Limb Deformity Gene. Development 1994, 120, 3367–3377. [Google Scholar] [CrossRef]
- Ryley, D.A.; Wu, H.-H.; Leader, B.; Zimon, A.; Reindollar, R.H.; Gray, M.R. Characterization and Mutation Analysis of the Human Formin-2 (FMN2) Gene in Women with Unexplained Infertility. Fertil. Steril. 2005, 83, 1363–1371. [Google Scholar] [CrossRef]
- Tšuiko, O.; Nõukas, M.; Žilina, O.; Hensen, K.; Tapanainen, J.S.; Mägi, R.; Kals, M.; Kivistik, P.A.; Haller-Kikkatalo, K.; Salumets, A.; et al. Copy Number Variation Analysis Detects Novel Candidate Genes Involved in Follicular Growth and Oocyte Maturation in a Cohort of Premature Ovarian Failure Cases. Hum. Reprod. 2016, 31, 1913–1925. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Liu, F.; Zhang, Z.; Zou, Y.; Yang, B.; Luo, Y.; Wang, L.; Huang, O. Inhibition of Formin like 2 Promotes the Transition of Ectopic Endometrial Stromal Cells to Epithelial Cells in Adenomyosis through a MET-like Process. Gene 2019, 710, 186–192. [Google Scholar] [CrossRef]
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical Roles for Multiple Formins during Cardiac Myofibril Development and Repair. Mol. Biol. Cell 2014, 25, 811–827. [Google Scholar] [CrossRef]
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian Formin Fhod3 Regulates Actin Assembly and Sarcomere Organization in Striated Muscles. J. Biol. Chem. 2009, 284, 29873–29881. [Google Scholar] [CrossRef]
- Ehler, E. Actin-Associated Proteins and Cardiomyopathy-the “unknown” beyond Troponin and Tropomyosin. Biophys. Rev. 2018, 10, 1121–1128. [Google Scholar] [CrossRef]
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated Cardiomyopathy-Associated FHOD3 Variant Impairs the Ability to Induce Activation of Transcription Factor Serum Response Factor. Circ. J. 2013, 77, 2990–2996. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, U.; Garnier, S.; Korniat, A.; Proust, C.; Kararigas, G.; Müller-Nurasyid, M.; Empana, J.-P.; Morley, M.P.; Perret, C.; Stark, K.; et al. Exome-Wide Association Study Reveals Novel Susceptibility Genes to Sporadic Dilated Cardiomyopathy. PLoS ONE 2017, 12, e0172995. [Google Scholar] [CrossRef]
- Kan, O.M.; Takeya, R.; Abe, T.; Kitajima, N.; Nishida, M.; Tominaga, R.; Kurose, H.; Sumimoto, H. Mammalian Formin Fhod3 Plays an Essential Role in Cardiogenesis by Organizing Myofibrillogenesis. Biol. Open 2012, 1, 889–896. [Google Scholar] [CrossRef]
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan, O.M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The Actin-Organizing Formin Protein Fhod3 Is Required for Postnatal Development and Functional Maintenance of the Adult Heart in Mice. J. Biol. Chem. 2018, 293, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef]
- Zhou, Q.; Wei, S.-S.; Wang, H.; Wang, Q.; Li, W.; Li, G.; Hou, J.-W.; Chen, X.-M.; Chen, J.; Xu, W.-P.; et al. Crucial Role of ROCK2-Mediated Phosphorylation and Upregulation of FHOD3 in the Pathogenesis of Angiotensin II-Induced Cardiac Hypertrophy. Hypertension 2017, 69, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Hilton-Jones, D. Myotonic Dystrophy: Diagnosis, Management and New Therapies. Curr. Opin. Neurol. 2014, 27, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Oliveira, R.; Sznajder, Ł.J.; Swanson, M.S. Myotonic Dystrophy and Developmental Regulation of RNA Processing. In Comprehensive Physiology; Terjung, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 509–553. ISBN 978-0-470-65071-4. [Google Scholar]
- Dixon, D.M.; Choi, J.; El-Ghazali, A.; Park, S.Y.; Roos, K.P.; Jordan, M.C.; Fishbein, M.C.; Comai, L.; Reddy, S. Loss of Muscleblind-like 1 Results in Cardiac Pathology and Persistence of Embryonic Splice Isoforms. Sci. Rep. 2015, 5, 9932. [Google Scholar] [CrossRef]
- Perfetti, A.; Greco, S.; Fasanaro, P.; Bugiardini, E.; Cardani, R.; Garcia-Manteiga, J.M.; Manteiga, J.M.G.; Riba, M.; Cittaro, D.; Stupka, E.; et al. Genome Wide Identification of Aberrant Alternative Splicing Events in Myotonic Dystrophy Type 2. PLoS ONE 2014, 9, e93983. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Zhang, L.; Hu, H.; Yin, S.; Liang, Z. Deletion of a Single-Copy DAAM1 Gene in Congenital Heart Defect: A Case Report. BMC Med. Genet. 2012, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Lehalle, D.; Sanlaville, D.; Guimier, A.; Plouvier, E.; Leblanc, T.; Galmiche, L.; Radford, I.; Romana, S.; Colleaux, L.; de Pontual, L.; et al. Multiple Congenital Anomalies-Intellectual Disability (MCA-ID) and Neuroblastoma in a Patient Harboring a de Novo 14q23.1q23.3 Deletion. Am. J. Med. Genet. 2014, 164, 1310–1317. [Google Scholar] [CrossRef]
- Li, D.; Hallett, M.A.; Zhu, W.; Rubart, M.; Liu, Y.; Yang, Z.; Chen, H.; Haneline, L.S.; Chan, R.J.; Schwartz, R.J.; et al. Dishevelled-Associated Activator of Morphogenesis 1 (Daam1) Is Required for Heart Morphogenesis. Development 2011, 138, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Caporale, N.; Testa, G. Autism Spectrum Disorder at the Crossroad between Genes and Environment: Contributions, Convergences, and Interactions in ASD Developmental Pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef]
- Vorstman, J.; van Daalen, E.; Jalali, G.; Schmidt, E.; Pasterkamp, R.; de Jonge, M.; Hennekam, E.; Janson, E.; Staal, W.; van der Zwaag, B.; et al. A Double Hit Implicates DIAPH3 as an Autism Risk Gene. Mol. Psychiatry 2011, 16, 442–451. [Google Scholar] [CrossRef]
- Xie, J.; Li, H.; Zhu, H.; Huang, L.; Li, H.; Zhang, X.; Zhou, Y.; Zhou, Q.; Xu, W. Analysis of DIAPH3 gene mutation in a boy with autism spectrum disorder. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2016, 33, 481–484. [Google Scholar] [CrossRef]
- Liu, L.; Liu, F.; Wang, Q.; Xie, H.; Li, Z.; Lu, Q.; Wang, Y.; Zhang, M.; Zhang, Y.; Picker, J.; et al. Confirming the Contribution and Genetic Spectrum of de Novo Mutation in Infantile Spasms: Evidence from a Chinese Cohort. Mol. Genet. Genom. Med. 2021, 9, e1689. [Google Scholar] [CrossRef]
- Abramov, D.; Guiberson, N.G.L.; Burré, J. STXBP1 Encephalopathies: Clinical Spectrum, Disease Mechanisms, and Therapeutic Strategies. J. Neurochem. 2020, 157, 165–178. [Google Scholar] [CrossRef]
- Lesca, G.; Rudolf, G.; Labalme, A.; Hirsch, E.; Arzimanoglou, A.; Genton, P.; Motte, J.; de Saint Martin, A.; Valenti, M.-P.; Boulay, C.; et al. Epileptic Encephalopathies of the Landau-Kleffner and Continuous Spike and Waves during Slow-Wave Sleep Types: Genomic Dissection Makes the Link with Autism. Epilepsia 2012, 53, 1526–1538. [Google Scholar] [CrossRef]
- Lau, E.O.-C.; Damiani, D.; Chehade, G.; Ruiz-Reig, N.; Saade, R.; Jossin, Y.; Aittaleb, M.; Schakman, O.; Tajeddine, N.; Gailly, P.; et al. DIAPH3 Deficiency Links Microtubules to Mitotic Errors, Defective Neurogenesis, and Brain Dysfunction. eLife 2021, 10, e61974. [Google Scholar] [CrossRef]
- Matsunami, N.; Hensel, C.H.; Baird, L.; Stevens, J.; Otterud, B.; Leppert, T.; Varvil, T.; Hadley, D.; Glessner, J.T.; Pellegrino, R.; et al. Identification of Rare DNA Sequence Variants in High-Risk Autism Families and Their Prevalence in a Large Case/Control Population. Mol. Autism 2014, 5, 5. [Google Scholar] [CrossRef]
- Cappi, C.; Hounie, A.G.; Mariani, D.B.; Diniz, J.B.; Silva, A.R.T.; Reis, V.N.S.; Busso, A.F.; Silva, A.G.; Fidalgo, F.; Rogatto, S.R.; et al. An Inherited Small Microdeletion at 15q13.3 in a Patient with Early- Onset Obsessive-Compulsive Disorder. PLoS ONE 2014, 9, e110198. [Google Scholar] [CrossRef]
- Giacopuzzi, E.; Gennarelli, M.; Minelli, A.; Gardella, R.; Valsecchi, P.; Traversa, M.; Bonvicini, C.; Vita, A.; Sacchetti, E.; Magri, C. Exome Sequencing in Schizophrenic Patients with High Levels of Homozygosity Identifies Novel and Extremely Rare Mutations in the GABA/Glutamatergic Pathways. PLoS ONE 2017, 12, e0182778. [Google Scholar] [CrossRef] [PubMed]
- Simon-Areces, J.; Dopazo, A.; Dettenhofer, M.; Rodriguez-Tebar, A.; Garcia-Segura, L.M.; Arevalo, M.-A. Formin1 Mediates the Induction of Dendritogenesis and Synaptogenesis by Neurogenin3 in Mouse Hippocampal Neurons. PLoS ONE 2011, 6, e21825. [Google Scholar] [CrossRef] [PubMed]
- Proitsi, P.; Li, T.; Hamilton, G.; Di Forti, M.; Collier, D.; Killick, R.; Chen, R.; Sham, P.; Murray, R.; Powell, J.; et al. Positional Pathway Screen of Wnt Signaling Genes in Schizophrenia: Association with DKK4. Biol. Psychiatry 2008, 63, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Kuzman, M.R.; Medved, V.; Terzic, J.; Krainc, D. Genome-Wide Expression Analysis of Peripheral Blood Identifies Candidate Biomarkers for Schizophrenia. J. Psychiatr. Res. 2009, 43, 1073–1077. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, Q.; Xie, P. Correlation Between Daam2 Expression Changes and Demyelination in Guillain–Barre Syndrome. Cell. Mol. Neurobiol. 2016, 36, 683–688. [Google Scholar] [CrossRef]
- Kumar, R.; Corbett, M.A.; Smith, N.J.C.; Jolly, L.A.; Tan, C.; Keating, D.J.; Duffield, M.D.; Utsumi, T.; Moriya, K.; Smith, K.R.; et al. Homozygous Mutation of STXBP5L Explains an Autosomal Recessive Infantile-Onset Neurodegenerative Disorder. Hum. Mol. Genet. 2015, 24, 2000–2010. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wallingford, J.B. Planar Cell Polarity in Development and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Torban, E.; Sokol, S.Y. Planar Cell Polarity Pathway in Kidney Development, Function and Disease. Nat. Rev. Nephrol. 2021, 17, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef]
- Liu, W.; Sato, A.; Khadka, D.; Bharti, R.; Diaz, H.; Runnels, L.W.; Habas, R. Mechanism of Activation of the Formin Protein Daam1. Proc. Natl. Acad. Sci. USA 2008, 105, 210–215. [Google Scholar] [CrossRef]
- Nicolaou, N.; Pulit, S.L.; Nijman, I.J.; Monroe, G.R.; Feitz, W.F.J.; Schreuder, M.F.; van Eerde, A.M.; de Jong, T.P.V.M.; Giltay, J.C.; van der Zwaag, B.; et al. Prioritization and Burden Analysis of Rare Variants in 208 Candidate Genes Suggest They Do Not Play a Major Role in CAKUT. Kidney Int. 2016, 89, 476–486. [Google Scholar] [CrossRef]
- McMichael, G.; Girirajan, S.; Moreno-De-Luca, A.; Gecz, J.; Shard, C.; Nguyen, L.S.; Nicholl, J.; Gibson, C.; Haan, E.; Eichler, E.; et al. Rare Copy Number Variation in Cerebral Palsy. Eur. J. Hum. Genet. 2014, 22, 40–45. [Google Scholar] [CrossRef]
- Habas, R.; Kato, Y.; He, X. Wnt/Frizzled Activation of Rho Regulates Vertebrate Gastrulation and Requires a Novel Formin Homology Protein Daam1. Cell 2001, 107, 843–854. [Google Scholar] [CrossRef]
- Miller, R.K.; de la Torre Canny, S.G.; Jang, C.-W.; Cho, K.; Ji, H.; Wagner, D.S.; Jones, E.A.; Habas, R.; McCrea, P.D. Pronephric Tubulogenesis Requires Daam1-Mediated Planar Cell Polarity Signaling. J. Am. Soc. Nephrol. 2011, 22, 1654–1664. [Google Scholar] [CrossRef]
- Corkins, M.E.; Krneta-Stankic, V.; Kloc, M.; McCrea, P.D.; Gladden, A.B.; Miller, R.K. Divergent Roles of the Wnt/PCP Formin Daam1 in Renal Ciliogenesis. PLoS ONE 2019, 14, e0221698. [Google Scholar] [CrossRef]
- Kida, Y.; Shiraishi, T.; Ogura, T. Identification of Chick and Mouse Daam1 and Daam2 Genes and Their Expression Patterns in the Central Nervous System. Dev. Brain Res. 2004, 153, 143–150. [Google Scholar] [CrossRef]
- Sun, S.-W.; Zhou, M.; Chen, L.; Wu, J.-H.; Meng, Z.-J.; Miao, S.-Y.; Han, H.-L.; Zhu, C.-C.; Xiong, X.-Z. Whole Exome Sequencing Identifies a Rare Variant in DAAM2 as a Potential Candidate in Idiopathic Pulmonary Ossification. Ann. Transl. Med. 2019, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Uehara, S.; Udagawa, N.; Mukai, H.; Ishihara, A.; Maeda, K.; Yamashita, T.; Murakami, K.; Nishita, M.; Nakamura, T.; Kato, S.; et al. Protein Kinase N3 Promotes Bone Resorption by Osteoclasts in Response to Wnt5a-Ror2 Signaling. Sci. Signal. 2017, 10, eaan0023. [Google Scholar] [CrossRef] [PubMed]
- Calvel, P.; Kusz-Zamelczyk, K.; Makrythanasis, P.; Janecki, D.; Borel, C.; Conne, B.; Vannier, A.; Béna, F.; Gimelli, S.; Fichna, P.; et al. A Case of Wiedemann-Steiner Syndrome Associated with a 46, XY Disorder of Sexual Development and Gonadal Dysgenesis. Sex Dev. 2015, 9, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Cenani, A.; Lenz, W. Total syndactylia and total radioulnar synostosis in 2 brothers. A contribution on the genetics of syndactylia. Z. Kinder-Heilk. 1967, 101, 181–190. [Google Scholar] [CrossRef]
- Dimitrov, B.I.; Voet, T.; De Smet, L.; Vermeesch, J.R.; Devriendt, K.; Fryns, J.-P.; Debeer, P. Genomic Rearrangements of the GREM1-FMN1 Locus Cause Oligosyndactyly, Radio-Ulnar Synostosis, Hearing Loss, Renal Defects Syndrome and Cenani-Lenz-like Non-Syndromic Oligosyndactyly. J. Med. Genet. 2010, 47, 569–574. [Google Scholar] [CrossRef]
- Pavel, E.; Zhao, W.; Powell, K.A.; Weinstein, M.; Kirschner, L.S. Analysis of a New Allele of Limb Deformity (Ld) Reveals Tissue- and Age-Specific Transcriptional Effects of the Ld Global Control Region. Int. J. Dev. Biol. 2007, 51, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Leder, P.; Zuniga, A.; Dettenhofer, M. Formin1 Disruption Confers Oligodactylism and Alters Bmp Signaling. Hum. Mol. Genet. 2009, 18, 2472–2482. [Google Scholar] [CrossRef]
- Jiang, R.; Dong, J.; Joo, J.; Geller, N.L.; Zheng, G. Simple Strategies for Haplotype Analysis of the X Chromosome with Application to Age-Related Macular Degeneration. Eur. J. Hum. Genet. 2011, 19, 801–806. [Google Scholar] [CrossRef][Green Version]
- Zheng, G.; Joo, J.; Zhang, C.; Geller, N.L. Testing association for markers on the X chromosome. Genet. Epidemiol. 2007, 31, 834–843. [Google Scholar] [CrossRef]
- Burri, A.; Maercker, A.; Krammer, S.; Simmen-Janevska, K. Childhood Trauma and PTSD Symptoms Increase the Risk of Cognitive Impairment in a Sample of Former Indentured Child Laborers in Old Age. PLoS ONE 2013, 8, e57826. [Google Scholar] [CrossRef]
- Yaffe, K.; Vittinghoff, E.; Lindquist, K.; Barnes, D.; Covinsky, K.E.; Neylan, T.; Kluse, M.; Marmar, C. Posttraumatic Stress Disorder and Risk of Dementia Among US Veterans. Arch. Gen. Psychiatry 2010, 67, 608. [Google Scholar] [CrossRef] [PubMed]
- Agís-Balboa, R.C.; Pinheiro, P.S.; Rebola, N.; Kerimoglu, C.; Benito, E.; Gertig, M.; Bahari-Javan, S.; Jain, G.; Burkhardt, S.; Delalle, I.; et al. Formin 2 Links Neuropsychiatric Phenotypes at Young Age to an Increased Risk for Dementia. EMBO J. 2017, 36, 2815–2828. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Derk, J.; Hernandez, K.B.; Rodriguez, M.; He, M.; Koh, H.; Abedini, A.; Li, H.; Fenyö, D.; Schmidt, A.M. Diaphanous 1 (DIAPH1) Is Highly Expressed in the Aged Human Medial Temporal Cortex and Upregulated in Myeloid Cells During Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 995. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for Vascular and Neuroinflammatory Dysfunction in Disorders of the Central Nervous System. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Chen, J.; Gao, X.-M.; Zhao, H.; Cai, H.; Zhang, L.; Cao, X.-X.; Zhou, D.-B.; Li, J. A Highly Heterogeneous Mutational Pattern in POEMS Syndrome. Leukemia 2021, 35, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.J.; Harbord, M.W.; MacAllister, R.; Rahman, F.Z.; Young, J.; Al-Lazikani, B.; Lees, W.; Novelli, M.; Bloom, S.; Segal, A.W. Defective Acute Inflammation in Crohn’s Disease: A Clinical Investigation. Lancet 2006, 367, 668–678. [Google Scholar] [CrossRef]
- Trefzer, R.; Elpeleg, O.; Gabrusskaya, T.; Stepensky, P.; Mor-Shaked, H.; Grosse, R.; Brandt, D.T. Characterization of a L136P Mutation in Formin-like 2 (FMNL2) from a Patient with Chronic Inflammatory Bowel Disease. PLoS ONE 2021, 16, e0252428. [Google Scholar] [CrossRef]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Yan, S.F.; Schmidt, A.M.; Shekhtman, A. Signal Transduction in Receptor for Advanced Glycation End Products (RAGE): Solution Structure of C-Terminal Rage (CtRAGE) and Its Binding to mDia1. J. Biol. Chem. 2012, 287, 5133–5144. [Google Scholar] [CrossRef]
- Egaña-Gorroño, L.; López-Díez, R.; Yepuri, G.; Ramirez, L.S.; Reverdatto, S.; Gugger, P.F.; Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Receptor for Advanced Glycation End Products (RAGE) and Mechanisms and Therapeutic Opportunities in Diabetes and Cardiovascular Disease: Insights from Human Subjects and Animal Models. Front. Cardiovasc. Med. 2020, 7, 37. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Friedman, R.A.; Ramasamy, R.; D’Agati, V.; Schmidt, A.M. Deletion of the Formin Diaph1 Protects from Structural and Functional Abnormalities in the Murine Diabetic Kidney. Am. J. Physiol. Ren. Physiol. 2018, 315, F1601–F1612. [Google Scholar] [CrossRef]
- Cooper, J.D.; Walker, N.M.; Smyth, D.J.; Downes, K.; Healy, B.C.; Todd, J.A. Type I Diabetes Genetics Consortium Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium Genome-Wide Association Study in Type I Diabetes Families. Genes Immun. 2009, 10 (Suppl. 1), S85–S94. [Google Scholar] [CrossRef]
- Qi, C.; Al Somali, F.; Zhong, J.; Harris, R.C.; Kon, V.; Yang, H.; Fogo, A.B. Increased DAAM2, a New Podocyte-Associated Protein, in Diabetic Nephropathy. Nephrol. Dial. Transplant. 2021, 36, 1006–1016. [Google Scholar] [CrossRef]
- De Alwis, N.; Beard, S.; Binder, N.K.; Pritchard, N.; Kaitu’u-Lino, T.J.; Walker, S.P.; Stock, O.; Groom, K.; Petersen, S.; Henry, A.; et al. DAAM2 Is Elevated in the Circulation and Placenta in Pregnancies Complicated by Fetal Growth Restriction and Is Regulated by Hypoxia. Sci. Rep. 2021, 11, 5540. [Google Scholar] [CrossRef]
- Nakaya, M.-A.; Gudmundsson, K.O.; Komiya, Y.; Keller, J.R.; Habas, R.; Yamaguchi, T.P.; Ajima, R. Placental Defects Lead to Embryonic Lethality in Mice Lacking the Formin and PCP Proteins Daam1 and Daam2. PLoS ONE 2020, 15, e0232025. [Google Scholar] [CrossRef]
- Morris, J.A.; Kemp, J.P.; Youlten, S.E.; Laurent, L.; Logan, J.G.; Chai, R.C.; Vulpescu, N.A.; Forgetta, V.; Kleinman, A.; Mohanty, S.T.; et al. An Atlas of Genetic Influences on Osteoporosis in Humans and Mice. Nat. Genet. 2019, 51, 258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deng, H.-W.; Shen, H.; Ehrlich, M. Prioritization of Osteoporosis-Associated Genome-wide Association Study (GWAS) Single-Nucleotide Polymorphisms (SNPs) Using Epigenomics and Transcriptomics. JBMR Plus 2021, 5, e10481. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ledo, A.; Jenny, A.; Marlow, F.L. Comparative Gene Expression Analysis of the Fmnl Family of Formins during Zebrafish Development and Implications for Tissue Specific Functions. Gene Expr. Patterns 2013, 13, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, X.; Tang, W.; Li, J.; Yang, S.; Chen, Y.; Zhao, X.; Zong, H.; Liu, C.; Shen, C. Association of DIAPH1 Gene Polymorphisms with Ischemic Stroke. Aging 2020, 12, 416–435. [Google Scholar] [CrossRef]
- Kundishora, A.J.; Peters, S.T.; Pinard, A.; Duran, D.; Panchagnula, S.; Barak, T.; Miyagishima, D.F.; Dong, W.; Smith, H.; Ocken, J.; et al. DIAPH1 Variants in Non-East Asian Patients With Sporadic Moyamoya Disease. JAMA Neurol. 2021, 78, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, J. Nurr1 Exacerbates Cerebral Ischemia-Reperfusion Injury via Modulating YAP-INF2-Mitochondrial Fission Pathways. Int. J. Biochem. Cell Biol. 2018, 104, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, W.; Chen, L.; Luo, Y.; Xu, R. Nur77 Promotes Cerebral Ischemia-Reperfusion Injury via Activating INF2-Mediated Mitochondrial Fragmentation. J. Mol. Histol. 2018, 49, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.R.; Polk, D.E.; Wang, X.; Feingold, E.; Weeks, D.E.; Lee, M.-K.; Cuenco, K.T.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; et al. Genome-Wide Association Study of Periodontal Health Measured by Probing Depth in Adults Ages 18–49 Years. G3 Genes Genomes Genet. 2014, 4, 307–314. [Google Scholar] [CrossRef]
- Choquet, H.; Thai, K.K.; Yin, J.; Hoffmann, T.J.; Kvale, M.N.; Banda, Y.; Schaefer, C.; Risch, N.; Nair, K.S.; Melles, R.; et al. A Large Multi-Ethnic Genome-Wide Association Study Identifies Novel Genetic Loci for Intraocular Pressure. Nat. Commun. 2017, 8, 2108. [Google Scholar] [CrossRef]
- Shin, H.-T.; Yoon, B.W.; Seo, J.H. Analysis of Risk Allele Frequencies of Single Nucleotide Polymorphisms Related to Open-Angle Glaucoma in Different Ethnic Groups. BMC Med. Genom. 2021, 14, 80. [Google Scholar] [CrossRef]
- Li, Z.; Xu, Y.; Zhang, C.; Liu, X.; Jiang, L.; Chen, F. Mammalian Diaphanous-Related Formin 1 Is Required for Motility and Invadopodia Formation in Human U87 Glioblastoma Cells. Int. J. Mol. Med. 2014, 33, 383–391. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, Y.; Ren, W.; Gong, J.; Chen, F. An Identification of Stem Cell-Resembling Gene Expression Profiles in High-Grade Astrocytomas. Mol. Carcinog. 2008, 47, 893–903. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, L.; Chen, J.; Liang, J.; Xu, Y.; Li, Z.; Chen, F.; Du, D. Knockdown of Diaph1 Expression Inhibits Migration and Decreases the Expression of MMP2 and MMP9 in Human Glioma Cells. Biomed. Pharmacother. 2017, 96, 596–602. [Google Scholar] [CrossRef]
- Yamana, N.; Arakawa, Y.; Nishino, T.; Kurokawa, K.; Tanji, M.; Itoh, R.E.; Monypenny, J.; Ishizaki, T.; Bito, H.; Nozaki, K.; et al. The Rho-MDia1 Pathway Regulates Cell Polarity and Focal Adhesion Turnover in Migrating Cells through Mobilizing Apc and c-Src. Mol. Cell. Biol. 2006, 26, 6844–6858. [Google Scholar] [CrossRef]
- Hager, M.H.; Morley, S.; Bielenberg, D.R.; Gao, S.; Morello, M.; Holcomb, I.N.; Liu, W.; Mouneimne, G.; Demichelis, F.; Kim, J.; et al. DIAPH3 Governs the Cellular Transition to the Amoeboid Tumour Phenotype. EMBO Mol. Med. 2012, 4, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yan, T.; Li, X.; Sun, J.; Zhang, B.; Wang, H.; Zhu, Y. Daam1 Activates RhoA to Regulate Wnt5a-induced Glioblastoma Cell Invasion. Oncol. Rep. 2018, 39, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, V.; Arnotskaya, N.; Zaitsev, S.; Sharma, A.; Sharma, H.S.; Bryukhovetskiy, A.; Pak, O.; Khotimchenko, Y.; Bryukhovetskiy, I. Proteins of Wnt Signaling Pathway in Cancer Stem Cells of Human Glioblastoma. Int. Rev. Neurobiol. 2020, 151, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Krishna, S.; Garcia, C.; Lin, C.-C.J.; Mitchell, B.D.; Scott, K.L.; Mohila, C.A.; Creighton, C.J.; Yoo, S.-H.; Lee, H.K.; et al. Daam2 Driven Degradation of VHL Promotes Gliomagenesis. eLife 2017, 6, e31926. [Google Scholar] [CrossRef] [PubMed]
- Higa, N.; Shinsato, Y.; Kamil, M.; Hirano, T.; Takajo, T.; Shimokawa, M.; Minami, K.; Yamamoto, M.; Kawahara, K.; Yonezawa, H.; et al. Formin-like 1 (FMNL1) Is Associated with Glioblastoma Multiforme Mesenchymal Subtype and Independently Predicts Poor Prognosis. Int. J. Mol. Sci. 2019, 20, 6355. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Monzo, P.; Chong, Y.K.; Guetta-Terrier, C.; Krishnasamy, A.; Sathe, S.R.; Yim, E.K.F.; Ng, W.H.; Ang, B.T.; Tang, C.; Ladoux, B.; et al. Mechanical Confinement Triggers Glioma Linear Migration Dependent on Formin FHOD3. Mol. Biol. Cell 2016, 27, 1246–1261. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Shi, W.; Zhao, R.; Shen, W.; Li, H. FHOD3 Promotes Carcinogenesis by Regulating RhoA/ROCK1/LIMK1 Signaling Pathway in Medulloblastoma. Clin. Transl. Oncol. 2020, 22, 2312–2323. [Google Scholar] [CrossRef]
- Heuser, V.D.; Kiviniemi, A.; Lehtinen, L.; Munthe, S.; Kristensen, B.W.; Posti, J.P.; Sipilä, J.O.T.; Vuorinen, V.; Carpén, O.; Gardberg, M. Multiple Formin Proteins Participate in Glioblastoma Migration. BMC Cancer 2020, 20, 710. [Google Scholar] [CrossRef]
- Kitzing, T.M.; Sahadevan, A.S.; Brandt, D.T.; Knieling, H.; Hannemann, S.; Fackler, O.T.; Großhans, J.; Grosse, R. Positive Feedback between Dia1, LARG, and RhoA Regulates Cell Morphology and Invasion. Genes Dev. 2007, 21, 1478–1483. [Google Scholar] [CrossRef]
- Kitzing, T.M.; Wang, Y.; Pertz, O.; Copeland, J.W.; Grosse, R. Formin-like 2 Drives Amoeboid Invasive Cell Motility Downstream of RhoC. Oncogene 2010, 29, 2441–2448. [Google Scholar] [CrossRef]
- Lizárraga, F.; Poincloux, R.; Romao, M.; Montagnac, G.; Le Dez, G.; Bonne, I.; Rigaill, G.; Raposo, G.; Chavrier, P. Diaphanous-Related Formins Are Required for Invadopodia Formation and Invasion of Breast Tumor Cells. Cancer Res. 2009, 69, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Jung, J.; You, E.; Ko, P.; Oh, S.; Rhee, S. MDia1 Regulates Breast Cancer Invasion by Controlling Membrane Type 1-Matrix Metalloproteinase Localization. Oncotarget 2016, 7, 17829–17843. [Google Scholar] [CrossRef]
- Jiang, J. Diaphanous-Related Formin-3 Overexpression Inhibits the Migration and Invasion of Triple-Negative Breast Cancer by Inhibiting RhoA-GTP Expression. Biomed. Pharmacother. 2017, 94, 439–445. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Chen, J.-F.; Novitskaya, T.; You, S.; Morley, S.; Steadman, K.; Gill, N.K.; Eskaros, A.; Rotinen, M.; Chu, C.-Y.; et al. Emerin Deregulation Links Nuclear Shape Instability to Metastatic Potential. Cancer Res. 2018, 78, 6086–6097. [Google Scholar] [CrossRef]
- Hao, L.; Liu, Y.; Yu, X.; Zhu, Y.; Zhu, Y. Formin Homology Domains of Daam1 Bind to Fascin and Collaboratively Promote Pseudopodia Formation and Cell Migration in Breast Cancer. Cell Prolif. 2021, 54, e12994. [Google Scholar] [CrossRef]
- Mei, J.; Xu, B.; Hao, L.; Xiao, Z.; Liu, Y.; Yan, T.; Zhu, Y. Overexpressed DAAM1 Correlates with Metastasis and Predicts Poor Prognosis in Breast Cancer. Pathol. Res. Pract. 2020, 216, 152736. [Google Scholar] [CrossRef]
- Yan, T.; Zhang, A.; Shi, F.; Chang, F.; Mei, J.; Liu, Y.; Zhu, Y. Integrin Avβ3-Associated DAAM1 Is Essential for Collagen-Induced Invadopodia Extension and Cell Haptotaxis in Breast Cancer Cells. J. Biol. Chem. 2018, 293, 10172–10185. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tian, Y.; Du, J.; Hu, Z.; Yang, L.; Liu, J.; Gu, L. Dvl2-Dependent Activation of Daam1 and RhoA Regulates Wnt5a-Induced Breast Cancer Cell Migration. PLoS ONE 2012, 7, e37823. [Google Scholar] [CrossRef] [PubMed]
- Heuser, V.D.; Mansuri, N.; Mogg, J.; Kurki, S.; Repo, H.; Kronqvist, P.; Carpén, O.; Gardberg, M. Formin Proteins FHOD1 and INF2 in Triple-Negative Breast Cancer: Association With Basal Markers and Functional Activities. Breast Cancer 2018, 12, 1178223418792247. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Shen, Y.; Yan, T.; Zhang, A. Wnt5a/ROR1 Activates DAAM1 and Promotes the Migration in Osteosarcoma Cells. Oncol. Rep. 2020, 43, 601–608. [Google Scholar] [CrossRef]
- Wang, Q. Identification of Biomarkers for Metastatic Osteosarcoma Based on DNA Microarray Data. Neoplasma 2015, 62, 365–371. [Google Scholar] [CrossRef][Green Version]
- Mei, J.; Xing, Y.; Lv, J.; Gu, D.; Pan, J.; Zhang, Y.; Liu, J. Construction of an Immune-Related Gene Signature for Prediction of Prognosis in Patients with Cervical Cancer. Int. Immunopharmacol. 2020, 88, 106882. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Izbicki, J.R.; König, A.; Habermann, J.K.; Blechner, C.; Lange, T.; Schumacher, U.; Windhorst, S. Expression of DIAPH1 Is Up-Regulated in Colorectal Cancer and Its down-Regulation Strongly Reduces the Metastatic Capacity of Colon Carcinoma Cells. Int. J. Cancer 2014, 134, 1571–1582. [Google Scholar] [CrossRef]
- Miao, S.; Schäfer, P.; Nojszewski, J.; Meyer, F.; Windhorst, S. DIAPH1 Regulates Chromosomal Instability of Cancer Cells by Controlling Microtubule Dynamics. Eur. J. Cell Biol. 2021, 100, 151156. [Google Scholar] [CrossRef] [PubMed]
- Sho, S.; Court, C.M.; Winograd, P.; Russell, M.M.; Tomlinson, J.S. A Prognostic Mutation Panel for Predicting Cancer Recurrence in Stages II and III Colorectal Cancer. J. Surg. Oncol. 2017, 116, 996–1004. [Google Scholar] [CrossRef]
- Grueb, S.S.; Muhs, S.; Popp, Y.; Schmitt, S.; Geyer, M.; Lin, Y.-N.; Windhorst, S. The Formin Drosophila Homologue of Diaphanous2 (Diaph2) Controls Microtubule Dynamics in Colorectal Cancer Cells Independent of Its FH2-Domain. Sci. Rep. 2019, 9, 5352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, J.; Liu, Z.; Liu, X.; Ma, Y.; Zhang, B.; Chen, Y.; Li, X.; Feng, Z.; Yang, N.; et al. Identification of Hub Genes in Colorectal Cancer Based on Weighted Gene Co-Expression Network Analysis and Clinical Data from The Cancer Genome Atlas. Biosci. Rep. 2021, 41, BSR20211280. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Albers, C.G.; Holcombe, R.F. Differentiation of Tubular and Villous Adenomas Based on Wnt Pathway-Related Gene Expression Profiles. Int. J. Mol. Med. 2010, 26, 121–125. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, A.; Liu, Z.; Wu, Q.; Li, H. Disheveled-Associated Activator of Morphogenesis 2 Promotes Invasion of Colorectal Cancer by Activating PAK1 and Promoting MMP7 Expression. Genes Genom. 2021, 43, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, X.; Zeng, Y.; Wang, J.; Zhang, X.; Ding, Y.; Liang, L. FMNL2 Enhances Invasion of Colorectal Carcinoma by Inducing Epithelial-Mesenchymal Transition. Mol. Cancer Res. 2010, 8, 1579–1590. [Google Scholar] [CrossRef]
- Ren, X.L.; Qiao, Y.D.; Li, J.Y.; Li, X.M.; Zhang, D.; Zhang, X.J.; Zhu, X.H.; Zhou, W.J.; Shi, J.; Wang, W.; et al. Cortactin Recruits FMNL2 to Promote Actin Polymerization and Endosome Motility in Invadopodia Formation. Cancer Lett. 2018, 419, 245–256. [Google Scholar] [CrossRef]
- Yang, S.S.; Li, X.M.; Yang, M.; Ren, X.L.; Hu, J.L.; Zhu, X.H.; Wang, F.F.; Zeng, Z.C.; Li, J.Y.; Cheng, Z.Q.; et al. FMNL2 Destabilises COMMD10 to Activate NF-ΚB Pathway in Invasion and Metastasis of Colorectal Cancer. Br. J. Cancer 2017, 117, 1164–1175. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Zeng, Y.-F.; Guan, J.; Li, Y.-F.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q.; Liang, L. FMNL2 Is a Positive Regulator of Cell Motility and Metastasis in Colorectal Carcinoma. J. Pathol. 2011, 224, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-L.; Liang, L.; Ding, Y.-Q. Overexpression of FMNL2 Is Closely Related to Metastasis of Colorectal Cancer. Int. J. Colorectal Dis. 2008, 23, 1041–1047. [Google Scholar] [CrossRef]
- Zeng, Y.-F.; Xiao, Y.-S.; Liu, Y.; Luo, X.-J.; Wen, L.-D.; Liu, Q.; Chen, M. Formin-like 3 Regulates RhoC/FAK Pathway and Actin Assembly to Promote Cell Invasion in Colorectal Carcinoma. World J. Gastroenterol. 2018, 24, 3884–3897. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-F.; Xiao, Y.-S.; Lu, M.-Z.; Luo, X.-J.; Hu, G.-Z.; Deng, K.-Y.; Wu, X.-M.; Xin, H.-B. Increased Expression of Formin-like 3 Contributes to Metastasis and Poor Prognosis in Colorectal Carcinoma. Exp. Mol. Pathol. 2015, 98, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-J.; Feng, Z.-C.; Li, X.-R.; Hu, G. Involvement of Methylation-Associated Silencing of Formin 2 in Colorectal Carcinogenesis. World J. Gastroenterol. 2018, 24, 5013–5024. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Wang, H.; Jiang, D.; Li, Y.; Feng, L.; Tian, C.; Pu, M.; Wang, X.; Zhang, J.; Hu, Y.; et al. Multi Gene Mutation Signatures in Colorectal Cancer Patients: Predict for the Diagnosis, Pathological Classification, Staging and Prognosis. BMC Cancer 2021, 21, 380. [Google Scholar] [CrossRef]
- Kawamata, H.; Furihata, T.; Omotehara, F.; Sakai, T.; Horiuchi, H.; Shinagawa, Y.; Imura, J.; Ohkura, Y.; Tachibana, M.; Kubota, K.; et al. Identification of Genes Differentially Expressed in a Newly Isolated Human Metastasizing Esophageal Cancer Cell Line, T.Tn-AT1, by CDNA Microarray. Cancer Sci. 2003, 94, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yan, T.; Hao, L.; Zhu, Y. Wnt5a Induces ROR1 and ROR2 to Activate RhoA in Esophageal Squamous Cell Carcinoma Cells. Cancer Manag. Res. 2019, 11, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, Y.; Mao, X.; Cheng, Z.; Wu, S.; Wu, S.; Zhou, L. Expression of Formin-like 2 and Cortactin in Gallbladder Adenocarcinoma and Their Clinical Significance. Int. J. Clin. Exp. Pathol. 2020, 13, 1655–1661. [Google Scholar] [PubMed]
- Yang, J.; Zhou, L.; Zhang, Y.; Zheng, J.; Zhou, J.; Wei, Z.; Zou, J. DIAPH1 Is Upregulated and Inhibits Cell Apoptosis through ATR/P53/Caspase-3 Signaling Pathway in Laryngeal Squamous Cell Carcinoma. Dis. Markers 2019, 2019, 6716472. Available online: https://www.hindawi.com/journals/dm/2019/6716472 (accessed on 30 August 2021). [CrossRef] [PubMed]
- Kostrzewska-Poczekaj, M.; Byzia, E.; Soloch, N.; Jarmuz-Szymczak, M.; Janiszewska, J.; Kowal, E.; Paczkowska, J.; Kiwerska, K.; Wierzbicka, M.; Bartochowska, A.; et al. DIAPH2 Alterations Increase Cellular Motility and May Contribute to the Metastatic Potential of Laryngeal Squamous Cell Carcinoma. Carcinogenesis 2019, 40, 1251–1259. [Google Scholar] [CrossRef]
- Śnit, M.; Misiołek, M.; Ścierski, W.; Koniewska, A.; Stryjewska-Makuch, G.; Okła, S.; Grzeszczak, W. DIAPH2, PTPRD and HIC1 Gene Polymorphisms and Laryngeal Cancer Risk. Int. J. Environ. Res. Public Health 2021, 18, 7486. [Google Scholar] [CrossRef]
- Chen, W.-H.; Cai, M.-Y.; Zhang, J.-X.; Wang, F.-W.; Tang, L.-Q.; Liao, Y.-J.; Jin, X.-H.; Wang, C.-Y.; Guo, L.; Jiang, Y.-G.; et al. FMNL1 Mediates Nasopharyngeal Carcinoma Cell Aggressiveness by Epigenetically Upregulating MTA1. Oncogene 2018, 37, 6243–6258. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Chen, Y.; Geng, N.; Feng, C. High Expression of FMNL3 Associates with Cancer Cell Migration, Invasion, and Unfavorable Prognosis in Tongue Squamous Cell Carcinoma. J. Oral Pathol. Med. 2019, 48, 459–467. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, Z.; Wang, K.; Ha, Y.; Lei, H.; Jia, Y.; Ding, R.; Wu, D.; Gan, S.; Li, R.; et al. High FMNL3 Expression Promotes Nasopharyngeal Carcinoma Cell Metastasis: Role in TGF-Β1-Induced Epithelia-to-Mesenchymal Transition. Sci. Rep. 2017, 7, 42507. [Google Scholar] [CrossRef]
- Gardberg, M.; Kaipio, K.; Lehtinen, L.; Mikkonen, P.; Heuser, V.D.; Talvinen, K.; Iljin, K.; Kampf, C.; Uhlen, M.; Grénman, R.; et al. FHOD1, a Formin Upregulated in Epithelial-Mesenchymal Transition, Participates in Cancer Cell Migration and Invasion. PLoS ONE 2013, 8, e74923. [Google Scholar] [CrossRef]
- Zhang, M.-F.; Li, Q.-L.; Yang, Y.-F.; Cao, Y.; Zhang, C.Z. FMNL1 Exhibits Pro-Metastatic Activity via CXCR2 in Clear Cell Renal Cell Carcinoma. Front. Oncol. 2020, 10, 564614. [Google Scholar] [CrossRef]
- Hirata, H.; Hinoda, Y.; Nakajima, K.; Kikuno, N.; Yamamura, S.; Kawakami, K.; Suehiro, Y.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Wnt Antagonist Gene Polymorphisms and Renal Cancer. Cancer 2009, 115, 4488–4503. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.B.; Wigton, E.J.; Krovi, S.H.; Chung, J.W.; Long, R.A.; Jacobelli, J. The Formin mDia1 Regulates Acute Lymphoblastic Leukemia Engraftment, Migration, and Progression in Vivo. Front. Oncol. 2018, 8, 389. [Google Scholar] [CrossRef] [PubMed]
- Favaro, P.; Traina, F.; Machado-Neto, J.A.; Lazarini, M.; Lopes, M.R.; Pereira, J.K.N.; Costa, F.F.; Infante, E.; Ridley, A.J.; Saad, S.T.O. FMNL1 Promotes Proliferation and Migration of Leukemia Cells. J. Leukoc. Biol. 2013, 94, 503–512. [Google Scholar] [CrossRef]
- Bergamo Favaro, P.; de Souza Medina, S.; Traina, F.; Sanchez Bassères, D.; Ferreira Costa, F.; Olalla Saad, S.T. Human Leukocyte Formin: A Novel Protein Expressed in Lymphoid Malignancies and Associated with Akt. Available online: https://pubmed.ncbi.nlm.nih.gov/14592423/ (accessed on 26 January 2021).
- French, D.; Yang, W.; Cheng, C.; Raimondi, S.C.; Mullighan, C.G.; Downing, J.R.; Evans, W.E.; Pui, C.-H.; Relling, M.V. Acquired Variation Outweighs Inherited Variation in whole Genome Analysis of Methotrexate Polyglutamate Accumulation in Leukemia. Blood 2009, 113, 4512–4520. [Google Scholar] [CrossRef] [PubMed]
- Favaro, P.M.B.; Traina, F.; Vassallo, J.; Brousset, P.; Delsol, G.; Costa, F.F.; Saad, S.T.O. High Expression of FMNL1 Protein in T Non-Hodgkin’s Lymphomas. Leuk. Res. 2006, 30, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Z.; Xue, L.; Li, G.; Zhang, C.; Cai, Z.; Li, H.; Guo, R. DIAPH3 Promoted the Growth, Migration and Metastasis of Hepatocellular Carcinoma Cells by Activating Beta-Catenin/TCF Signaling. Mol. Cell. Biochem. 2018, 438, 183–190. [Google Scholar] [CrossRef]
- Fang, X.; Zhang, D.; Zhao, W.; Gao, L.; Wang, L. Dishevelled Associated Activator Of Morphogenesis (DAAM) Facilitates Invasion of Hepatocellular Carcinoma by Upregulating Hypoxia-Inducible Factor 1α (HIF-1α) Expression. Med. Sci. Monit. 2020, 26, e924670. [Google Scholar] [CrossRef]
- Liang, L.; Guan, J.; Zeng, Y.; Wang, J.; Li, X.; Zhang, X.; Ding, Y. Down-Regulation of Formin-like 2 Predicts Poor Prognosis in Hepatocellular Carcinoma. Hum. Pathol. 2011, 42, 1603–1612. [Google Scholar] [CrossRef]
- Xiang, G.; Weiwei, H.; Erji, G.; Haitao, M. DIAPH3 Promotes the Tumorigenesis of Lung Adenocarcinoma. Exp. Cell Res. 2019, 385, 111662. [Google Scholar] [CrossRef]
- Liu, Y.; Lusk, C.M.; Cho, M.H.; Silverman, E.K.; Qiao, D.; Zhang, R.; Scheurer, M.E.; Kheradmand, F.; Wheeler, D.A.; Tsavachidis, S.; et al. Rare Variants in Known Susceptibility Loci and Their Contribution to Risk of Lung Cancer. J. Thorac. Oncol. 2018, 13, 1483–1495. [Google Scholar] [CrossRef]
- Yang, X.-Y.; Liao, J.-J.; Xue, W.-R. FMNL1 Down-Regulation Suppresses Bone Metastasis through Reducing TGF-Β1 Expression in Non-Small Cell Lung Cancer (NSCLC). Biomed. Pharmacother. 2019, 117, 109126. [Google Scholar] [CrossRef]
- Hałas-Wiśniewska, M.; Izdebska, M.; Zielińska, W.; Grzanka, A. Downregulation of FHOD1 Inhibits Metastatic Potential in A549 Cells. Cancer Manag. Res. 2021, 13, 91–106. [Google Scholar] [CrossRef]
- Schiewek, J.; Schumacher, U.; Lange, T.; Joosse, S.A.; Wikman, H.; Pantel, K.; Mikhaylova, M.; Kneussel, M.; Linder, S.; Schmalfeldt, B.; et al. Clinical Relevance of Cytoskeleton Associated Proteins for Ovarian Cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 2195–2205. [Google Scholar] [CrossRef]
- Pettee, K.M.; Dvorak, K.M.; Nestor-Kalinoski, A.L.; Eisenmann, K.M. An MDia2/ROCK Signaling Axis Regulates Invasive Egress from Epithelial Ovarian Cancer Spheroids. PLoS ONE 2014, 9, e90371. [Google Scholar] [CrossRef]
- Paul, N.R.; Allen, J.L.; Chapman, A.; Morlan-Mairal, M.; Zindy, E.; Jacquemet, G.; Fernandez del Ama, L.; Ferizovic, N.; Green, D.M.; Howe, J.D.; et al. A5β1 Integrin Recycling Promotes Arp2/3-Independent Cancer Cell Invasion via the Formin FHOD3. J. Cell Biol. 2015, 210, 1013–1031. [Google Scholar] [CrossRef]
- Rong, Y.; Gao, J.; Kuang, T.; Chen, J.; Li, J.-A.; Huang, Y.; Xin, H.; Fang, Y.; Han, X.; Sun, L.-Q.; et al. DIAPH3 Promotes Pancreatic Cancer Progression by Activating Selenoprotein TrxR1-Mediated Antioxidant Effects. J. Cell. Mol. Med. 2020, 25, 2163–2175. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, H.; Luo, S.; Moorman, P.G.; Walsh, K.M.; Li, W.; Wei, Q. Associations between Genetic Variants of KIF5B, FMN1, and MGAT3 in the Cadherin Pathway and Pancreatic Cancer Risk. Cancer Med. 2020, 9, 9620–9631. [Google Scholar] [CrossRef]
- Raja, A.; Malik, M.F.A.; Haq, F. Genomic Relevance of FGF14 and Associated Genes on the Prognosis of Pancreatic Cancer. PLoS ONE 2021, 16, e0252344. [Google Scholar] [CrossRef]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome Formation in Prostate Cancer: Association with a Region of Frequent Chromosomal Deletion in Metastatic Disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC Have Distinct Roles in Migration and Invasion by Acting through Different Targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef]
- Lisitskaia, K.V.; Krakhmaleva, I.N.; Shishkin, S.S. A study of the single nucleotide polymorphism in seven genes (GHR, IGFBP3, IGFR1, IRS1, FMN1, ANXA2, TaGLN) in ethnic Russians and in patients with prostate cancer. Mol. Gen. Mikrobiol. Virusol. 2010, 2, 34–37. [Google Scholar] [CrossRef]
- Jin, X.; Wang, J.; Gao, K.; Zhang, P.; Yao, L.; Tang, Y.; Tang, L.; Ma, J.; Xiao, J.; Zhang, E.; et al. Dysregulation of INF2-Mediated Mitochondrial Fission in SPOP-Mutated Prostate Cancer. PLoS Genet. 2017, 13, e1006748. [Google Scholar] [CrossRef]
- Rodriguez-Hernandez, I.; Maiques, O.; Kohlhammer, L.; Cantelli, G.; Perdrix-Rosell, A.; Monger, J.; Fanshawe, B.; Bridgeman, V.L.; Karagiannis, S.N.; Penin, R.M.; et al. WNT11-FZD7-DAAM1 Signalling Supports Tumour Initiating Abilities and Melanoma Amoeboid Invasion. Nat. Commun. 2020, 11, 5315. [Google Scholar] [CrossRef]
- Gardberg, M.; Heuser, V.D.; Koskivuo, I.; Koivisto, M.; Carpén, O. FMNL2/FMNL3 Formins Are Linked with Oncogenic Pathways and Predict Melanoma Outcome. J. Pathol. Clin. Res. 2016, 2, 41–52. [Google Scholar] [CrossRef]
- Péladeau, C.; Heibein, A.; Maltez, M.T.; Copeland, S.J.; Copeland, J.W. A Specific FMNL2 Isoform Is Up-Regulated in Invasive Cells. BMC Cell Biol. 2016, 17, 32. [Google Scholar] [CrossRef]
- Koka, S.; Neudauer, C.L.; Li, X.; Lewis, R.E.; McCarthy, J.B.; Westendorf, J.J. The Formin-Homology-Domain-Containing Protein FHOD1 Enhances Cell Migration. J. Cell Sci. 2003, 116, 1745–1755. [Google Scholar] [CrossRef]
- Peippo, M.; Gardberg, M.; Lamminen, T.; Kaipio, K.; Carpén, O.; Heuser, V.D. FHOD1 Formin Is Upregulated in Melanomas and Modifies Proliferation and Tumor Growth. Exp. Cell Res. 2017, 350, 267–278. [Google Scholar] [CrossRef]
- Nie, H.; Mei, J.; Zhang, Q.; An, F.; Zhan, Q. Systematic Characterization of the Expression and Prognostic Values of Formin-Like Gene Family in Gastric Cancer. DNA Cell Biol. 2020, 39, 1664–1677. [Google Scholar] [CrossRef]
- Zhong, B.; Wang, K.; Xu, H.; Kong, F. Silencing Formin-like 2 Inhibits Growth and Metastasis of Gastric Cancer Cells through Suppressing Internalization of Integrins. Cancer Cell Int. 2018, 18, 79. [Google Scholar] [CrossRef]
- Mansuri, N.; Heuser, V.D.; Birkman, E.-M.; Lintunen, M.; Ålgars, A.; Sundström, J.; Ristamäki, R.; Carpén, O.; Lehtinen, L. FHOD1 and FMNL1 Formin Proteins in Intestinal Gastric Cancer: Correlation with Tumor-Infiltrating T Lymphocytes and Molecular Subtypes. Gastric Cancer 2021, 1–10. [Google Scholar] [CrossRef]
- Liang, L.; Ye, L.; Cui, D.; Yang, D. Gene Expression Signature Associated with Metastasis of Stomach Adenocarcinoma. Int. J. Clin. Exp. Med. 2017, 10, 3016–3026. [Google Scholar]
- Chai, L.; Li, J.; Lv, Z. An Integrated Analysis of Cancer Genes in Thyroid Cancer. Oncol. Rep. 2016, 35, 962–970. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Marshall, C.J. The Plasticity of Cytoskeletal Dynamics Underlying Neoplastic Cell Migration. Curr. Opin. Cell Biol. 2010, 22, 690–696. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions That Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Compact Spheroid Formation by Ovarian Cancer Cells Is Associated with Contractile Behavior and an Invasive Phenotype. Int. J. Cancer 2009, 124, 2060–2070. [Google Scholar] [CrossRef]
- Creekmore, A.L.; Silkworth, W.T.; Cimini, D.; Jensen, R.V.; Roberts, P.C.; Schmelz, E.M. Changes in Gene Expression and Cellular Architecture in an Ovarian Cancer Progression Model. PLoS ONE 2011, 6, e17676. [Google Scholar] [CrossRef]
- Ketene, A.N.; Schmelz, E.M.; Roberts, P.C.; Agah, M. The Effects of Cancer Progression on the Viscoelasticity of Ovarian Cell Cytoskeleton Structures. Nanomedicine 2012, 8, 391. [Google Scholar] [CrossRef]
- Wyse, M.M.; Lei, J.; Nestor-Kalinoski, A.L.; Eisenmann, K.M. Dia-Interacting Protein (DIP) Imposes Migratory Plasticity in MDia2-Dependent Tumor Cells in Three-Dimensional Matrices. PLoS ONE 2012, 7, e45085. [Google Scholar] [CrossRef]
- Chakrabarti, R.; Ji, W.-K.; Stan, R.V.; de Juan Sanz, J.; Ryan, T.A.; Higgs, H.N. INF2-Mediated Actin Polymerization at the ER Stimulates Mitochondrial Calcium Uptake, Inner Membrane Constriction, and Division. J. Cell Biol. 2018, 217, 251–268. [Google Scholar] [CrossRef]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Clark, A.; Burleson, M. SPOP and Cancer: A Systematic Review. Am. J. Cancer Res. 2020, 10, 704–726. [Google Scholar]
- Da Silva, A.F.; Mariotti, F.R.; Máximo, V.; Campello, S. Mitochondria Dynamism: Of Shape, Transport and Cell Migration. Cell. Mol. Life Sci. 2014, 71, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, M.; An, D.; Yuan, H.; Li, Z.; Song, Y.; Liu, Z. Suppression of Tafazzin Promotes Thyroid Cancer Apoptosis via Activating the JNK Signaling Pathway and Enhancing INF2-Mediated Mitochondrial Fission. J. Cell. Physiol. 2019, 234, 16238–16251. [Google Scholar] [CrossRef]
- Yasuda, S.; Oceguera-Yanez, F.; Kato, T.; Okamoto, M.; Yonemura, S.; Terada, Y.; Ishizaki, T.; Narumiya, S. Cdc42 and mDia3 Regulate Microtubule Attachment to Kinetochores. Nature 2004, 428, 767–771. [Google Scholar] [CrossRef]
- Morley, S.; You, S.; Pollan, S.; Choi, J.; Zhou, B.; Hager, M.H.; Steadman, K.; Spinelli, C.; Rajendran, K.; Gertych, A.; et al. Regulation of Microtubule Dynamics by DIAPH3 Influences Amoeboid Tumor Cell Mechanics and Sensitivity to Taxanes. Sci. Rep. 2015, 5, 12136. [Google Scholar] [CrossRef]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef]
- Dvorak, K.M.; Pettee, K.M.; Rubinic-Minotti, K.; Su, R.; Nestor-Kalinoski, A.; Eisenmann, K.M. Carcinoma Associated Fibroblasts (CAFs) Promote Breast Cancer Motility by Suppressing Mammalian Diaphanous-Related Formin-2 (mDia2). PLoS ONE 2018, 13, e0195278. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Kersten, K.; de Visser, K.E.; van Miltenburg, M.H.; Jonkers, J. Genetically Engineered Mouse Models in Oncology Research and Cancer Medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef]
- Antsiferova, M.; Huber, M.; Meyer, M.; Piwko-Czuchra, A.; Ramadan, T.; MacLeod, A.S.; Havran, W.L.; Dummer, R.; Hohl, D.; Werner, S. Activin Enhances Skin Tumourigenesis and Malignant Progression by Inducing a Pro-Tumourigenic Immune Cell Response. Nat. Commun. 2011, 2, 576. [Google Scholar] [CrossRef] [PubMed]
- Cangkrama, M.; Wietecha, M.; Mathis, N.; Okumura, R.; Ferrarese, L.; Al-Nuaimi, D.; Antsiferova, M.; Dummer, R.; Innocenti, M.; Werner, S. A Paracrine Activin A-mDia2 Axis Promotes Squamous Carcinogenesis via Fibroblast Reprogramming. EMBO Mol. Med. 2020, 12, e11466. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a Guardian of the Cancer Cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Kim, J.; Morley, S.; Le, M.; Bedoret, D.; Umetsu, D.T.; Di Vizio, D.; Freeman, M.R. Enhanced Shedding of Extracellular Vesicles from Amoeboid Prostate Cancer Cells: Potential Effects on the Tumor Microenvironment. Cancer Biol. Ther. 2014, 15, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Ziske, M.A.; Pettee, K.M.; Khaing, M.; Rubinic, K.; Eisenmann, K.M. SMIFH2-Mediated mDia Formin Functional Inhibition Potentiates Chemotherapeutic Targeting of Human Ovarian Cancer Spheroids. Biochem. Biophys. Res. Commun. 2016, 472, 33–39. [Google Scholar] [CrossRef]
- Isogai, T.; van der Kammen, R.; Innocenti, M. SMIFH2 has Effects on Formins and p53 that Perturb the Cell Cytoskeleton. Sci. Rep. 2015, 5, 9802. [Google Scholar] [CrossRef]
- Nishimura, Y.; Shi, S.; Zhang, F.; Liu, R.; Takagi, Y.; Bershadsky, A.D.; Viasnoff, V.; Sellers, J.R. The Formin Inhibitor SMIFH2 Inhibits Members of the Myosin Superfamily. J. Cell Sci. 2021, 134, jcs253708. [Google Scholar] [CrossRef]
- Monroe, J.D.; Moolani, S.A.; Irihamye, E.N.; Speed, J.S.; Gibert, Y.; Smith, M.E. RNA-Seq Analysis of Cisplatin and the Monofunctional Platinum(II) Complex, Phenanthriplatin, in A549 Non-Small Cell Lung Cancer and IMR90 Lung Fibroblast Cell Lines. Cells 2020, 9, 2637. [Google Scholar] [CrossRef] [PubMed]
- Fabre, I.; Fabre, G.; Goldman, I.D. Polyglutamylation, an Important Element in Methotrexate Cytotoxicity and Selectivity in Tumor versus Murine Granulocytic Progenitor Cells in Vitro. Cancer Res. 1984, 44, 3190–3195. [Google Scholar] [PubMed]
- Schuster, I.G.; Busch, D.H.; Eppinger, E.; Kremmer, E.; Milosevic, S.; Hennard, C.; Kuttler, C.; Ellwart, J.W.; Frankenberger, B.; Nössner, E.; et al. Allorestricted T Cells with Specificity for the FMNL1-Derived Peptide PP2 Have Potent Antitumor Activity against Hematologic and Other Malignancies. Blood 2007, 110, 2931–2939. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Fang, D.; Lu, H.; Cao, Y.; Zhang, J.; Ding, R.; Li, L.; Huo, J. Tanshinone IIA Promotes IL2-Mediated SW480 Colorectal Cancer Cell Apoptosis by Triggering INF2-Related Mitochondrial Fission and Activating the Mst1-Hippo Pathway. Biomed. Pharmacother. 2018, 108, 1658–1669. [Google Scholar] [CrossRef]
- Fernandez-Barrera, J.; Bernabe-Rubio, M.; Casares-Arias, J.; Rangel, L.; Fernandez-Martin, L.; Correas, I.; Alonso, M.A. The Actin-MRTF-SRF Transcriptional Circuit Controls Tubulin Acetylation via α-TAT1 Gene Expression. J. Cell Biol. 2018, 217, 929–944. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking Actin Dynamics and Gene Transcription to Drive Cellular Motile Functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Thurston, S.F.; Kulacz, W.A.; Shaikh, S.; Lee, J.M.; Copeland, J.W. The Ability to Induce Microtubule Acetylation Is a General Feature of Formin Proteins. PLoS ONE 2012, 7, e48041. [Google Scholar] [CrossRef]
- Gualdrini, F.; Esnault, C.; Horswell, S.; Stewart, A.; Matthews, N.; Treisman, R. SRF Co-Factors Control the Balance between Cell Proliferation and Contractility. Mol. Cell 2016, 64, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-Actin Signaling to the MRTF Coactivators Dominates the Immediate Transcriptional Response to Serum in Fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef]
- Kim, H.-C.; Jo, Y.-J.; Kim, N.-H.; Namgoong, S. Small Molecule Inhibitor of Formin Homology 2 Domains (SMIFH2) Reveals the Roles of the Formin Family of Proteins in Spindle Assembly and Asymmetric Division in Mouse Oocytes. PLoS ONE 2015, 10, e0123438. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Anamag, F.T.; Taheri, M. The Role of Non-Coding RNAs in Controlling Cell Cycle Related Proteins in Cancer Cells. Front. Oncol. 2020, 10, 608975. [Google Scholar] [CrossRef]
- Lin, C.-P.; He, L. Noncoding RNAs in Cancer Development. Annu. Rev. Cancer Biol. 2017, 1, 163–184. [Google Scholar] [CrossRef]




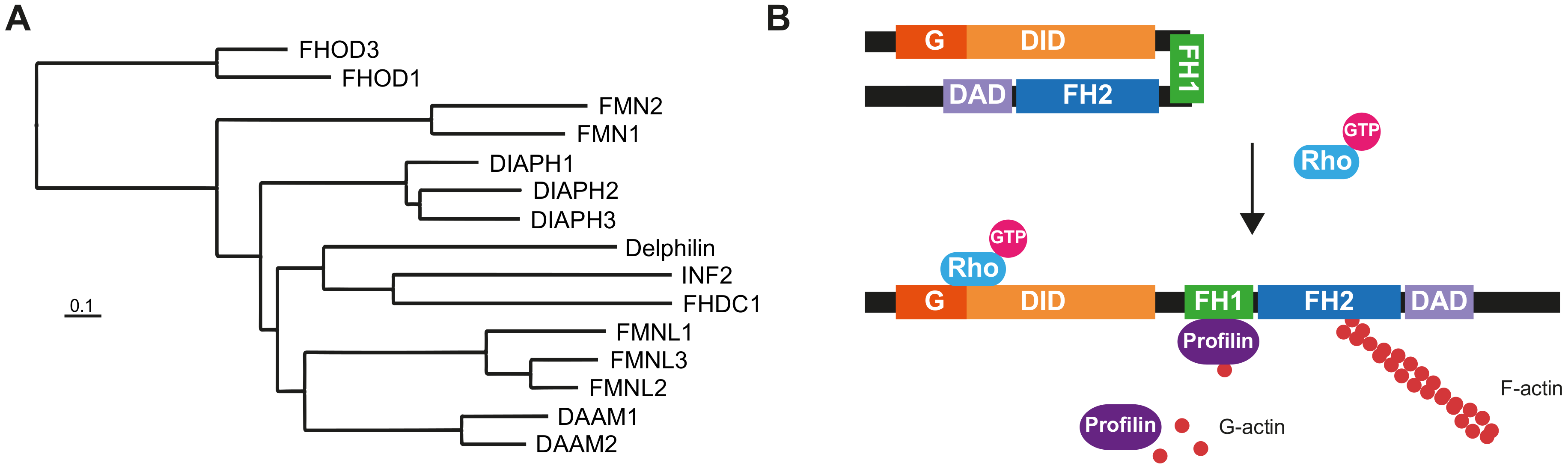{kind=link}
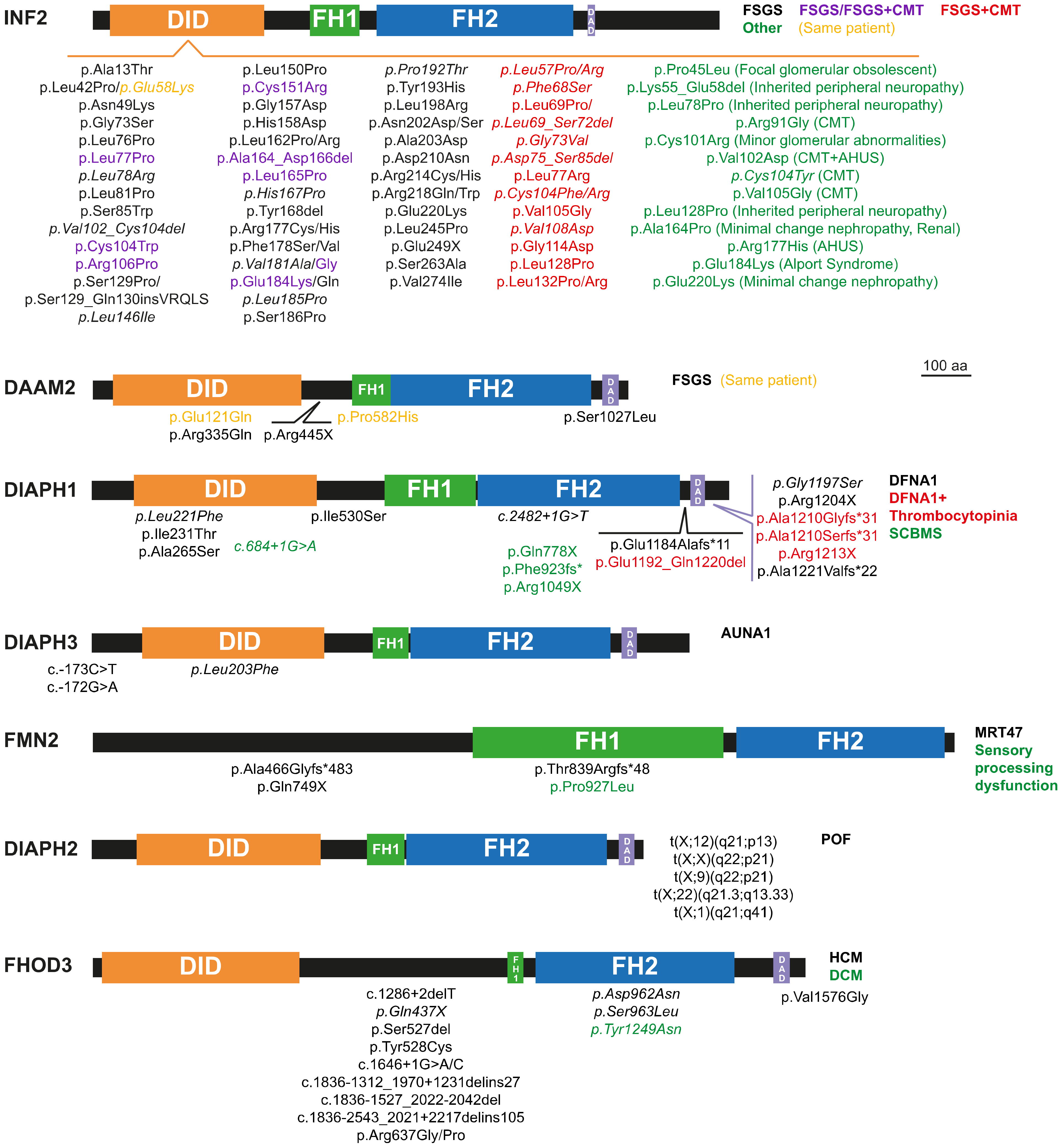{kind=link}
{kind=link}
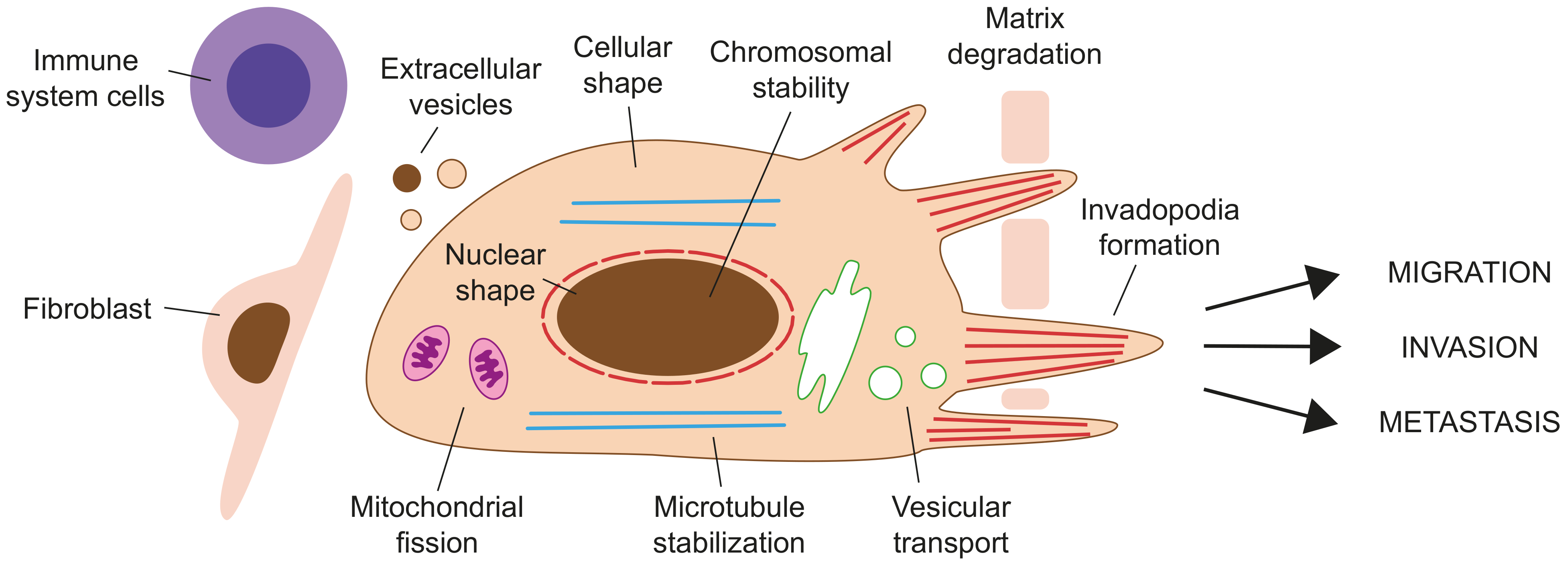{kind=link}
| Cancer | Formin | Expression Levels (Technique) or Mutations | In Vitro | In Vivo (Nude Mice) | Prognosis | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Matrix Degradation | Invadopodia Formation | Migration | Invasion | Metastasis | Tumor size/Weight | |||||
| Brain | DIAPH1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | ↑ | ↑ | [228,229,230] | ||
| DIAPH1 | ↑ | [231] | ||||||||
| DIAPH3 | [232] | |||||||||
| DAAM1 | ↑ | [233] | ||||||||
| DAAM1 | ↑ (Mass spectrometry) | [234] | ||||||||
| DAAM2 | ↑ (ISH/T) | ↑ | [235] | |||||||
| FMNL1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | Poor | [236] | ||||
| FMNL2 FMN2 | Mutants | [237] | ||||||||
| FHOD3 | ↑ (WB) | ↑ | [238] | |||||||
| FHOD3 | ↑ (IHC/T) | ↑ | ↑ | Poor | [239] | |||||
| FHOD1 INF2 | ↑ (IHC/T) | ↑ | Not clear | [240] | ||||||
| Breast | All | ↑ or ↓ depending on the specific formin | [241,242] | |||||||
| DIAPH1/2/3 | ↑ | ↑ | ↑ | [243] | ||||||
| DIAPH1 | ↑ (T/WB) | ↑ | ↑ | [244] | ||||||
| DIAPH3 | Deletions | ↑ | ↑ | [232] | ||||||
| DIAPH3 | ↓ (IHC/T/WB) | ↑ | ↑ | ↑ | [245] | |||||
| DIAPH3 | [246] | |||||||||
| DAAM1 | ↑ (IHC) | ↑ | ↑ | ↑ | No effect | Poor | [247,248,249,250] | |||
| FHOD1 INF2 | ↑ (IHC/T) | ↑ | ↑ | No effect | [251] | |||||
| Bone | DAAM1 | ↑ | [252] | |||||||
| FMNL1 | ↑ (T) | ↑ Lung metastasis | [253] | |||||||
| Cervix | DAAM2 | ↑ (T) | Poor | [254] | ||||||
| Colorectal | DIAPH1 | ↑ (IHC) | ↑ | ↑ | ↑ | ↑ | [255] | |||
| DIAPH1 | [256] | |||||||||
| DIAPH2 | Mutants | Poor | [257] | |||||||
| DIAPH2 | [258] | |||||||||
| DIAPH3 | ↑ (T) Mutants | Poor | [259] | |||||||
| DAAM1/2 | ↑ (T) | [260] | ||||||||
| DAAM2 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [261] | |||||
| FMNL2 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | [262,263,264,265,266] | ||
| FMNL3 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | ↑ | Poor | [267,268] | ||
| FMN2 | ↓ (T) | No effect | [269] | |||||||
| FMN2 | Mutants | [270] | ||||||||
| Esophagus | DIAPH2 | ↓ (T) | [271] | |||||||
| DAAM1 | ↑ | [272] | ||||||||
| Gallbladder | FMNL2 | ↑ (IHC) | Poor | [273] | ||||||
| Head & neck | DIAPH1*/2/3 | ↑* (IHC/T/WB) Mutants | Poor | [274] | ||||||
| DIAPH2*/3 | ↓* (T) Mutants in DIAPH2 SNP in DIAPH3 | [275] | ||||||||
| DIAPH2 | SNP | Protective | [276] | |||||||
| FMNL1 | ↑ (IHC/ISH/WB) Amplifications | ↑ | ↑ | ↑ | ↑ | Poor | [277] | |||
| FMNL3 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [278] | |||||
| FMNL3 | ↑ (IHC) | ↑ | ↑ | No effect | No effect | [279] | ||||
| FHOD1 | ↑ (IHC/T) | ↑ | ↑ | ↑ | ↑ | [280] | ||||
| Kidney | FMNL1 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | Poor | [281] | |||
| DAAM2 | SNP | [282] | ||||||||
| Leukemia | DIAPH1 | ↑ | ↑ | [283] | ||||||
| FMNL1 | ↑ (T/WB) | ↑ | ↑ | [284,285] | ||||||
| FHOD3 | Mutants | [286] | ||||||||
| Lymphoma | FMNL1 | ↑ (IHC/WB) | [287] | |||||||
| Liver | DIAPH3 | ↑ (IHC/T/WB) | ↑ | ↑ | [288] | |||||
| DIAPH3 | Deletions | [232] | ||||||||
| DAAM2 | ↑ (IHC/T/WB) | ↑ | Poor | [289] | ||||||
| FMNL2 | ↓ (IHC/T/WB) | ↑ | ↑ | Poor | [290] | |||||
| Lung | DIAPH3 | ↑ (IHC/T/WB) | ↑ | Poor | [291] | |||||
| DAAM2 | SNP | Protective | [292] | |||||||
| FMNL1 | ↑ (IHC/T/WB) | ↑ | ↑ | ↑ | ↑ | Poor | [293] | |||
| FHOD1 | ↑ | ↑ | Poor | [294] | ||||||
| Ovary | DIAPH1 | ↑ (IHC/T/WB) | Protective | [295] | ||||||
| DIAPH3 | ↑ | ↓ | [296] | |||||||
| FMNL1/2/3 FHOD3 | ↑ | ↑ (in vivo zebrafish) | [297] | |||||||
| Pancreas | DIAPH3 | ↑ (IHC/T/WB) | ↑ | ↑ | Poor | [298] | ||||
| FMNL1/3 FMN2 | Mutants | [299] | ||||||||
| FMN1 | SNP | Higher risk | [300] | |||||||
| FMN2 | ↑ (T) | Protective | [301] | |||||||
| Prostate | DIAPH3 | Deletions (IHC/ISH) | ↑ | ↑ | ↑ | ↑ | [232,302] | |||
| DIAPH3 | Knockdown | ↑ | ↑ | [246] | ||||||
| FMNL3 | ↑ | [303] | ||||||||
| FMN1 | SNP | Higher risk | [304] | |||||||
| INF2 | ↑ | ↑ | [305] | |||||||
| Skin | DIAPH1 | ↑ | [241] | |||||||
| DAAM1 | ↑ (IHC) | ↑ | ↑ | ↑ | [306] | |||||
| FMNL2/3 | ↑ (IHC/WB) | ↑ | Poor (FMNL2) | [307] | ||||||
| FMNL2 | ↑ (T/WB) | ↑ | ↑ | [308] | ||||||
| FHOD1 | ↑ | [309] | ||||||||
| FHOD1 | ↑ (IHC/WB) | ↑ | ↑ | ↑ | [310] | |||||
| Stomach | FMNL1/2/3 | ↑ (T) | Poor (FMNL1/3) No effect (FMNL2) | [311] | ||||||
| FMNL2 | ↑ (WB) | ↑ | ↑ | [312] | ||||||
| FMNL1 FHOD1 | [313] | |||||||||
| FHOD1 | ↑ (T) | [314] | ||||||||
| Thyroid | FHOD3 | ↑ (T) Copy number variations | [315] | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labat-de-Hoz, L.; Alonso, M.A. Formins in Human Disease. Cells 2021, 10, 2554. https://doi.org/10.3390/cells10102554
Labat-de-Hoz L, Alonso MA. Formins in Human Disease. Cells. 2021; 10(10):2554. https://doi.org/10.3390/cells10102554
Chicago/Turabian StyleLabat-de-Hoz, Leticia, and Miguel A. Alonso. 2021. "Formins in Human Disease" Cells 10, no. 10: 2554. https://doi.org/10.3390/cells10102554
APA StyleLabat-de-Hoz, L., & Alonso, M. A. (2021). Formins in Human Disease. Cells, 10(10), 2554. https://doi.org/10.3390/cells10102554