
Comparative Review of Microglia and Monocytes in CNS Phagocytosis

Abstract
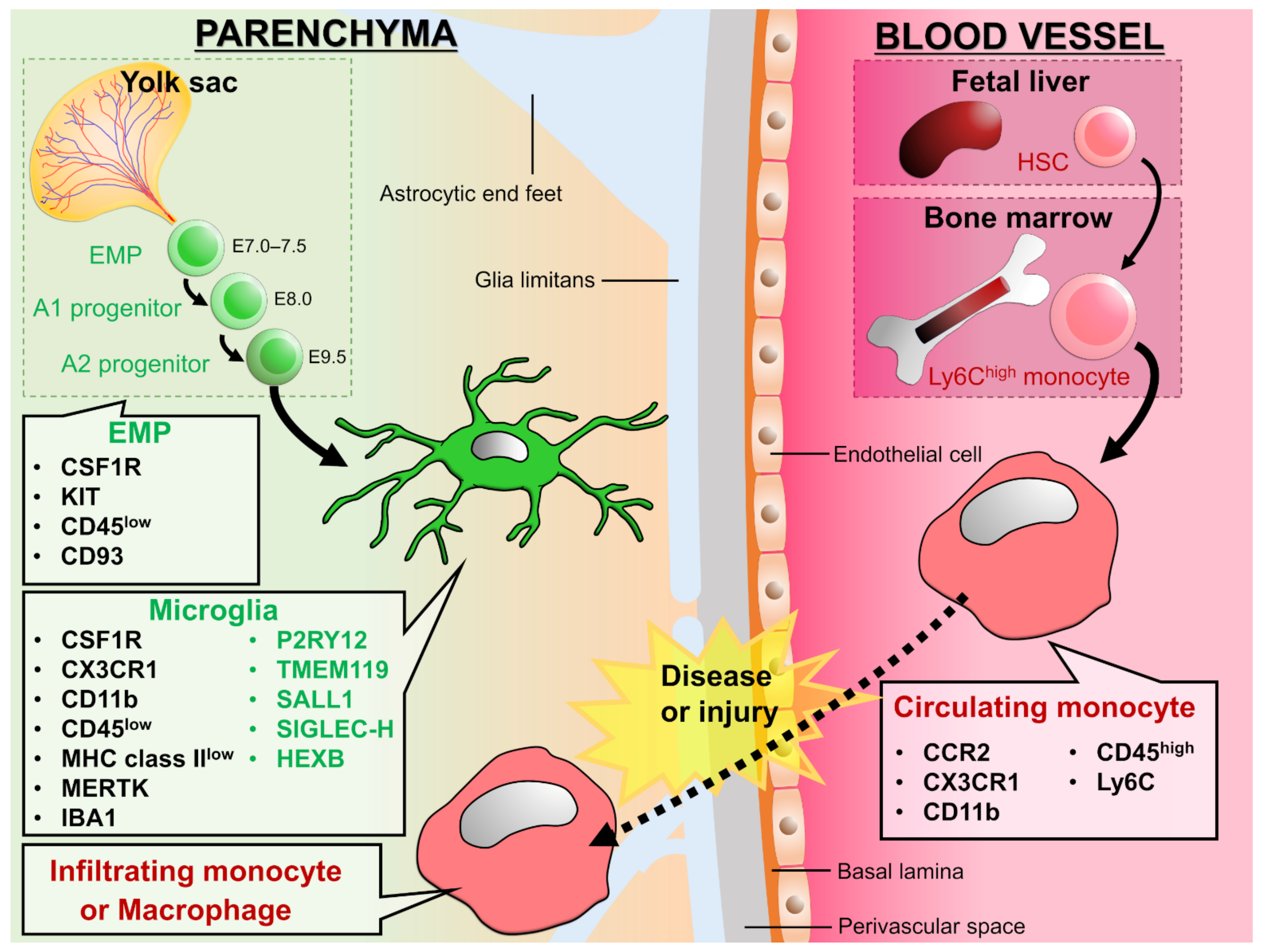
:1. Introduction
2. Phagocytosis in the Intact Brain
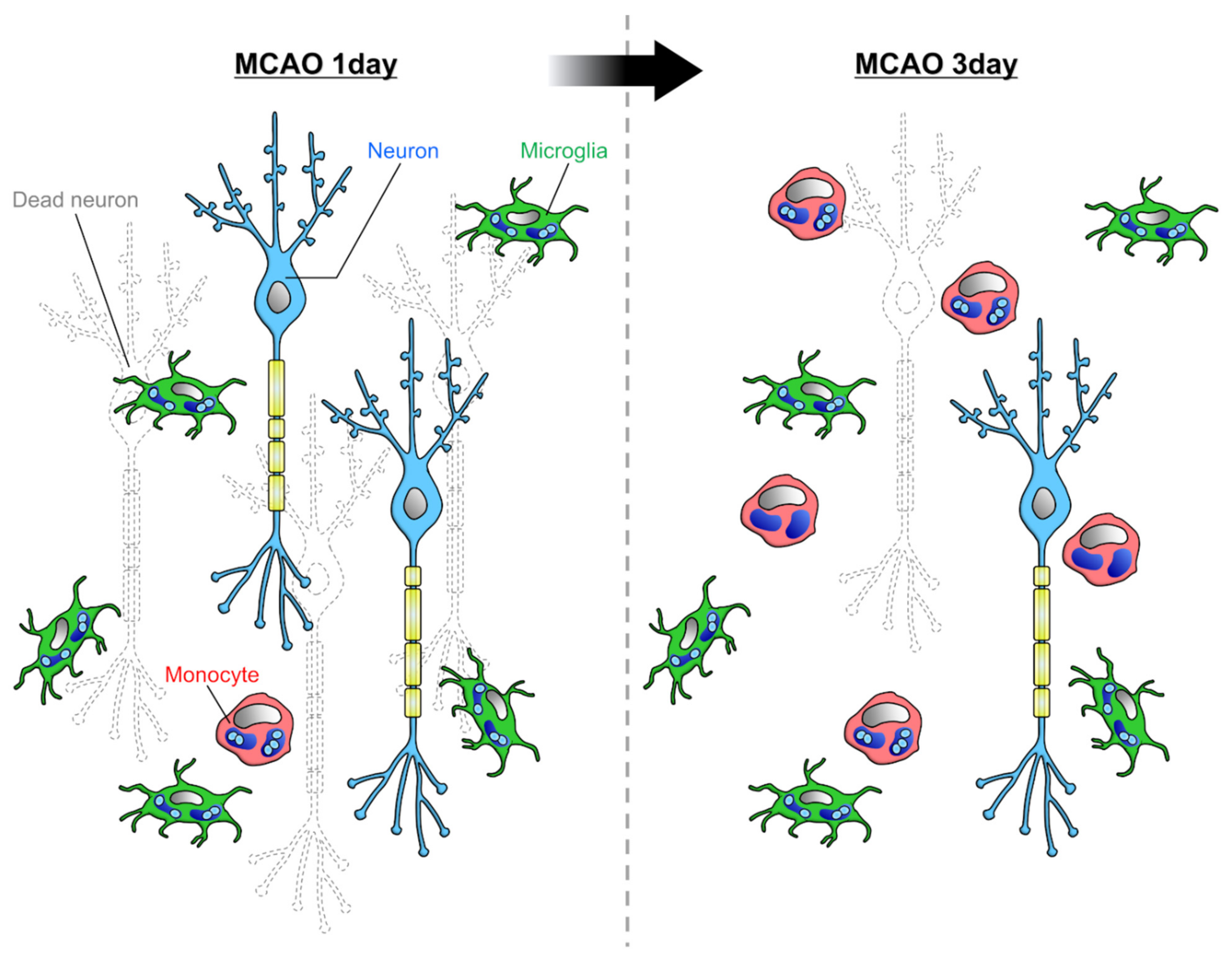
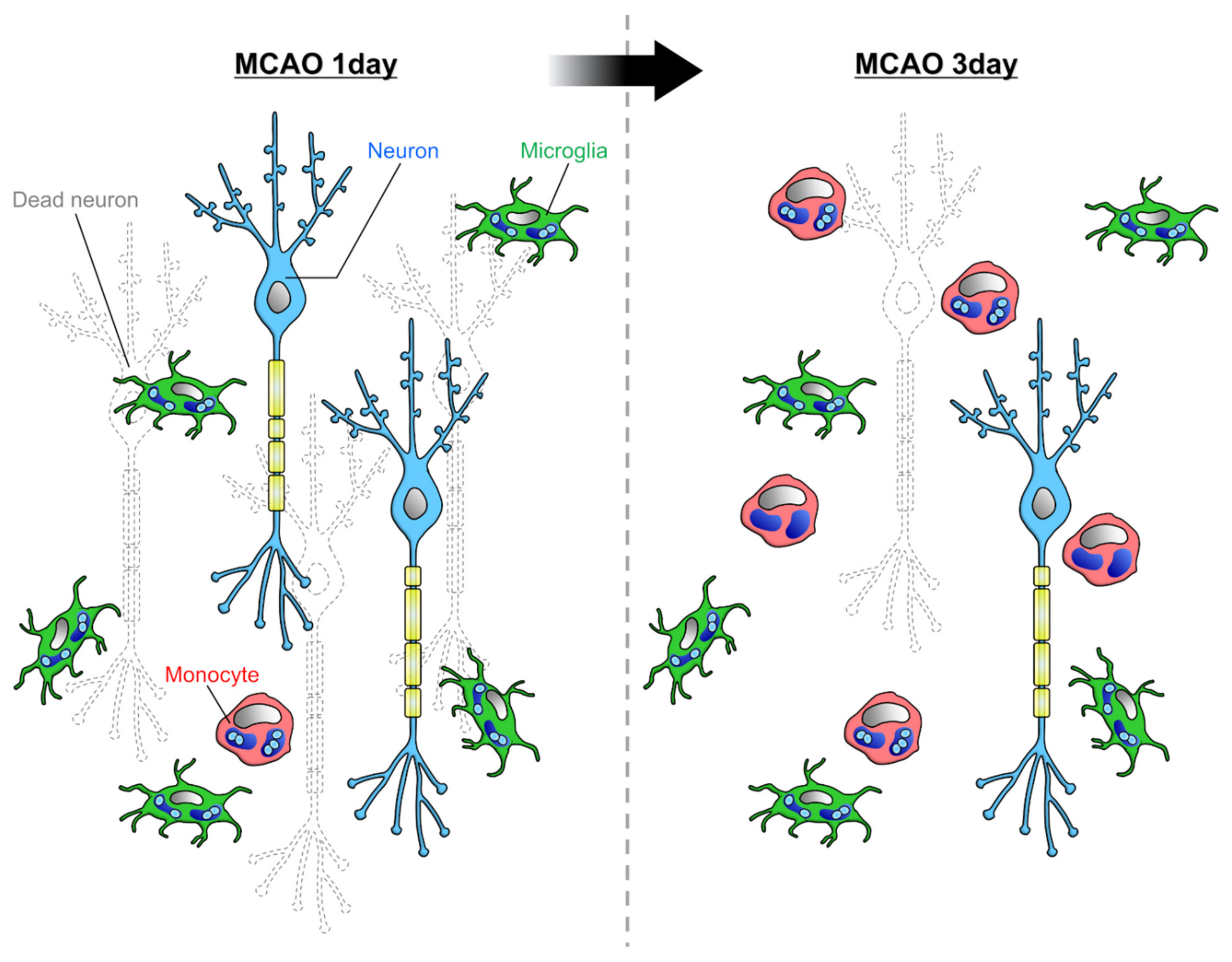
3. Phagocytosis by Microglia and Infiltrating Monocytes in the Diseased Brain
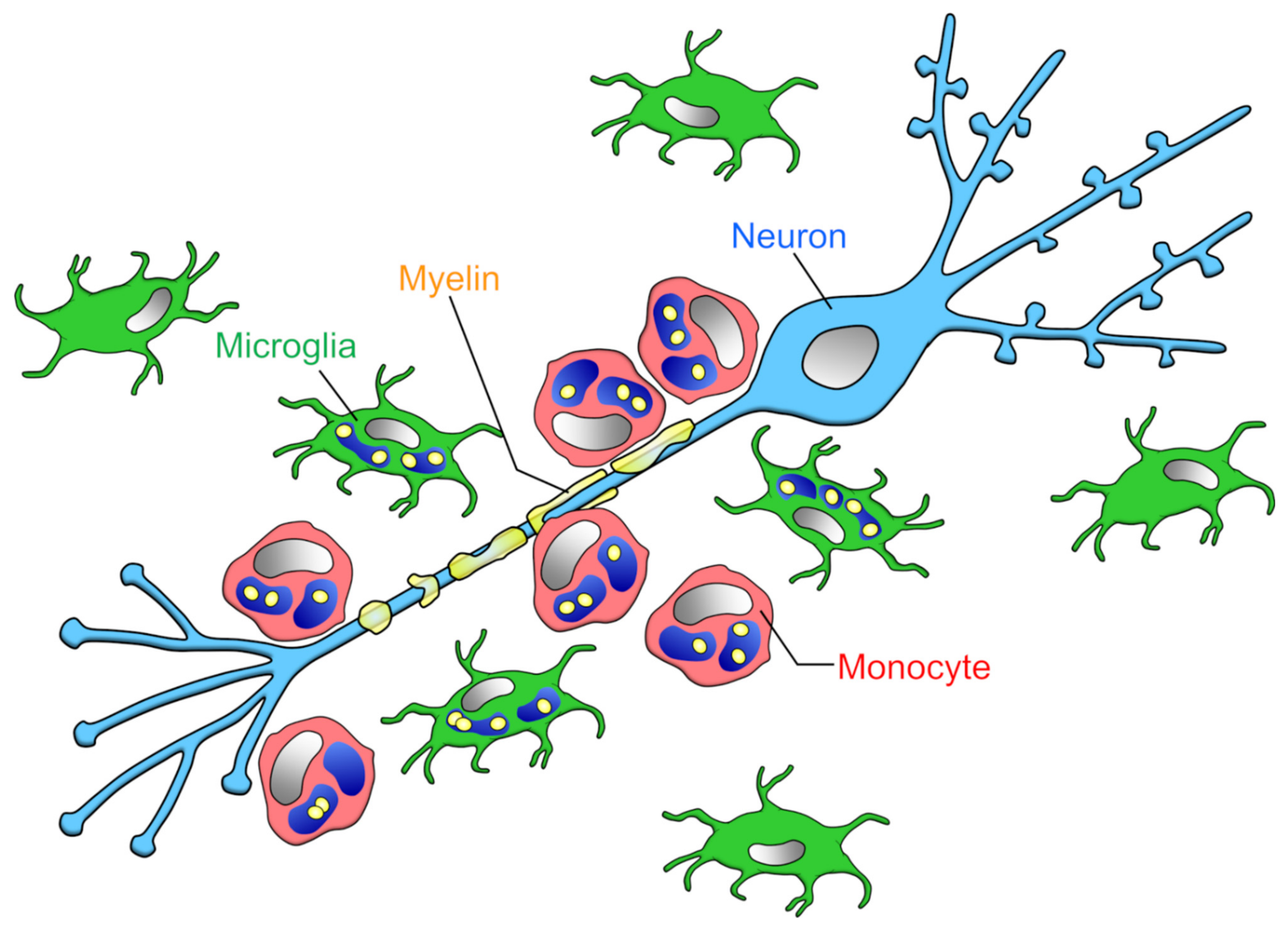
3.1. Myelin
3.2. Apoptotic Cells
3.3. Tumor Cells
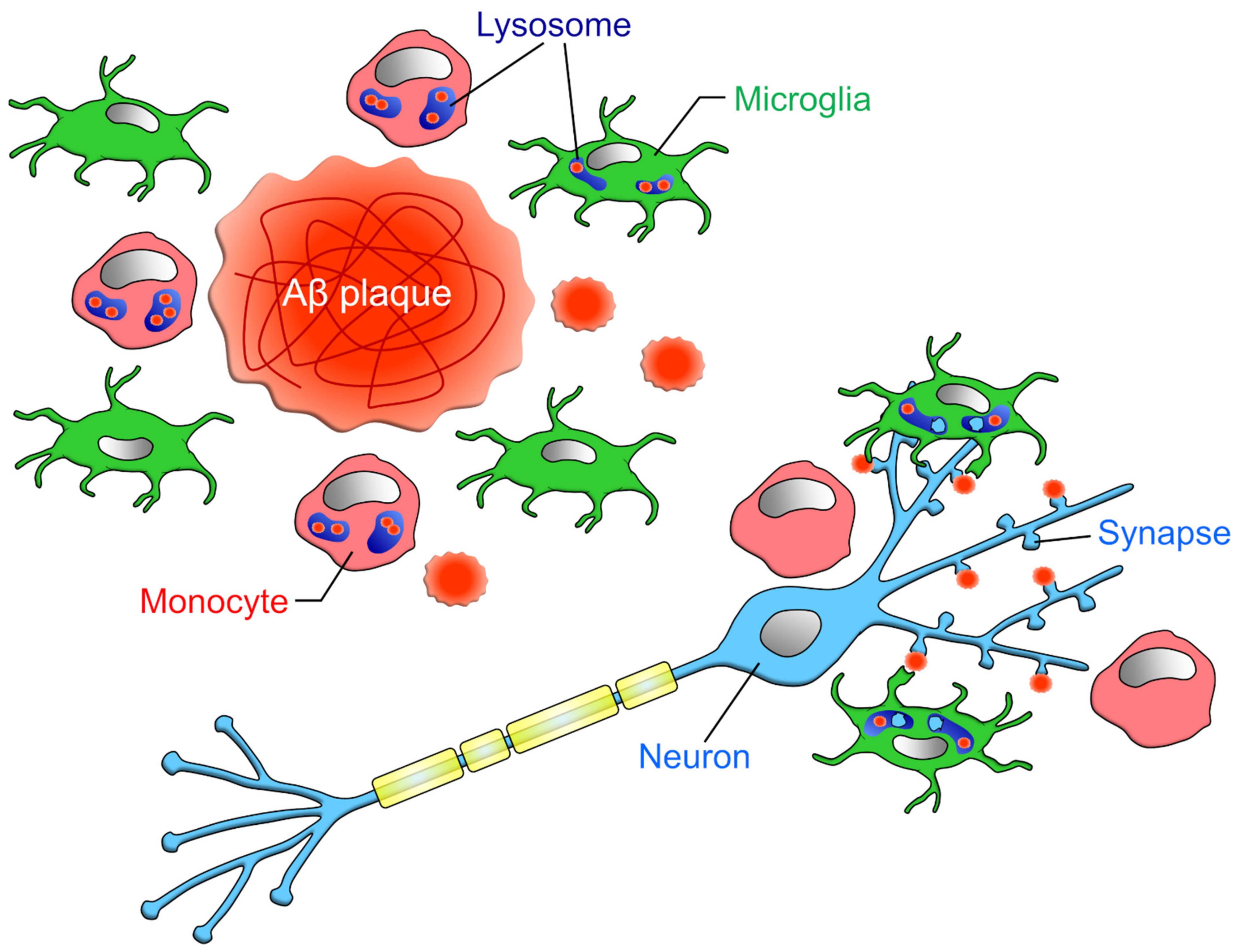
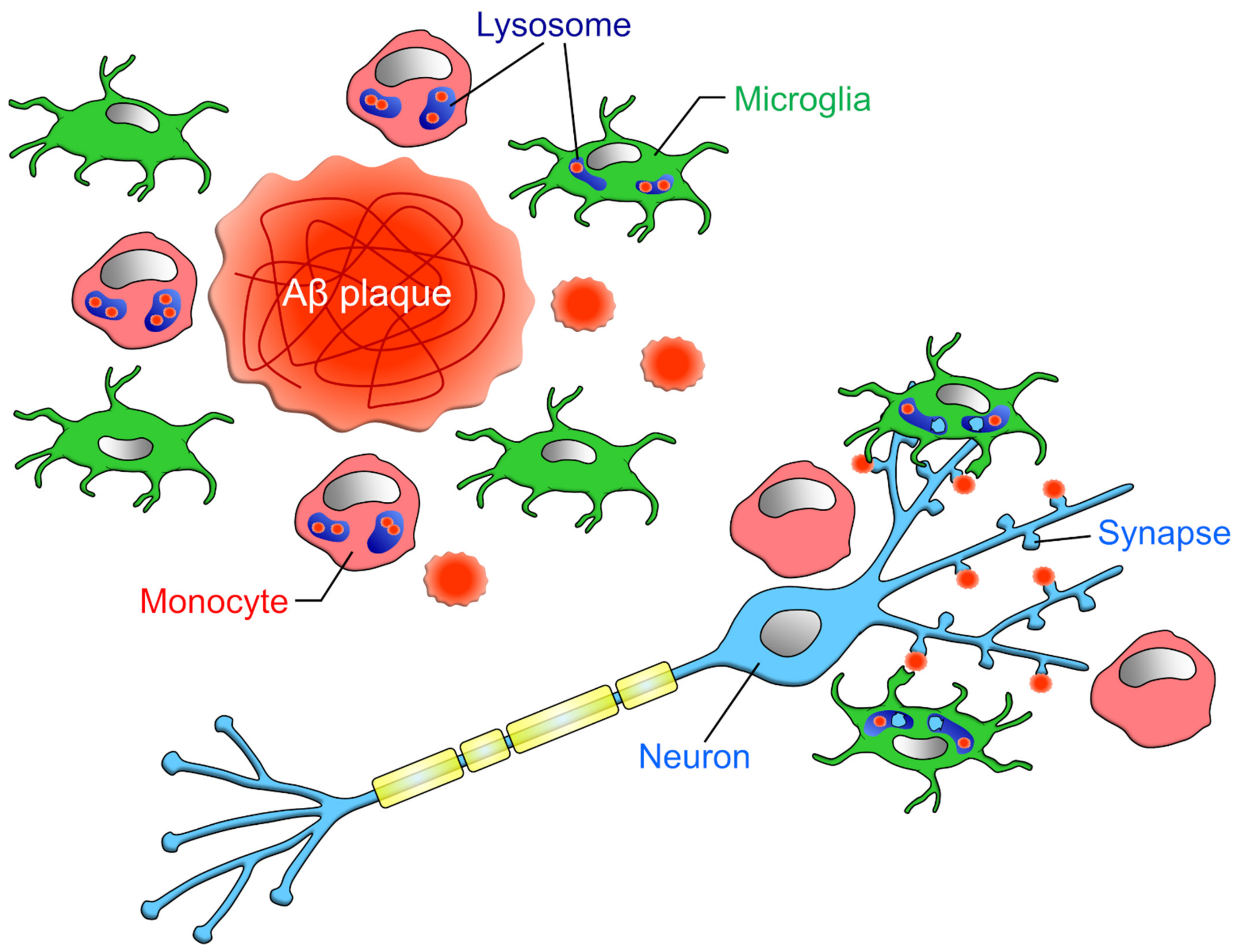
3.4. Amyloid-β (Aβ)
3.5. Synapses
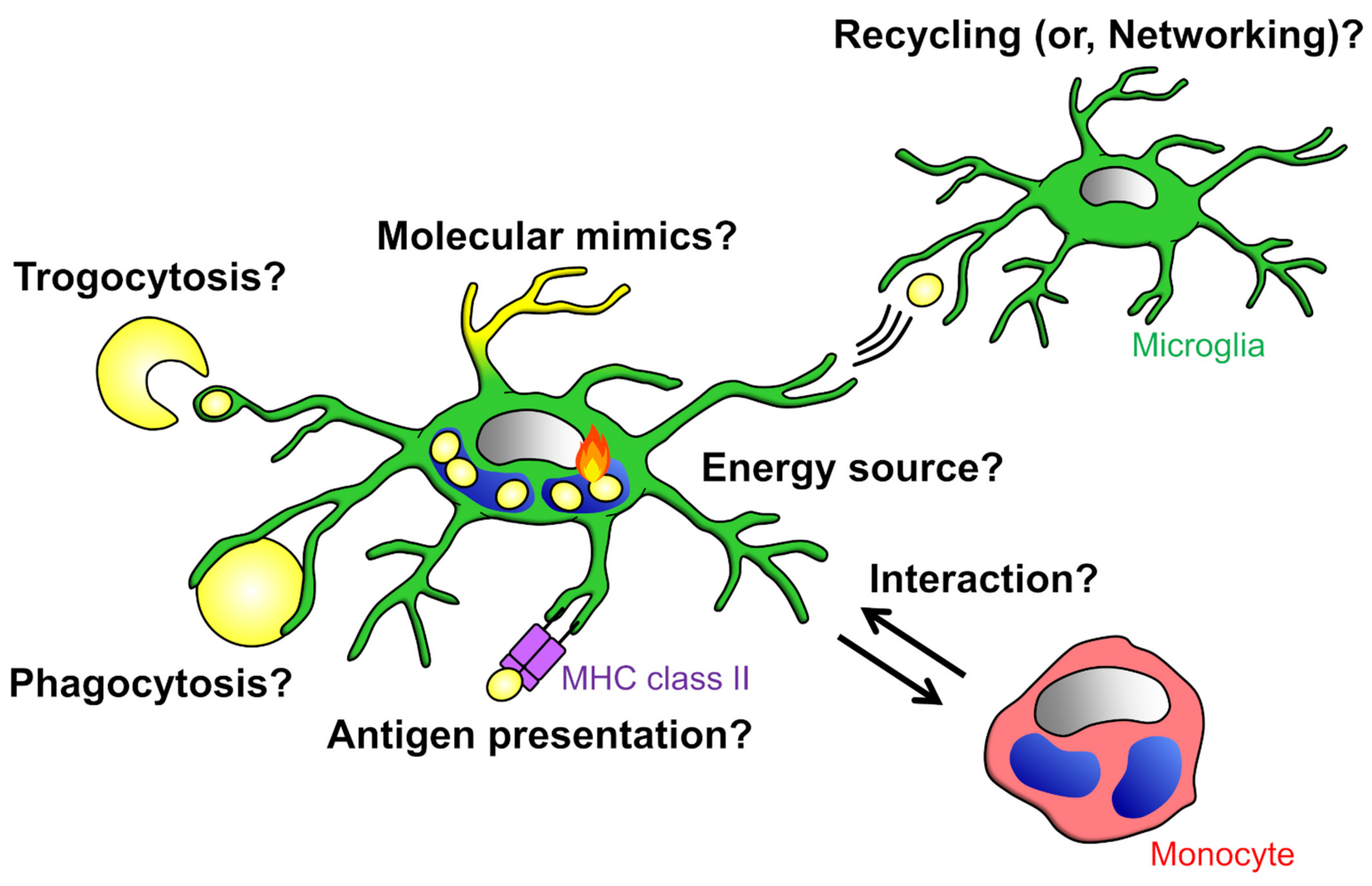
4. Significance of Phagocytosis on Phagocytes
4.1. Suppression of Inflammatory Response
4.2. Acquisition of Nutrients
4.3. Survival in Unfamiliar Environments
4.4. Antigen Presentation
4.5. Networking
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid-β |
| ABCA1 | ATP-binding cassette transporter A1 |
| AD | Alzheimer’s disease |
| BAMs | border-associated macrophages |
| BBB | blood brain barrier |
| BM | bone marrow |
| BrdU | bromodeoxyuridine |
| C1q | complement component 1q |
| C3 | complement component 3 |
| C3aR | C3a anaphylatoxin receptor |
| C4 | complement component 4 |
| CAG | CMV early enhancer/chicken β actin |
| CCL2 | CC chemokine ligand 2 |
| CCR2 | CC chemokine receptor 2 |
| CD8 | cluster of differentiation 8 |
| CD11b | cluster of differentiation 11b |
| CD11c | cluster of differentiation 11c |
| CD11c-DNR | CD11c promoter–driven dominant-negative TGF-b receptor type II |
| CD36 | cluster of differentiation 36 |
| CD45 | cluster of differentiation 45 |
| CD47 | cluster of differentiation 47 |
| CD68 | cluster of differentiation 68 |
| CD93 | cluster of differentiation 93 |
| CD115 | cluster of differentiation 115 |
| CD163 | cluster of differentiation 163 |
| CD206 | cluster of differentiation206 |
| CNS | central nervous system |
| CR3 | complement component 3 receptor |
| CSF1R | colony-stimulating factor 1 receptor |
| CX3CR1 | CX3C chemokine receptor 1 |
| EAE | experimental autoimmune encephalitis |
| EMP | erythro-myeloid progenitors |
| FBS | fetal bovine serum |
| GA | glatiramer acetate |
| GCV | ganciclovir |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GTP | guanosine triphosphate |
| HexB | hexosaminidase subunit beta |
| HSC | hematopoietic stem cell |
| HSVTK | herpes simplex virus thymidine kinase |
| Iba1 | ionized calcium-binding adapter molecule 1 |
| IFN-γ | interferon-γ |
| IL-4 | interleukin-4 |
| IL-10 | interleukin-10 |
| IL-12 | interleukin-12 |
| KO | knockout |
| Ly6C | lymphocyte antigen 6 complex locus C |
| LPS | Lipopolysaccharide |
| LXRs | liver X receptors |
| MBP | myelin binding protein |
| MCAO | middle cerebral artery occlusion |
| M-CSF | Macrophage Colony-Stimulating Factor |
| MERTK | Mer tyrosine kinase |
| MS | multiple sclerosis |
| NAD | nicotinamide adenine dinucleotide |
| NPCs | neural progenitor cells |
| Nr4a1 | nuclear receptor subfamily 4 group A member 1 |
| P2Y12 | P2Y purinoceptor 12 |
| Pbx1 | PBX Homeobox 1 |
| RPMI-1640 | Roswell Park Memorial Institute-1640 |
| Sall1 | SAL-like 1 |
| SCD1 | stearoyl-CoA desaturase 1 |
| Siglec-H | sialic acid-binding immunoglobulin-like lectin H |
| SIRPα | signal regulatory protein α |
| SIRT1 | Sirtuin 1 |
| SLCs | solute carriers |
| Smad | mothers against decapentaplegic homolog |
| SR-A | scavenger receptor class A |
| STAT1 | signal transducer and activator of transcription 1 |
| TAMs | tumor-associated macrophages and microglia |
| TGF-β | transforming growth factor β |
| TMEM119 | transmembrane protein 119 |
| TNF-α | tumor necrosis factor-α |
| WNV | West Nile virus |
References
- Galea, I.; Bechmann, I.; Perry, V.H. What is immune privilege (not)? Trends Immunol. 2007, 28, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Niederkorn, J.Y. See no evil, hear no evil, do no evil: The lessons of immune privilege. Nat. Immunol. 2006, 7, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B. Regulation of immune cell entry into the central nervous system. Results Probl. Cell Differ. 2006, 43, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Pösel, C.; Möller, K.; Boltze, J.; Wagner, D.C.; Weise, G. Isolation and Flow Cytometric Analysis of Immune Cells from the Ischemic Mouse Brain. J. Vis. Exp. 2016, 108, 53658. [Google Scholar] [CrossRef] [Green Version]
- Korin, B.; Ben-Shaanan, T.L.; Schiller, M.; Dubovik, T.; Azulay-Debby, H.; Boshnak, N.T.; Koren, T.; Rolls, A. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat. Neurosci. 2017, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [Green Version]
- Kono, R.; Ikegaya, Y.; Koyama, R. Phagocytic Glial Cells in Brain Homeostasis. Cells 2021, 10, 1348. [Google Scholar] [CrossRef]
- Green, D.R.; Oguin, T.H.; Martinez, J. The clearance of dying cells: Table for two. Cell Death Differ. 2016, 23, 915–926. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Cavallucci, V.; D’Amelio, M.; Cecconi, F. Aβ toxicity in Alzheimer’s disease. Mol. Neurobiol. 2012, 45, 366–378. [Google Scholar] [CrossRef]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fani Maleki, A.; Rivest, S. Innate Immune Cells: Monocytes, Monocyte-Derived Macrophages and Microglia as Therapeutic Targets for Alzheimer’s Disease and Multiple Sclerosis. Front. Cell Neurosci. 2019, 13, 355. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Thelin, E.P.; Tajsic, T.; Khan, D.Z.; Khellaf, A.; Patani, R.; Helmy, A. Cellular infiltration in traumatic brain injury. J. Neuroinflamm. 2020, 17, 328. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Schilling, M.; Besselmann, M.; Müller, M.; Strecker, J.K.; Ringelstein, E.B.; Kiefer, R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2005, 196, 290–297. [Google Scholar] [CrossRef]
- Unger, M.S.; Schernthaner, P.; Marschallinger, J.; Mrowetz, H.; Aigner, L. Microglia prevent peripheral immune cell invasion and promote an anti-inflammatory environment in the brain of APP-PS1 transgenic mice. J. Neuroinflamm. 2018, 15, 274. [Google Scholar] [CrossRef] [Green Version]
- Di Liberto, G.; Pantelyushin, S.; Kreutzfeldt, M.; Page, N.; Musardo, S.; Coras, R.; Steinbach, K.; Vincenti, I.; Klimek, B.; Lingner, T.; et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell 2018, 175, 458–471.e419. [Google Scholar] [CrossRef] [Green Version]
- Prinz, M.; Erny, D.; Hagemeyer, N. Ontogeny and homeostasis of CNS myeloid cells. Nat. Immunol. 2017, 18, 385–392. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Eme-Scolan, E.; Dando, S.J. Tools and Approaches for Studying Microglia. Front. Immunol. 2020, 11, 583647. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, A.G.; Wishart, C.L.; King, N.J.C. Immovable Object Meets Unstoppable Force? Dialogue Between Resident and Peripheral Myeloid Cells in the Inflamed Brain. Front. Immunol. 2020, 11, 600822. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.P.; Bellavance, M.A.; Préfontaine, P.; Rivest, S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep. 2013, 5, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Kucharova, K.; Stallcup, W.B. Distinct NG2 proteoglycan-dependent roles of resident microglia and bone marrow-derived macrophages during myelin damage and repair. PLoS ONE 2017, 12, e0187530. [Google Scholar] [CrossRef] [Green Version]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, A.D.; David, S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J. Neurosci. 2014, 34, 6316–6322. [Google Scholar] [CrossRef]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 1847–1860. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Laouar, Y.; Pittenger, C.; Mori, T.; Szekely, C.A.; Tan, J.; Duman, R.S.; Flavell, R.A. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 2008, 14, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Rinner, W.A.; Bauer, J.; Schmidts, M.; Lassmann, H.; Hickey, W.F. Resident microglia and hematogenous macrophages as phagocytes in adoptively transferred experimental autoimmune encephalomyelitis: An investigation using rat radiation bone marrow chimeras. Glia 1995, 14, 257–266. [Google Scholar] [CrossRef]
- Plemel, J.R.; Stratton, J.A.; Michaels, N.J.; Rawji, K.S.; Zhang, E.; Sinha, S.; Baaklini, C.S.; Dong, Y.; Ho, M.; Thorburn, K.; et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci. Adv. 2020, 6, eaay6324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hayden, E.Y.; Garcia, V.J.; Fuchs, D.T.; Sheyn, J.; Daley, D.A.; Rentsendorj, A.; Torbati, T.; Black, K.L.; Rutishauser, U.; et al. Activated Bone Marrow-Derived Macrophages Eradicate Alzheimer’s-Related Aβ. Front. Immunol. 2020, 11, 49. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Grenier, J.M.; Crapser, J.; Verma, R.; Jellison, E.R.; McCullough, L.D. Functional differences between microglia and monocytes after ischemic stroke. J. Neuroinflamm. 2015, 12, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Gieryng, A.; Zawadzka, M.; Kaminska, B. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia 2019, 67, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bonilla, L.; Faraco, G.; Moore, J.; Murphy, M.; Racchumi, G.; Srinivasan, J.; Brea, D.; Iadecola, C.; Anrather, J. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J. Neuroinflamm. 2016, 13, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo, G.L.; Ballard, V.A.; Glausen, T.; Boone, Z.; Teamer, J.; Hinkson, C.L.; Wohlfert, E.A.; Blader, I.J.; Fox, M.A. Toxoplasma infection induces microglia-neuron contact and the loss of perisomatic inhibitory synapses. Glia 2020, 68, 1968–1986. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Schönheit, J.; Giladi, A.; David, E.; Lara-Astiaso, D.; Lorenzo-Vivas, E.; Paul, F.; Chappell-Maor, L.; Priller, J.; Leutz, A.; et al. Genomic Characterization of Murine Monocytes Reveals C/EBPβ Transcription Factor Dependence of Ly6C. Immunity 2017, 46, 849–862.e847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Chen, H.R.; Sun, Y.Y.; Chen, C.W.; Kuo, Y.M.; Kuan, I.S.; Tiger Li, Z.R.; Short-Miller, J.C.; Smucker, M.R.; Kuan, C.Y. Fate mapping via CCR2-CreER mice reveals monocyte-to-microglia transition in development and neonatal stroke. Sci. Adv. 2020, 6, eabb2119. [Google Scholar] [CrossRef]
- Masuda, T.; Amann, L.; Sankowski, R.; Staszewski, O.; Lenz, M.; Errico, P.D.; Snaidero, N.; Costa Jordão, M.J.; Böttcher, C.; Kierdorf, K.; et al. Novel Hexb-based tools for studying microglia in the CNS. Nat. Immunol. 2020, 21, 802–815. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pouchoulen, M.; VanRyzin, J.W.; McCarthy, M.M. Morphological and Phagocytic Profile of Microglia in the Developing Rat Cerebellum. eNeuro 2015, 2, e0036-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanRyzin, J.W.; Marquardt, A.E.; Argue, K.J.; Vecchiarelli, H.A.; Ashton, S.E.; Arambula, S.E.; Hill, M.N.; McCarthy, M.M. Microglial Phagocytosis of Newborn Cells Is Induced by Endocannabinoids and Sculpts Sex Differences in Juvenile Rat Social Play. Neuron 2019, 102, 435–449.e436. [Google Scholar] [CrossRef] [Green Version]
- Fourgeaud, L.; Través, P.G.; Tufail, Y.; Leal-Bailey, H.; Lew, E.D.; Burrola, P.G.; Callaway, P.; Zagórska, A.; Rothlin, C.V.; Nimmerjahn, A.; et al. TAM receptors regulate multiple features of microglial physiology. Nature 2016, 532, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Abiega, O.; Shahraz, A.; Neumann, H. Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front. Cell Neurosci. 2013, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Mazaheri, F.; Breus, O.; Durdu, S.; Haas, P.; Wittbrodt, J.; Gilmour, D.; Peri, F. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 2014, 5, 4046. [Google Scholar] [CrossRef] [Green Version]
- Marín-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; van Rooijen, N.; Mallat, M. Microglia promote the death of developing Purkinje cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e210. [Google Scholar] [CrossRef] [Green Version]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Hill, R.A.; Li, A.M.; Grutzendler, J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat. Neurosci. 2018, 21, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron number and size in prefrontal cortex of children with autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Cho, M.H.; Shim, W.H.; Kim, J.K.; Jeon, E.Y.; Kim, D.H.; Yoon, S.Y. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 2017, 22, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e978. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Jin, X.; Parada, I.; Pesic, A.; Stevens, B.; Barres, B.; Prince, D.A. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl. Acad. Sci. USA 2010, 107, 7975–7980. [Google Scholar] [CrossRef] [Green Version]
- Andoh, M.; Shibata, K.; Okamoto, K.; Onodera, J.; Morishita, K.; Miura, Y.; Ikegaya, Y.; Koyama, R. Exercise Reverses Behavioral and Synaptic Abnormalities after Maternal Inflammation. Cell Rep. 2019, 27, 2817–2825.e2815. [Google Scholar] [CrossRef] [Green Version]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef]
- Peters, A.; Moss, M.B.; Sethares, C. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J. Comp. Neurol. 2000, 419, 364–376. [Google Scholar] [CrossRef]
- Jeon, S.B.; Yoon, H.J.; Park, S.H.; Kim, I.H.; Park, E.J. Sulfatide, a major lipid component of myelin sheath, activates inflammatory responses as an endogenous stimulator in brain-resident immune cells. J. Immunol. 2008, 181, 8077–8087. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.M.; Sánchez, M.G.; Yong, V.W.; Stys, P.K.; Tremblay, M.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosley, K.; Cuzner, M.L. Receptor-mediated phagocytosis of myelin by macrophages and microglia: Effect of opsonization and receptor blocking agents. Neurochem. Res. 1996, 21, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Durafourt, B.A.; Moore, C.S.; Zammit, D.A.; Johnson, T.A.; Zaguia, F.; Guiot, M.C.; Bar-Or, A.; Antel, J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 2012, 60, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palle, P.; Monaghan, K.L.; Milne, S.M.; Wan, E.C.K. Cytokine Signaling in Multiple Sclerosis and Its Therapeutic Applications. Med. Sci. 2017, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.E. Phagocytosis of myelin in demyelinative disease: A review. Neurochem. Res. 1999, 24, 261–268. [Google Scholar] [CrossRef]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-β peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef] [Green Version]
- Frackowiak, J.; Wisniewski, H.M.; Wegiel, J.; Merz, G.S.; Iqbal, K.; Wang, K.C. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992, 84, 225–233. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef]
- Fiala, M.; Lin, J.; Ringman, J.; Kermani-Arab, V.; Tsao, G.; Patel, A.; Lossinsky, A.S.; Graves, M.C.; Gustavson, A.; Sayre, J.; et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J. Alzheimer’s Dis. 2005, 7, 221–232, discussion 255–262. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Brück, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [Green Version]
- Tichauer, J.E.; von Bernhardi, R. Transforming growth factor-β stimulates β amyloid uptake by microglia through Smad3-dependent mechanisms. J. Neurosci. Res. 2012, 90, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, V.; Schurr, J.; Ball, M.J.; Pelaez, R.P.; Bazan, N.G.; Lukiw, W.J. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: Transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J. Neurosci Res. 2002, 70, 462–473. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Flores, B.; Soler, B.; Eugenín-von Bernhardi, L.; Ramírez, G.; von Bernhardi, R. Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Von Bernhardi, R.; Cornejo, F.; Parada, G.E.; Eugenín, J. Role of TGFβ signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 2015, 9, 426. [Google Scholar] [CrossRef] [Green Version]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Li, S.; Ljubimov, V.; Moyseyev, M.; Daley, D.; Fuchs, D.T.; Pham, M.; et al. Therapeutic effects of glatiramer acetate and grafted CD115⁺ monocytes in a mouse model of Alzheimer’s disease. Brain 2015, 138, 2399–2422. [Google Scholar] [CrossRef] [Green Version]
- Rentsendorj, A.; Sheyn, J.; Fuchs, D.T.; Daley, D.; Salumbides, B.C.; Schubloom, H.E.; Hart, N.J.; Li, S.; Hayden, E.Y.; Teplow, D.B.; et al. A novel role for osteopontin in macrophage-mediated amyloid-β clearance in Alzheimer’s models. Brain Behav. Immun. 2018, 67, 163–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.L.; Reeves, T.M.; Phillips, L.L. Osteopontin expression in acute immune response mediates hippocampal synaptogenesis and adaptive outcome following cortical brain injury. Exp. Neurol. 2014, 261, 757–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Severance, E.G.; Viscidi, R.P.; Yolken, R.H.; Xiao, J. Persistent Toxoplasma Infection of the Brain Induced Neurodegeneration Associated with Activation of Complement and Microglia. Infect. Immun. 2019, 87, e00139-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Li, Y.; Gressitt, K.L.; He, H.; Kannan, G.; Schultz, T.L.; Svezhova, N.; Carruthers, V.B.; Pletnikov, M.V.; Yolken, R.H.; et al. Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav. Immun. 2016, 58, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saederup, N.; Cardona, A.E.; Croft, K.; Mizutani, M.; Cotleur, A.C.; Tsou, C.L.; Ransohoff, R.M.; Charo, I.F. Correction: Selective Chemokine Receptor Usage by Central Nervous System Myeloid Cells in CCR2-Red Fluorescent Protein Knock-In Mice. PLoS ONE 2017, 12, e0176931. [Google Scholar] [CrossRef]
- Kourtzelis, I.; Hajishengallis, G.; Chavakis, T. Phagocytosis of Apoptotic Cells in Resolution of Inflammation. Front. Immunol. 2020, 11, 553. [Google Scholar] [CrossRef]
- Kim, S.; Elkon, K.B.; Ma, X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity 2004, 21, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Bogie, J.F.; Timmermans, S.; Huynh-Thu, V.A.; Irrthum, A.; Smeets, H.J.; Gustafsson, J.; Steffensen, K.R.; Mulder, M.; Stinissen, P.; Hellings, N.; et al. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS ONE 2012, 7, e44998. [Google Scholar] [CrossRef] [Green Version]
- Bogie, J.F.; Jorissen, W.; Mailleux, J.; Nijland, P.G.; Zelcer, N.; Vanmierlo, T.; Van Horssen, J.; Stinissen, P.; Hellings, N.; Hendriks, J.J. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol. Commun. 2013, 1, 43. [Google Scholar] [CrossRef] [Green Version]
- Bogie, J.F.J.; Grajchen, E.; Wouters, E.; Corrales, A.G.; Dierckx, T.; Vanherle, S.; Mailleux, J.; Gervois, P.; Wolfs, E.; Dehairs, J.; et al. Stearoyl-CoA desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morioka, S.; Perry, J.S.A.; Raymond, M.H.; Medina, C.B.; Zhu, Y.; Zhao, L.; Serbulea, V.; Onengut-Gumuscu, S.; Leitinger, N.; Kucenas, S.; et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 2018, 563, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e445. [Google Scholar] [CrossRef] [Green Version]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Assessing Microglial Dynamics by Live Imaging. Front. Immunol. 2021, 12, 617564. [Google Scholar] [CrossRef]
- Yurdagul, A.; Subramanian, M.; Wang, X.; Crown, S.B.; Ilkayeva, O.R.; Darville, L.; Kolluru, G.K.; Rymond, C.C.; Gerlach, B.D.; Zheng, Z.; et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020, 31, 518–533.e510. [Google Scholar] [CrossRef]
- Grajchen, E.; Wouters, E.; van de Haterd, B.; Haidar, M.; Hardonnière, K.; Dierckx, T.; Van Broeckhoven, J.; Erens, C.; Hendrix, S.; Kerdine-Römer, S.; et al. CD36-mediated uptake of myelin debris by macrophages and microglia reduces neuroinflammation. J. Neuroinflamm. 2020, 17, 224. [Google Scholar] [CrossRef] [PubMed]
- Ralston, K.S.; Solga, M.D.; Mackey-Lawrence, N.M.; Somlata; Bhattacharya, A.; Petri, W.A. Trogocytosis by Entamoeba histolytica contributes to cell killing and tissue invasion. Nature 2014, 508, 526–530. [Google Scholar] [CrossRef] [Green Version]
- Miller, H.W.; Suleiman, R.L.; Ralston, K.S. Trogocytosis by Entamoeba histolytica Mediates Acquisition and Display of Human Cell Membrane Proteins and Evasion of Lysis by Human Serum. MBio 2019, 10, e00068-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casano, A.M.; Albert, M.; Peri, F. Developmental Apoptosis Mediates Entry and Positioning of Microglia in the Zebrafish Brain. Cell Rep. 2016, 16, 897–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, T.; Wu, Y.; Jin, W.; Wen, Z. Microglia Colonization of Developing Zebrafish Midbrain Is Promoted by Apoptotic Neuron and Lysophosphatidylcholine. Dev. Cell 2016, 38, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2017, 8, 1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Zhu, K.; Pieber, M.; Han, J.; Blomgren, K.; Zhang, X.M.; Harris, R.A.; Lund, H. Absence of microglia or presence of peripherally-derived macrophages does not affect tau pathology in young or old hTau mice. Glia 2020, 68, 1466–1478. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Dou, Y.; Wu, H.J.; Li, H.Q.; Qin, S.; Wang, Y.E.; Li, J.; Lou, H.F.; Chen, Z.; Li, X.M.; Luo, Q.M.; et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res. 2012, 22, 1022–1033. [Google Scholar] [CrossRef] [Green Version]
- Pasqual, G.; Chudnovskiy, A.; Tas, J.M.J.; Agudelo, M.; Schweitzer, L.D.; Cui, A.; Hacohen, N.; Victora, G.D. Monitoring T cell-dendritic cell interactions in vivo by intercellular enzymatic labelling. Nature 2018, 553, 496–500. [Google Scholar] [CrossRef]
- Giladi, A.; Cohen, M.; Medaglia, C.; Baran, Y.; Li, B.; Zada, M.; Bost, P.; Blecher-Gonen, R.; Salame, T.M.; Mayer, J.U.; et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 2020, 38, 629–637. [Google Scholar] [CrossRef]
- Clark, I.C.; Gutiérrez-Vázquez, C.; Wheeler, M.A.; Li, Z.; Rothhammer, V.; Linnerbauer, M.; Sanmarco, L.M.; Guo, L.; Blain, M.; Zandee, S.E.J.; et al. Barcoded viral tracing of single-cell interactions in central nervous system inflammation. Science 2021, 372, eabf1230. [Google Scholar] [CrossRef]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e279. [Google Scholar] [CrossRef] [Green Version]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E.; et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e186. [Google Scholar] [CrossRef]
- Ohgidani, M.; Kato, T.A.; Setoyama, D.; Sagata, N.; Hashimoto, R.; Shigenobu, K.; Yoshida, T.; Hayakawa, K.; Shimokawa, N.; Miura, D.; et al. Direct induction of ramified microglia-like cells from human monocytes: Dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 2014, 4, 4957. [Google Scholar] [CrossRef] [Green Version]
- Sellgren, C.M.; Sheridan, S.D.; Gracias, J.; Xuan, D.; Fu, T.; Perlis, R.H. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 2017, 22, 170–177. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal | Manipulation | Objective | Limitation | Reference |
|---|---|---|---|---|
| CD11b-HSVTK mice | Injection of GCV into the ventricle | Microglia-specific depletion | Long-term administration of GCV causes microhemorrhages and artificial influx of peripheral macrophages into the CNS [23]. | [23] |
| Inhibition of monocyte infiltration into the brain parenchyma | [24] | |||
| unspecified | Injection of clodronate into the lateral ventricle | Perivascular-macrophage-specific depletion | - | [25] |
| Nr4a1−/− mice | BM derived monocyte transplantation | BM derived monocyte-specific labeling | - | [26] |
| CX3CR1CreERT2/+xR26-stop-RFPfl/+ mice | Intraperitoneal injection of tamoxifen | Microglia-specific labeling | It is necessary to distinguish between microglia and BAMs [27]. | [18] |
| β-actin-EGFP mice | BM derived macrophage transplantation | BM derived macrophage-specific labeling | - | [28] |
| CCR2-RFP::CX3CR1-GFP mice | - | Distinguishment between microglia and macrophage (microglia: GFP; monocyte: RFP) | - | [15,29] |
| lysozyme M EGFP-knockin mice | BM derived monocyte transplantation | BM derived monocyte-specific labeling | - | [30] |
| C57BL/6J-GFP mice | BM derived monocyte transplantation | BM derived monocyte-specific labeling | - | [16] |
| CD11c-DNR mice | BM derived monocyte-specific manipulation | CD11c expression is upregulated in microglia surrounding Aβ plaques [31]. | [32] |
| Marker | Observation System | Objective | Limitation | Reference |
|---|---|---|---|---|
| P2Y12 | Immunohistochemistry | Microglia-specific detection | In the disease conditions, P2Y12 is downregulated in microglia [33]. | [34] |
| TMEM119 | Immunohistochemistry | Microglia-specific detection | In the disease conditions, TMEM119 is downregulated in microglia [35]. | [17,18] |
| Il-69 | Immunohistochemistry | Hematogenous macrophage-specific detection | - | [36] |
| CD45 | Flow cytometry | Distinguishment between microglia and BM-derived monocyte (low: microglia; high: monocyte) | In the disease conditions, CD45 is upregulated in microglia [37]. | [38,39,40,41,42] |
| CX3CR1 | Flowcytometry | Distinguishment between microglia and BM-derived monocyte (low: monocyte; high: microglia) | In the disease conditions, CX3CR1 is downregulated in microglia [37]. | [39,43] |
| Immunohistochemistry | [44] | |||
| CCR2 | Flow cytometry | Distinguishment between microglia and BM-derived monocyte (low: microglia; high: monocyte) | - | [43] |
| Ly6C | Flow cytometry | Distinguishment between microglia and BM-derived monocyte (low: microglia; high: monocyte) | Ly6Chi monocytes give rise to Ly6Clo monocytes [45,46]. | [40,41,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andoh, M.; Koyama, R. Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells 2021, 10, 2555. https://doi.org/10.3390/cells10102555
Andoh M, Koyama R. Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells. 2021; 10(10):2555. https://doi.org/10.3390/cells10102555
Chicago/Turabian StyleAndoh, Megumi, and Ryuta Koyama. 2021. "Comparative Review of Microglia and Monocytes in CNS Phagocytosis" Cells 10, no. 10: 2555. https://doi.org/10.3390/cells10102555
APA StyleAndoh, M., & Koyama, R. (2021). Comparative Review of Microglia and Monocytes in CNS Phagocytosis. Cells, 10(10), 2555. https://doi.org/10.3390/cells10102555