
Caspase-Dependent HMGB1 Release from Macrophages Participates in Peripheral Neuropathy Caused by Bortezomib, a Proteasome-Inhibiting Chemotherapeutic Agent, in Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Major Chemicals
2.3. Assessment of Mechanical Nociceptive Threshold and Creation of a Mouse Model of CIPN Caused by Bortezomib
2.4. Drug Administration Schedules
2.5. Determination of Protein Levels of HMGB1 and Its Targeted Receptors
2.6. Depletion of Macrophages in Mice
2.7. Analysis of Macrophage Accumulation in the Sciatic Nerve and DRG by Immunofluorometry
2.8. Cell Culture
2.9. Determination of Expression, Release, and Cytoplasmic Translocation of Nuclear HMGB1 and Protein Levels of Cleaved Caspase-8 in RAW264.7 Cells
2.10. Detection of Nuclear Condensation or Fragmentation by Hoechst Staining
2.11. Statistics
3. Results
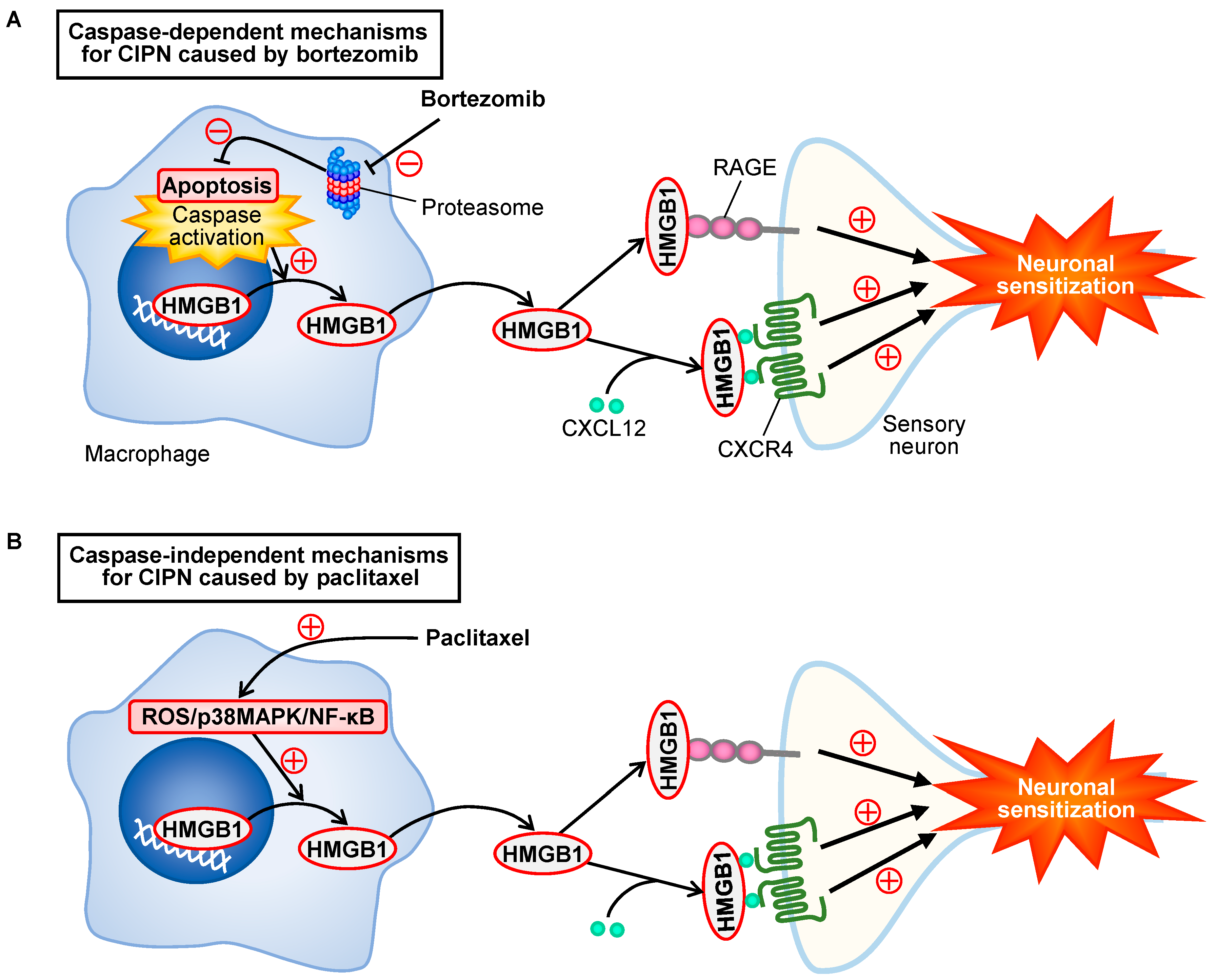
3.1. Involvement of HMGB1 in the CIPN Caused by Bortezomib in Mice
3.2. TM/TMα Prevents and Reverses the CIPN Caused by Bortezomib in a Thrombin-Dependent Manner in Mice
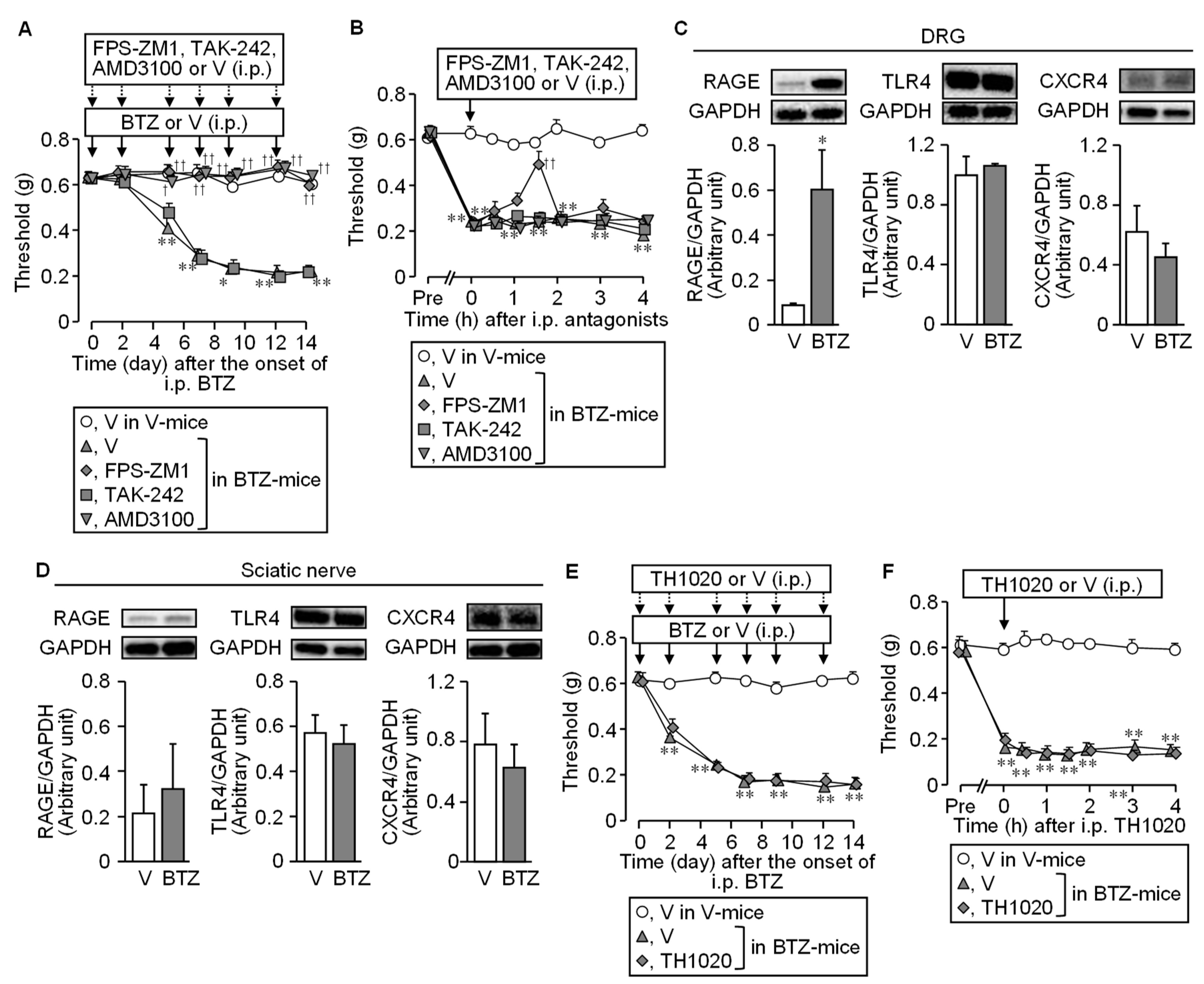
3.3. HMGB1-Targeted Receptors Involved in the CIPN Caused by Bortezomib in Mice
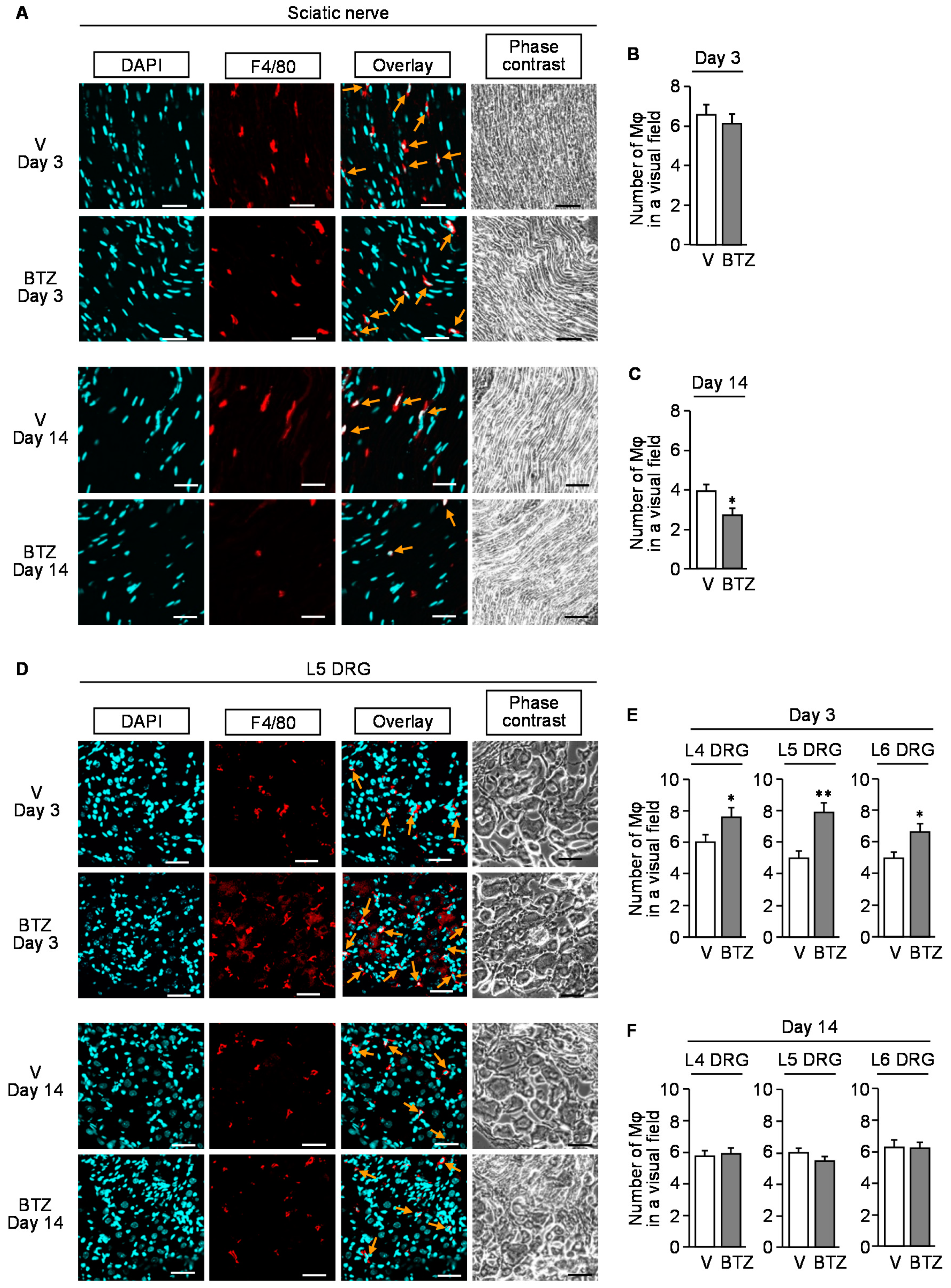
3.4. Involvement of Macrophages in the Developmental Stage of CIPN following Bortezomib Treatment in Mice
3.5. Bortezomib and MG132, Proteasome Inhibitors, Evoke HMGB1 Release from Macrophage-Like RAW264.7 Cells
3.6. Involvement of Caspase in Bortezomib-Induced HMGB1 Release from RAW264.7 Macrophages
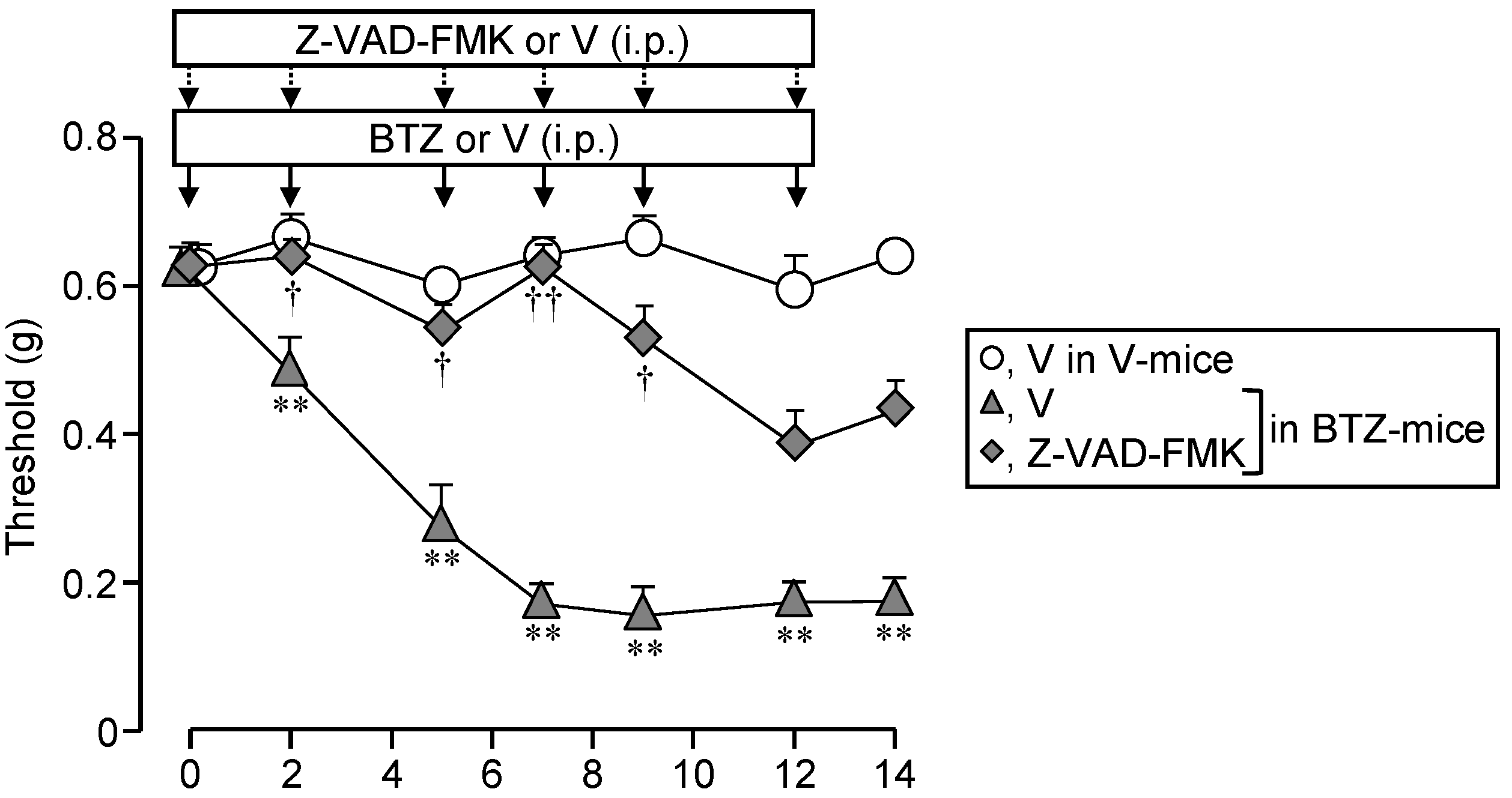
3.7. Systemic Administration of a Caspase Inhibitor Prevents the CIPN Caused by Bortezomib in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Lustberg, M.B.; Hu, S. Emerging pharmacological and non-pharmacological therapeutics for prevention and treatment of chemotherapy-induced peripheral neuropathy. Cancers 2021, 13, 766. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Kawabata, A. Role of HMGB1 in chemotherapy-induced peripheral neuropathy. Int. J. Mol. Sci. 2020, 22, 367. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Kelley, M.R.; Paice, J.A.; Lustberg, M.B.; et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3325–3348. [Google Scholar] [CrossRef]
- Nishida, T.; Tsubota, M.; Kawaishi, Y.; Yamanishi, H.; Kamitani, N.; Sekiguchi, F.; Ishikura, H.; Liu, K.; Nishibori, M.; Kawabata, A. Involvement of high mobility group box 1 in the development and maintenance of chemotherapy-induced peripheral neuropathy in rats. Toxicology 2016, 365, 48–58. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Domoto, R.; Nakashima, K.; Yamasoba, D.; Yamanishi, H.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacology 2018, 141, 201–213. [Google Scholar] [CrossRef]
- Tsubota, M.; Fukuda, R.; Hayashi, Y.; Miyazaki, T.; Ueda, S.; Yamashita, R.; Koike, N.; Sekiguchi, F.; Wake, H.; Wakatsuki, S.; et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: Negative impact of anticoagulants. J. Neuroinflammation 2019, 16, 199. [Google Scholar] [CrossRef] [PubMed]
- Tsujita, R.; Tsubota, M.; Sekiguchi, F.; Kawabata, A. Role of high-mobility group box 1 and its modulation by thrombomodulin/thrombin axis in neuropathic and inflammatory pain. Br. J. Pharmacol. 2021, 178, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Agalave, N.M.; Svensson, C.I. Extracellular high-mobility group box 1 protein (HMGB1) as a mediator of persistent pain. Mol Med. 2015, 20, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Lu, Y.Q. The regulatory role of high-mobility group protein 1 in sepsis-related immunity. Front. Immunol. 2020, 11, 601815. [Google Scholar] [CrossRef] [PubMed]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Maruyama, I. Thrombomodulin: Protectorate God of the vasculature in thrombosis and inflammation. J. Thromb. Haemost. 2011, 9, 168–173. [Google Scholar] [CrossRef]
- Kotaka, M.; Saito, Y.; Kato, T.; Satake, H.; Makiyama, A.; Tsuji, Y.; Shinozaki, K.; Fujiwara, T.; Fujiwara, T.; Harihara, Y.; et al. A placebo-controlled, double-blind, randomized study of recombinant thrombomodulin (ART-123) to prevent oxaliplatin-induced peripheral neuropathy. Cancer Chemother. Pharmacol. 2020, 86, 607–618. [Google Scholar] [CrossRef]
- Chang, K.C.; Ko, Y.S.; Kim, H.J.; Nam, D.Y.; Lee, D.U. 13-Methylberberine reduces HMGB1 release in LPS-activated RAW264.7 cells and increases the survival of septic mice through AMPK/P38 MAPK activation. Int. Immunopharmacol. 2016, 40, 269–276. [Google Scholar] [CrossRef]
- Lu, B.; Antoine, D.J.; Kwan, K.; Lundback, P.; Wahamaa, H.; Schierbeck, H.; Robinson, M.; Van Zoelen, M.A.; Yang, H.; Li, J.; et al. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc. Natl. Acad. Sci. USA 2014, 111, 3068–3073. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, L.; Collage, R.D.; Stripay, J.L.; Tsung, A.; Lee, J.S. Calcium/calmodulin-dependent protein kinase (CaMK) Ialpha mediates the macrophage inflammatory response to sepsis. J. Leukoc. Biol. 2011, 90, 249–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duggett, N.A.; Flatters, S.J.L. Characterization of a rat model of bortezomib-induced painful neuropathy. Br. J. Pharmacol. 2017, 174, 4812–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flatters, S.J.L.; Dougherty, P.M.; Colvin, L.A. Clinical and preclinical perspectives on Chemotherapy-Induced Peripheral Neuropathy (CIPN): A narrative review. Br. J. Anaesth. 2017, 119, 737–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, S.; Sekiguchi, F.; Deguchi, T.; Miyazaki, T.; Ikeda, Y.; Tsubota, M.; Yoshida, S.; Nguyen, H.D.; Okada, T.; Toyooka, N.; et al. Critical role of Cav3.2 T-type calcium channels in the peripheral neuropathy induced by bortezomib, a proteasome-inhibiting chemotherapeutic agent, in mice. Toxicology 2019, 413, 33–39. [Google Scholar] [CrossRef]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Nishibori, M.; Matsukawa, A.; Morishima, T. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [Green Version]
- Chaplan, S.R.; Bach, F.W.; Pogrel, J.W.; Chung, J.M.; Yaksh, T.L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 1994, 53, 55–63. [Google Scholar] [CrossRef]
- Boehmerle, W.; Huehnchen, P.; Peruzzaro, S.; Balkaya, M.; Endres, M. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Sci. Rep. 2014, 4, 6370. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.E.; Lunardi, N.; Boscolo, A.; Dong, X.; Erisir, A.; Jevtovic-Todorovic, V.; Todorovic, S.M. Immunohistological demonstration of CaV3.2 T-type voltage-gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 2013, 250, 263–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irie, Y.; Tsubota, M.; Ishikura, H.; Sekiguchi, F.; Terada, Y.; Tsujiuchi, T.; Liu, K.; Nishibori, M.; Kawabata, A. Macrophage-derived HMGB1 as a pain mediator in the early stage of acute pancreatitis in mice: Targeting RAGE and CXCL12/CXCR4 axis. J. Neuroimmune Pharmacol. 2017, 12, 693–707. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar]
- Zavrski, I.; Naujokat, C.; Niemoller, K.; Jakob, C.; Heider, U.; Langelotz, C. Proteasome inhibitors induce growth inhibition and apoptosis in myeloma cell lines and in human bone marrow myeloma cells irrespective of chromosome 13 deletion. J. Cancer Res. Clin. Oncol. 2003, 129, 383–391. [Google Scholar] [CrossRef]
- Das, N.; Dewan, V.; Grace, P.M.; Gunn, R.J.; Tamura, R.; Tzarum, N.; Watkins, R.; Wilson, I.A. HMGB1 activates proinflammatory signaling via TLR5 leading to allodynia. Cell Rep. 2016, 17, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front Physiol. 2015, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Bell, C.W.; Pisetsky, D.S. The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J. Immunol. 2007, 178, 6495–6503. [Google Scholar] [CrossRef] [Green Version]
- Tsujita, R.; Tsubota, M.; Hayashi, Y.; Saeki, H.; Sekiguchi, F.; Kawabata, A. Role of thrombin in soluble thrombomodulin-induced suppression of peripheral HMGB1-mediated allodynia in mice. J. Neuroimmune Pharmacol. 2018, 13, 179–188. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; de Carvalho-Barbosa, M.; Kavelaars, A.; Heijnen, C.J.; Albrecht, P.J.; Dougherty, P.M. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy-induced peripheral neuropathy. J. Pain 2016, 17, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Tsujita, R.; Tsubota, M.; Saeki, H.; Sekiguchi, F.; Honda, G. Human soluble thrombomodulin-induced blockade of peripheral HMGB1-dependent allodynia in mice requires both the lectin-like and EGF-like domains. Biochem. Biophys. Res. Commun. 2018, 495, 634–638. [Google Scholar] [CrossRef]
- Zavrski, I.; Jakob, C.; Kaiser, M.; Fleissner, C.; Heider, U.; Sezer, O. Molecular and clinical aspects of proteasome inhibition in the treatment of cancer. Recent Results Cancer Res. 2007, 176, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Barker, N.; Hoon, S.; He, P.; Thakur, T.; Abdeen, S.R.; Maruthappan, P.; Ghadessy, F.J.; Lane, D.P. Bortezomib stabilizes and activates p53 in proliferative compartments of both normal and tumor tissues in vivo. Cancer Res. 2019, 79, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, M.; Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Vassallo, B.; Carlisi, D.; Portanova, P.; Vento, R.; Tesoriere, G. JNK and AP-1 mediate apoptosis induced by bortezomib in HepG2 cells via FasL/caspase-8 and mitochondria-dependent pathways. Apoptosis 2006, 11, 607–625. [Google Scholar] [CrossRef]
- Cai, X.; Biswas, I.; Panicker, S.R.; Giri, H.; Rezaie, A.R. Activated protein C inhibits lipopolysaccharide-mediated acetylation and secretion of high-mobility group box 1 in endothelial cells. J. Thromb. Haemost. 2019, 17, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zeng, Q.; Silverman, H.A.; Gunasekaran, M.; George, S.J.; Devarajan, A.; Addorisio, M.E.; Li, J.; Tsaava, T.; Shah, V.; et al. HMGB1 released from nociceptors mediates inflammation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Wei, J.Y.; Liu, C.C.; Ouyang, H.D.; Ma, C.; Xie, M.X.; Liu, M.; Lei, W.L.; Ding, H.H.; Wu, S.L.; Xin, W.J. Activation of RAGE/STAT3 pathway by methylglyoxal contributes to spinal central sensitization and persistent pain induced by bortezomib. Exp. Neurol. 2017, 296, 74–82. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubota, M.; Miyazaki, T.; Ikeda, Y.; Hayashi, Y.; Aokiba, Y.; Tomita, S.; Sekiguchi, F.; Wang, D.; Nishibori, M.; Kawabata, A. Caspase-Dependent HMGB1 Release from Macrophages Participates in Peripheral Neuropathy Caused by Bortezomib, a Proteasome-Inhibiting Chemotherapeutic Agent, in Mice. Cells 2021, 10, 2550. https://doi.org/10.3390/cells10102550
Tsubota M, Miyazaki T, Ikeda Y, Hayashi Y, Aokiba Y, Tomita S, Sekiguchi F, Wang D, Nishibori M, Kawabata A. Caspase-Dependent HMGB1 Release from Macrophages Participates in Peripheral Neuropathy Caused by Bortezomib, a Proteasome-Inhibiting Chemotherapeutic Agent, in Mice. Cells. 2021; 10(10):2550. https://doi.org/10.3390/cells10102550
Chicago/Turabian StyleTsubota, Maho, Takaya Miyazaki, Yuya Ikeda, Yusuke Hayashi, Yui Aokiba, Shiori Tomita, Fumiko Sekiguchi, Dengli Wang, Masahiro Nishibori, and Atsufumi Kawabata. 2021. "Caspase-Dependent HMGB1 Release from Macrophages Participates in Peripheral Neuropathy Caused by Bortezomib, a Proteasome-Inhibiting Chemotherapeutic Agent, in Mice" Cells 10, no. 10: 2550. https://doi.org/10.3390/cells10102550
APA StyleTsubota, M., Miyazaki, T., Ikeda, Y., Hayashi, Y., Aokiba, Y., Tomita, S., Sekiguchi, F., Wang, D., Nishibori, M., & Kawabata, A. (2021). Caspase-Dependent HMGB1 Release from Macrophages Participates in Peripheral Neuropathy Caused by Bortezomib, a Proteasome-Inhibiting Chemotherapeutic Agent, in Mice. Cells, 10(10), 2550. https://doi.org/10.3390/cells10102550