Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy

 ,
,  and
and 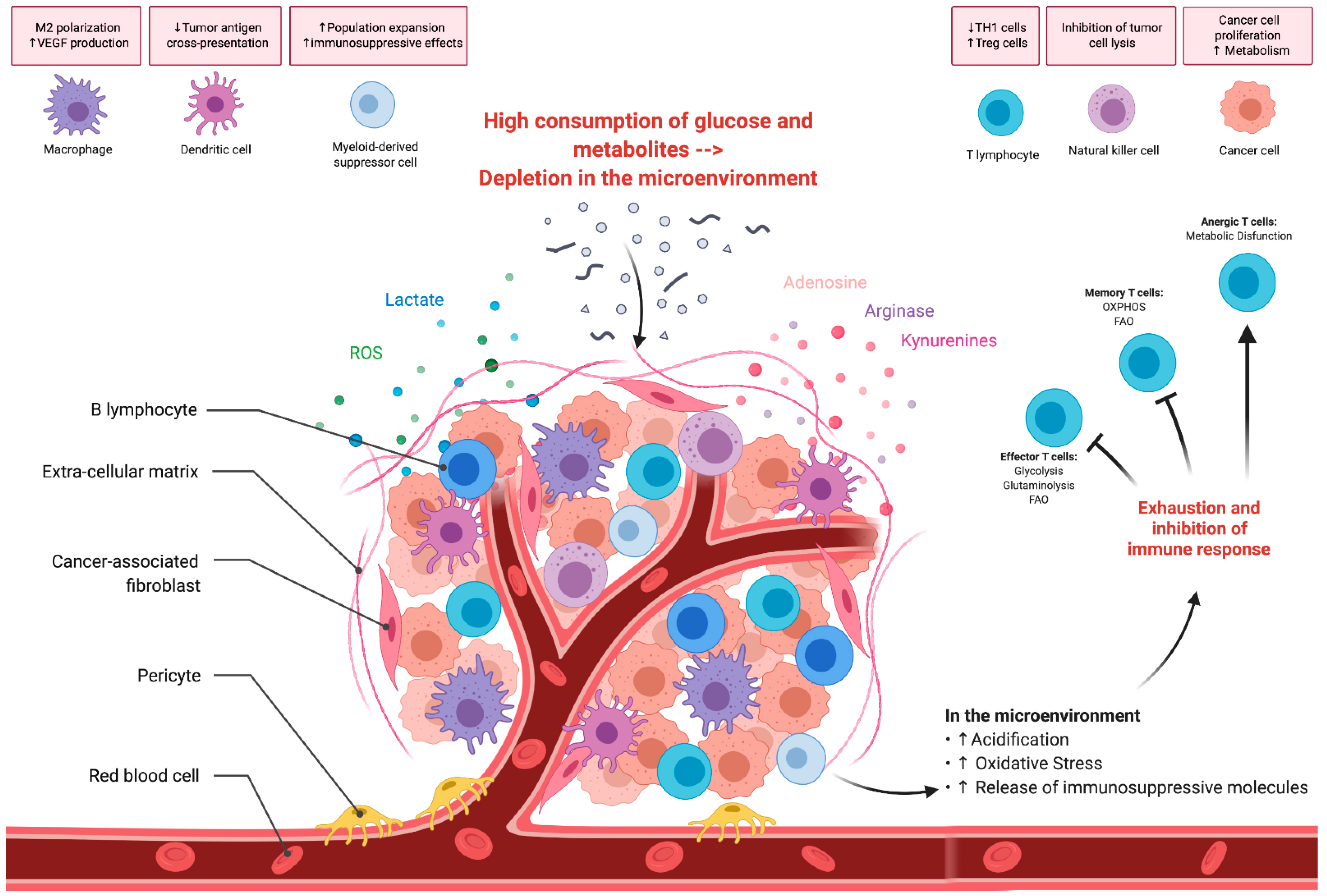{kind=link}
{kind=link}
Abstract
1. Introduction
2. Metabolism: The Energy Engine
3. Effector T Cells and Metabolic Reprogramming
4. Tumor Metabolism and Microenvironment
5. Armoring CAR T-cells: Improving the Intrinsic Anti-Tumor Activity through Improved Metabolic Fitness
5.1. Engineering of the CAR Module: Costimulation as Metabolic Support for T Cells
5.2. Exploiting Transcription Factors and Specific Genes Pathways to Promote Potent Antitumor Activity of CAR T-Cells
6. Pre-Conditioning CAR T-Cells to Increase Their Metabolic Fitness
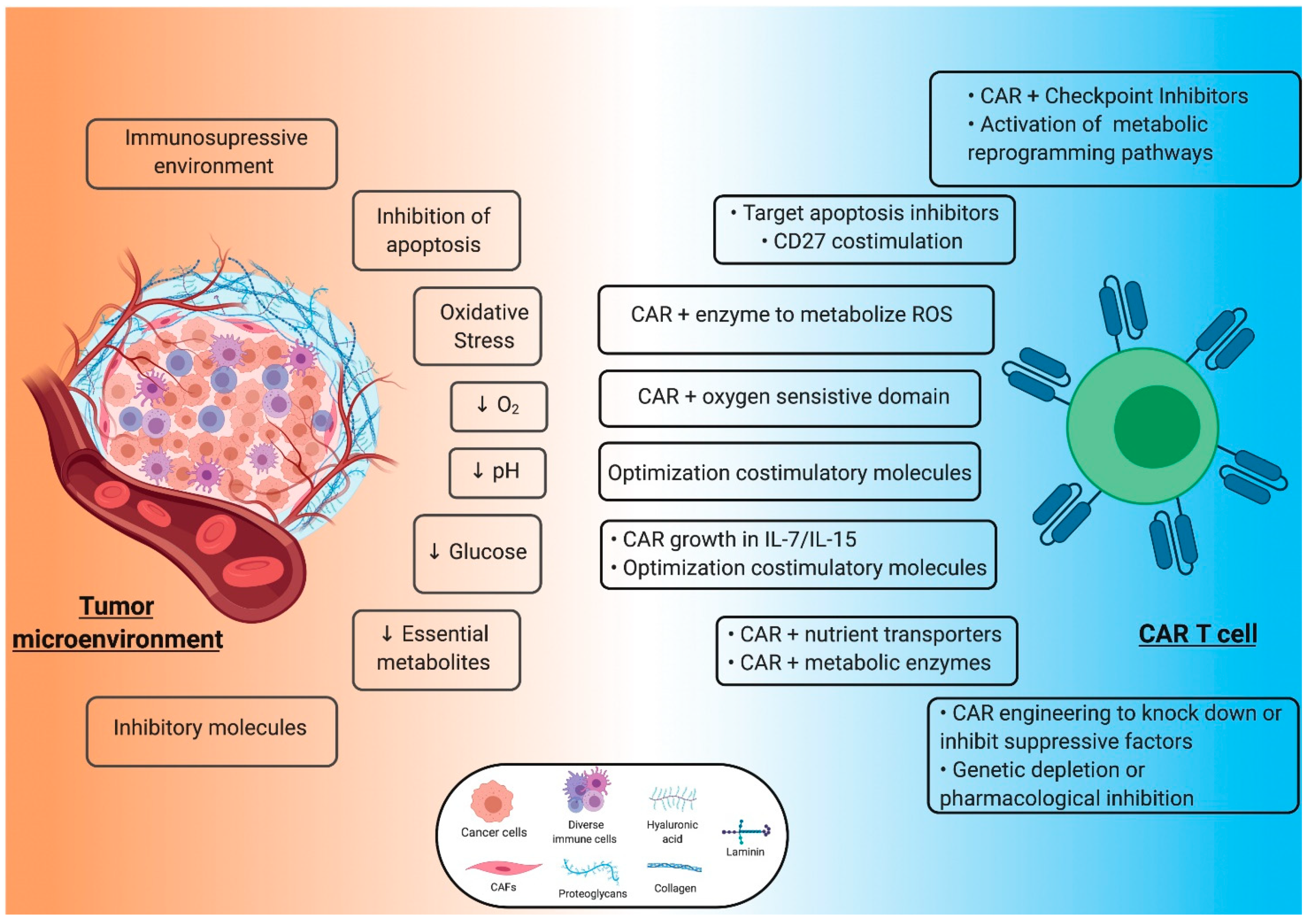
7. Molding the Tumor Microenvironment: Improving Extrinsic Factors to Contrast Adverse TME
7.1. Adapting the Strategy to the Specific Situation
7.2. Checkpoint Blockade and CAR T-Cell Therapy
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Diaconu, I.; Dotti, G. From monoclonal antibodies to chimeric antigen receptors for the treatment of human malignancies. Semin. Oncol. 2014, 41, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef]
- Caruana, I.; Simula, L.; Locatelli, F.; Campello, S. T lymphocytes against solid malignancies: Winning ways to defeat tumours. Cell Stress 2018, 2, 200–212. [Google Scholar] [CrossRef]
- Chang, C.H.; Pearce, E.L. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat. Immunol. 2016, 17, 364–368. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Domblides, C.; Lartigue, L.; Faustin, B. Control of the Antitumor Immune Response by Cancer Metabolism. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T cell metabolism in the tumor microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Dimeloe, S.; Burgener, A.V.; Grahlert, J.; Hess, C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology 2017, 150, 35–44. [Google Scholar] [CrossRef]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu Rev. Immunol 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef]
- Pearce, E.L. Metabolism in T cell activation and differentiation. Curr. Opin. Immunol. 2010, 22, 314–320. [Google Scholar] [CrossRef]
- Ma, E.H.; Verway, M.J.; Johnson, R.M.; Roy, D.G.; Steadman, M.; Hayes, S.; Williams, K.S.; Sheldon, R.D.; Samborska, B.; Kosinski, P.A.; et al. Metabolic Profiling Using Stable Isotope Tracing Reveals Distinct Patterns of Glucose Utilization by Physiologically Activated CD8(+) T Cells. Immunity 2019, 51, 856–870 e855. [Google Scholar] [CrossRef] [PubMed]
- van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Aksoylar, H.I.; Tijaro-Ovalle, N.M.; Boussiotis, V.A.; Patsoukis, N. T Cell Metabolism in Cancer Immunotherapy. Immunometabolism 2020, 2. [Google Scholar] [CrossRef]
- Balkhi, M.Y.; Ma, Q.; Ahmad, S.; Junghans, R.P. T cell exhaustion and Interleukin 2 downregulation. Cytokine 2015, 71, 339–347. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [CrossRef]
- Jiang, B. Aerobic glycolysis and high level of lactate in cancer metabolism and microenvironment. Genes Dis. 2017, 4, 25–27. [Google Scholar] [CrossRef]
- Fukumura, D.; Xu, L.; Chen, Y.; Gohongi, T.; Seed, B.; Jain, R.K. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res. 2001, 61, 6020–6024. [Google Scholar]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: Mechanism of low pH-induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-kappaB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fukuda, R.; Kumar, G.; Krishnamachary, B.; Zeller, K.I.; Dang, C.V.; Semenza, G.L. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Capece, D.; Verzella, D.; Di Francesco, B.; Alesse, E.; Franzoso, G.; Zazzeroni, F. NF-kappaB and mitochondria cross paths in cancer: Mitochondrial metabolism and beyond. Semin. Cell Dev. Biol. 2020, 98, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Rodrigues, V.; Silvestre, R.; Gautier, C.; Weil, R.; Corti, O.; Estaquier, J. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016, 23, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Hong, S.H.; Park, R.K.; Shin, T.; An, N.H.; Kim, H.M. Hypoxia-induced IL-6 production is associated with activation of MAP kinase, HIF-1, and NF-kappaB on HEI-OC1 cells. Hear. Res. 2005, 207, 59–67. [Google Scholar] [CrossRef]
- Meng, X.; Grotsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Simon, M.C. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 2010, 345, 105–120. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Zarour, H.M. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin. Cancer Res. 2016, 22, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Betzler, A.C.; Theodoraki, M.N.; Schuler, P.J.; Doscher, J.; Laban, S.; Hoffmann, T.K.; Brunner, C. NF-kappaB and Its Role in Checkpoint Control. Int. J. Mol. Sci. 2020, 21, 3949. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Zuazo, M.; Gato-Canas, M.; Llorente, N.; Ibanez-Vea, M.; Arasanz, H.; Kochan, G.; Escors, D. Molecular mechanisms of programmed cell death-1 dependent T cell suppression: Relevance for immunotherapy. Ann. Transl. Med. 2017, 5, 385. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Hui, E. Immune checkpoint inhibitors. J. Cell Biol. 2019, 218, 740–741. [Google Scholar] [CrossRef]
- Lee, M.J.; Yun, S.J.; Lee, B.; Jeong, E.; Yoon, G.; Kim, K.; Park, S. Association of TIM-3 expression with glucose metabolism in Jurkat T cells. BMC Immunol. 2020, 21, 48. [Google Scholar] [CrossRef]
- Previte, D.M.; Martins, C.P.; O’Connor, E.C.; Marre, M.L.; Coudriet, G.M.; Beck, N.W.; Menk, A.V.; Wright, R.H.; Tse, H.M.; Delgoffe, G.M.; et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4(+) T Cells. Cell Rep. 2019, 27, 129–141 e124. [Google Scholar] [CrossRef]
- Grohmann, U.; Orabona, C.; Fallarino, F.; Vacca, C.; Calcinaro, F.; Falorni, A.; Candeloro, P.; Belladonna, M.L.; Bianchi, R.; Fioretti, M.C.; et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol. 2002, 3, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8(+) T-cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, P.; Fallarino, F.; Italiano, A.; Soubeyran, I.; MacGrogan, G.; Debled, M.; Velasco, V.; Bodet, D.; Eimer, S.; Veldhoen, M.; et al. Accumulation of an endogenous tryptophan-derived metabolite in colorectal and breast cancers. PLoS ONE 2015, 10, e0122046. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- Murray, P.J. Amino acid auxotrophy as a system of immunological control nodes. Nat. Immunol. 2016, 17, 132–139. [Google Scholar] [CrossRef]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolanos, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Quintarelli, C.; Orlando, D.; Boffa, I.; Guercio, M.; Polito, V.A.; Petretto, A.; Lavarello, C.; Sinibaldi, M.; Weber, G.; Del Bufalo, F.; et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology 2018, 7, e1433518. [Google Scholar] [CrossRef]
- Tsurutani, N.; Mittal, P.; St Rose, M.C.; Ngoi, S.M.; Svedova, J.; Menoret, A.; Treadway, F.B.; Laubenbacher, R.; Suarez-Ramirez, J.E.; Cauley, L.S.; et al. Costimulation Endows Immunotherapeutic CD8 T Cells with IL-36 Responsiveness during Aerobic Glycolysis. J. Immunol. 2016, 196, 124–134. [Google Scholar] [CrossRef]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef]
- Hendriks, J.; Gravestein, L.A.; Tesselaar, K.; van Lier, R.A.; Schumacher, T.N.; Borst, J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 2000, 1, 433–440. [Google Scholar] [CrossRef]
- Peperzak, V.; Veraar, E.A.; Keller, A.M.; Xiao, Y.; Borst, J. The Pim kinase pathway contributes to survival signaling in primed CD8+ T cells upon CD27 costimulation. J. Immunol. 2010, 185, 6670–6678. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Paulos, C.M.; Carpenito, C.; Plesa, G.; Suhoski, M.M.; Varela-Rohena, A.; Golovina, T.N.; Carroll, R.G.; Riley, J.L.; June, C.H. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci. Transl. Med. 2010, 2, 55ra78. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Balic, J.J.; Albargy, H.; Luu, K.; Kirby, F.J.; Jayasekara, W.S.N.; Mansell, F.; Garama, D.J.; De Nardo, D.; Baschuk, N.; Louis, C.; et al. STAT3 serine phosphorylation is required for TLR4 metabolic reprogramming and IL-1beta expression. Nat. Commun. 2020, 11, 3816. [Google Scholar] [CrossRef]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Kondo, T.; Ando, M.; Nagai, N.; Tomisato, W.; Srirat, T.; Liu, B.; Mise-Omata, S.; Ikeda, M.; Chikuma, S.; Nishimasu, H.; et al. The NOTCH-FOXM1 Axis Plays a Key Role in Mitochondrial Biogenesis in the Induction of Human Stem Cell Memory-like CAR-T Cells. Cancer Res. 2020, 80, 471–483. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Oestreich, K.J.; Read, K.A.; Gilbertson, S.E.; Hough, K.P.; McDonald, P.W.; Krishnamoorthy, V.; Weinmann, A.S. Bcl-6 directly represses the gene program of the glycolysis pathway. Nat. Immunol. 2014, 15, 957–964. [Google Scholar] [CrossRef]
- Gacerez, A.T.; Sentman, C.L. T-bet promotes potent antitumor activity of CD4(+) CAR T cells. Cancer Gene 2018, 25, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8(+) T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141 e1125. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef]
- Urak, R.; Walter, M.; Lim, L.; Wong, C.W.; Budde, L.E.; Thomas, S.; Forman, S.J.; Wang, X. Ex vivo Akt inhibition promotes the generation of potent CD19CAR T cells for adoptive immunotherapy. J. Immunother. Cancer 2017, 5, 26. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842 e813. [Google Scholar] [CrossRef] [PubMed]
- Fultang, L.; Booth, S.; Yogev, O.; Martins da Costa, B.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Lynn, R.C.; Sotillo, E.; Lattin, J.; Xu, P.; Mackall, C.L. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019, 3, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia Selectively Impairs CAR-T Cells In Vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef]
- Devadas, S.; Zaritskaya, L.; Rhee, S.G.; Oberley, L.; Williams, M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: Selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002, 195, 59–70. [Google Scholar] [CrossRef]
- Schmielau, J.; Finn, O.J. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. 2001, 61, 4756–4760. [Google Scholar]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Soukupova, J.; Malfettone, A.; Hyrossova, P.; Hernandez-Alvarez, M.I.; Penuelas-Haro, I.; Bertran, E.; Junza, A.; Capellades, J.; Giannelli, G.; Yanes, O.; et al. Role of the Transforming Growth Factor-beta in regulating hepatocellular carcinoma oxidative metabolism. Sci. Rep. 2017, 7, 12486. [Google Scholar] [CrossRef]
- Young, V.J.; Brown, J.K.; Maybin, J.; Saunders, P.T.; Duncan, W.C.; Horne, A.W. Transforming growth factor-beta induced Warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, 3450–3459. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015, 6, 1. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Campello, S.; Lacalle, R.A.; Bettella, M.; Manes, S.; Scorrano, L.; Viola, A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 2006, 203, 2879–2886. [Google Scholar] [CrossRef]
- Simula, L.; Corrado, M.; Accordi, B.; Di Rita, A.; Nazio, F.; Antonucci, Y.; Di Daniele, A.; Caicci, F.; Caruana, I.; Soriano, M.E.; et al. JNK1 and ERK1/2 modulate lymphocyte homeostasis via BIM and DRP1 upon AICD induction. Cell Death Differ. 2020, 27, 2749–2767. [Google Scholar] [CrossRef]
- Mardiana, S.; Solomon, B.J.; Darcy, P.K.; Beavis, P.A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Suarez, E.R.; Chang de, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef] [PubMed]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef]
- Antonia, S.; Goldberg, S.B.; Balmanoukian, A.; Chaft, J.E.; Sanborn, R.E.; Gupta, A.; Narwal, R.; Steele, K.; Gu, Y.; Karakunnel, J.J.; et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016, 17, 299–308. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Rizvi, N.A.; Goldman, J.W.; Gettinger, S.N.; Borghaei, H.; Brahmer, J.R.; Ready, N.E.; Gerber, D.E.; Chow, L.Q.; Juergens, R.A.; et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): Results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017, 18, 31–41. [Google Scholar] [CrossRef]
- Gubens, M.A.; Sequist, L.V.; Stevenson, J.P.; Powell, S.F.; Villaruz, L.C.; Gadgeel, S.M.; Langer, C.J.; Patnaik, A.; Borghaei, H.; Jalal, S.I.; et al. Pembrolizumab in combination with ipilimumab as second-line or later therapy for advanced non-small-cell lung cancer: KEYNOTE-021 cohorts D and H. Lung Cancer 2019, 130, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Winer, A.; Ghatalia, P.; Bubes, N.; Anari, F.; Varshavsky, A.; Kasireddy, V.; Liu, Y.; El-Deiry, W.S. Dual Checkpoint Inhibition with Ipilimumab plus Nivolumab After Progression on Sequential PD-1/PDL-1 Inhibitors Pembrolizumab and Atezolizumab in a Patient with Lynch Syndrome, Metastatic Colon, and Localized Urothelial Cancer. Oncologist 2019, 24, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine Receptor 2A Blockade Increases the Efficacy of Anti-PD-1 through Enhanced Antitumor T-cell Responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrino, M.; Del Bufalo, F.; De Angelis, B.; Quintarelli, C.; Caruana, I.; de Billy, E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells 2021, 10, 14. https://doi.org/10.3390/cells10010014
Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells. 2021; 10(1):14. https://doi.org/10.3390/cells10010014
Chicago/Turabian StylePellegrino, Marsha, Francesca Del Bufalo, Biagio De Angelis, Concetta Quintarelli, Ignazio Caruana, and Emmanuel de Billy. 2021. "Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy" Cells 10, no. 1: 14. https://doi.org/10.3390/cells10010014
APA StylePellegrino, M., Del Bufalo, F., De Angelis, B., Quintarelli, C., Caruana, I., & de Billy, E. (2021). Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells, 10(1), 14. https://doi.org/10.3390/cells10010014