MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine

Abstract
1. Introduction
2. Diverse Distribution of MG53 and Cellular Mechanism of MG53-Mediated Regeneration
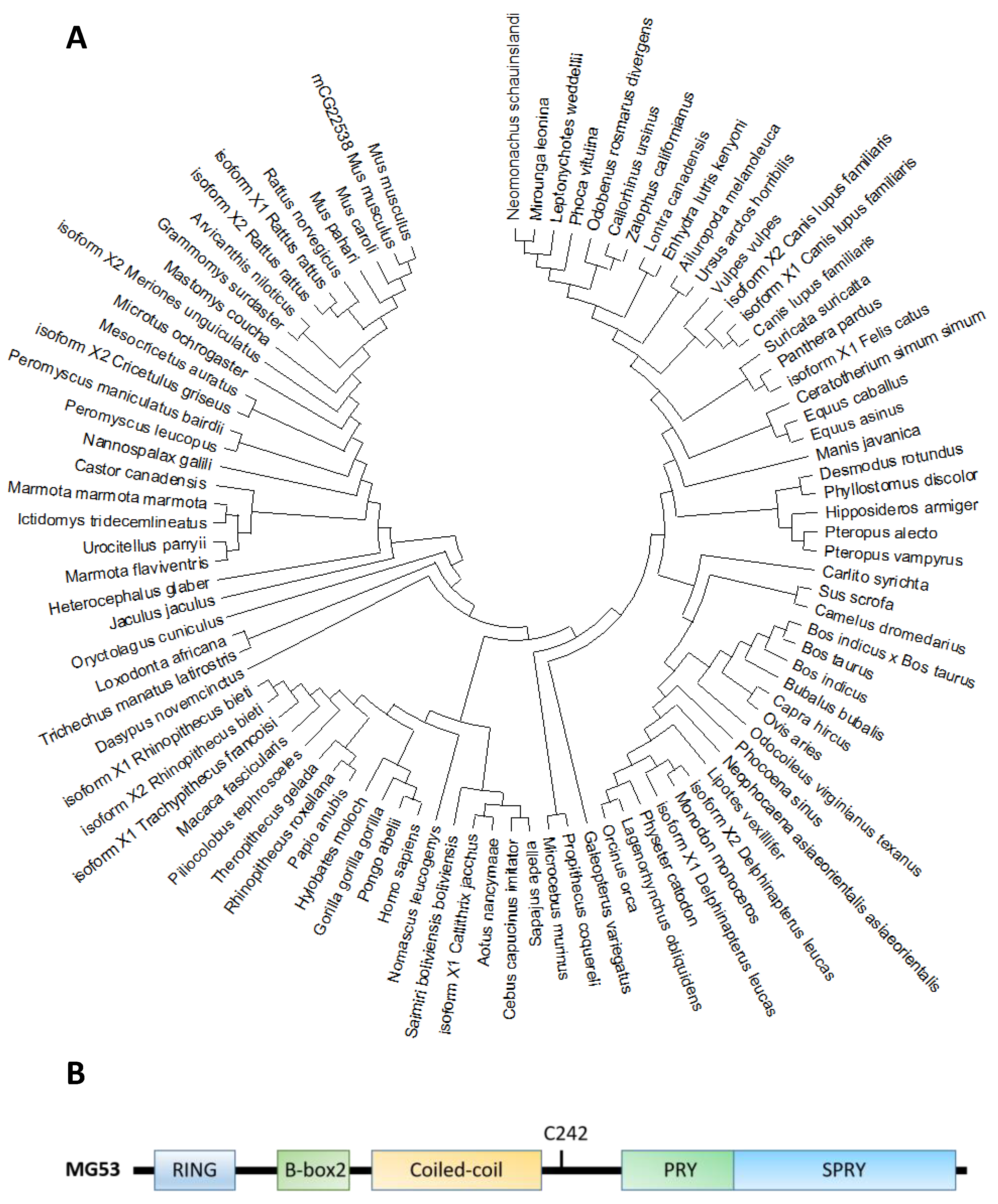
2.1. MG53 Protein Structure, Distribution, and Conservation across Species
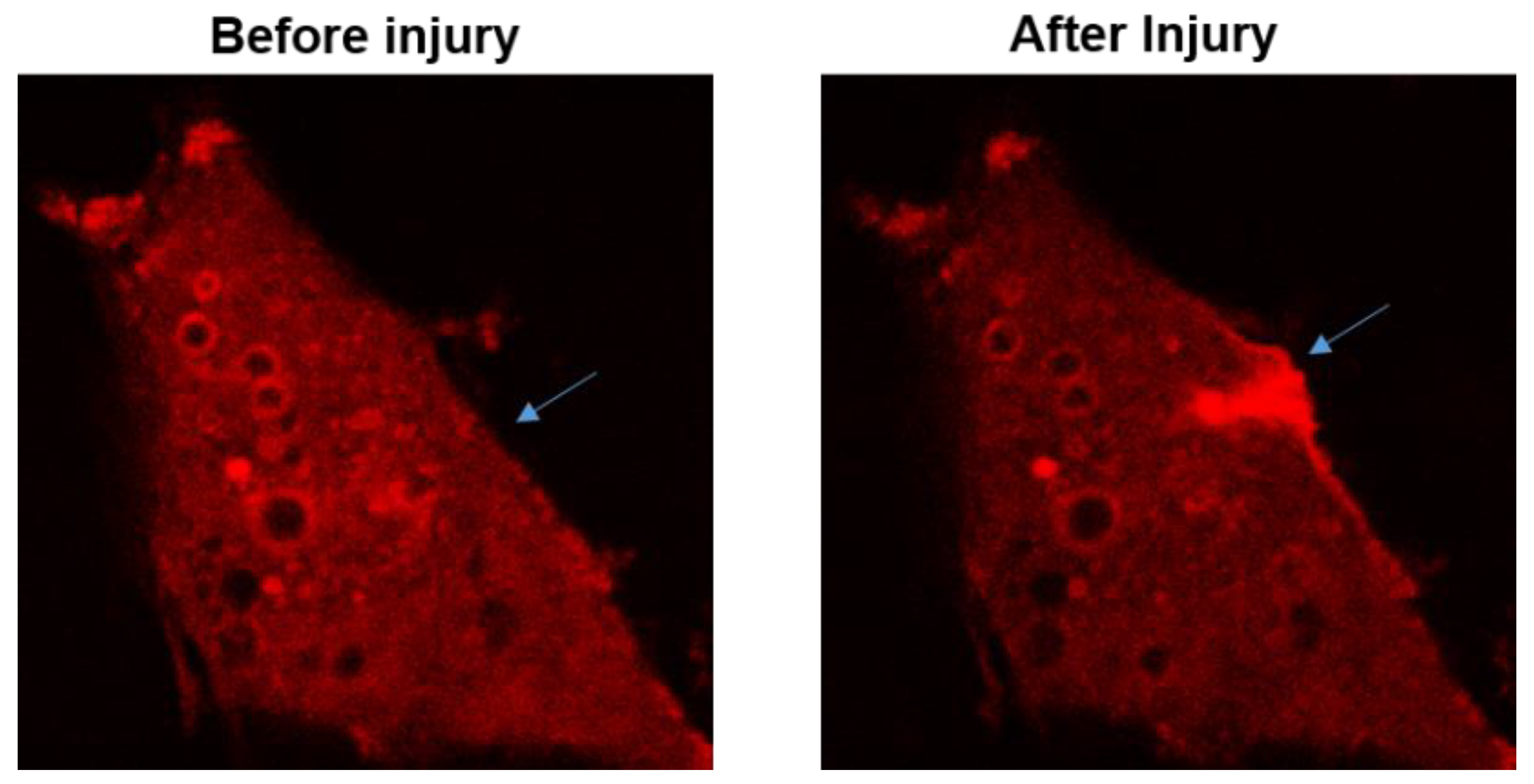
2.2. MG53 and Its Molecular Mechanism in Cell Membrane Repair
3. MG53 in Cardiac Protection against Injury from Ischemia/Reperfusion (I/R)
4. MG53’s Role in Repair and Regeneration of Skeletal Muscle
5. Injury Protection of MG53 in Non-Muscle Organs
5.1. MG53 on Acute Kidney Injury (AKI)
5.2. MG53-Mediated Repair in Wound Healing
5.3. MG53-Mediated Repair in Liver Protection
5.4. MG53-Mediated Repair in Cornea Protection
6. A Potential Role of MG53 in Treatment of Acute Lung Injury
7. Alteration of MG53 Expression Does Not Impact Glucose Metabolism
8. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- McPhail, M.J.; Janus, J.R.; Lott, D.G. Advances in Regenerative Medicine for Otolaryngology/Head and Neck Surgery. BMJ 2020, 369, m718. [Google Scholar] [CrossRef]
- Weisleder, N.; Takeshima, H.; Ma, J. Immuno-proteomic approach to excitation—Contraction coupling in skeletal and cardiac muscle: Molecular insights revealed by the mitsugumins. Cell Calcium 2008, 43, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, F.; Jin, L.; Peng, W.; Song, R.; Ma, J.; Cao, C.M.; Xiao, R.P. MG53 participates in ischaemic postconditioning through the RISK signalling pathway. Cardiovasc. Res. 2011, 91, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra236. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Chen, K.; Lin, P.; Lieber, G.; Nishi, M.; Yan, R.; Wang, Z.; Yao, Y.; Li, Y.; Whitson, B.A.; et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 2014, 5, 4387. [Google Scholar] [CrossRef]
- Chandler, H.L.; Tan, T.; Yang, C.; Gemensky-Metzler, A.J.; Wehrman, R.F.; Jiang, Q.; Peterson, C.; Geng, B.; Zhou, X.; Wang, Q.; et al. MG53 promotes corneal wound healing and mitigates fibrotic remodeling in rodents. Commun. Biol. 2019, 2, 71. [Google Scholar] [CrossRef]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in mus-cular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef]
- Bian, Z.; Wang, Q.; Zhou, X.; Tan, T.; Park, K.H.; Kramer, H.F.; McDougal, A.; Laping, N.J.; Kumar, S.; Adesanya, T.; et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic func-tion. Nat. Commun. 2019, 10, 4659. [Google Scholar] [CrossRef]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombi-nant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra85. [Google Scholar] [CrossRef]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 con-stitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell. Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Kellett, T.; Wang, S.; Nishi, M.; Nagre, N.; Zhou, B.; Flodby, P.; Shilo, K.; Ghadiali, S.N.; Takeshima, H.; et al. TRIM72 is required for effective repair of alveolar epithelial cell wounding. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L449–L459. [Google Scholar] [CrossRef] [PubMed]
- Nagre, N.; Cong, X.; Ji, H.L.; Schreiber, J.M.; Fu, H.; Pepper, I.; Warren, S.; Sill, J.M.; Hubmayr, R.D.; Zhao, X. Inhaled TRIM72 Protein Protects Ventilation Injury to the Lung through Injury-guided Cell Repair. Am. J. Respir. Cell Mol. Biol. 2018, 59, 635–647. [Google Scholar] [CrossRef]
- Liu, C.; Hu, Y.H.; Han, Y.; Wang, Y.B.; Zhang, Y.; Zhang, X.Q.; He, D.F.; Ren, H.M.; Liu, Y.K.; Wang, H.Y.; et al. MG53 pro-tects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis. Acta Pharmacol. Sin. 2020, 41, 1457–1464. [Google Scholar] [CrossRef]
- Wang, X.; Xie, W.; Zhang, Y.; Lin, P.; Han, L.; Han, P.; Wang, Y.; Chen, Z.; Ji, G.; Zheng, M.; et al. Cardioprotection of is-chemia/reperfusion injury by cholesterol-dependent MG53-mediated membrane repair. Circ. Res. 2010, 107, 76–83. [Google Scholar] [CrossRef]
- He, B.; Tang, R.H.; Weisleder, N.; Xiao, B.; Yuan, Z.; Cai, C.; Zhu, H.; Lin, P.; Qiao, C.; Li, J.; et al. Enhancing muscle mem-brane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in δ-Sarcoglycan-deficient hamsters. Mol. Ther. 2012, 20, 727–735. [Google Scholar] [CrossRef]
- Wu, Y.; Huang, J.; Liu, D.; Tan, J.; Peng, Y.; Yang, J.; Cui, Y.; He, W.; Luo, G.; Wu, J. Mitsugumin 53 protects the kidney from severe burn injury in mice. Burn. Trauma 2013, 1, 128–133. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474–22485. [Google Scholar] [CrossRef]
- Yao, W.; Li, H.; Han, X.; Chen, C.; Zhang, Y.; Tai, W.L.; Xia, Z.; Hei, Z. MG53 anchored by dysferlin to cell membrane reduc-es hepatocyte apoptosis which induced by ischaemia/reperfusion injury in vivo and in vitro. J. Cell. Mol. Med. 2017, 21, 2503–2513. [Google Scholar] [CrossRef]
- Li, H.; Duann, P.; Lin, P.H.; Zhao, L.; Fan, Z.; Tan, T.; Zhou, X.; Sun, M.; Fu, M.; Orange, M.; et al. Modulation of Wound Healing and Scar Formation by MG53 Protein-mediated Cell Membrane Repair. J. Biol. Chem. 2015, 290, 24592–24603. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.K.; Zhang, Y.; Cao, C.M.; Hu, X.; Fang, M.; Yao, Y.; Jin, L.; Chen, G.; Jiang, P.; Zhang, S.; et al. Glucose-Sensitive My-okine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019, 139, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Guo, S.; Wu, H.K.; Lv, F.; Jin, L.; Zhang, M.; Xie, P.; Wang, Y.; Song, Y.; Wu, F.; et al. Cardiac Ischemic Precondi-tioning Promotes MG53 Secretion Through H2O2-Activated PKC-δ Signaling. Circulation 2020, 142, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Bian, Z.; Jiang, Q.; Wang, X.; Zhou, X.; Park, K.H.; Hsueh, W.; Whitson, B.A.; Haggard, E.; Li, H.; et al. MG53 Does Not Manifest the Development of Diabetes in db/db Mice. Diabetes 2020, 69, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Gushchina, L.V.; Kwiatkowski, T.A.; Bhattacharya, S.; Weisleder, N.L. Conserved structural and functional aspects of the tripartite motif gene family point towards therapeutic applications in multiple diseases. Pharmacol. Ther. 2018, 185, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Meroni, G. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer for-mation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, H.K.; Lv, F.; Xiao, R.P. MG53: Biological Function and Potential as a Therapeutic Target. Mol. Pharmacol. 2017, 92, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.; Ko, Y.G.; Ma, J. Dual function of MG53 in membrane repair and insulin signaling. BMB Rep. 2016, 49, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Cai, C.; Lin, P.; Zhu, H.; Ko, J.K.; Hwang, M.; Tan, T.; Pan, Z.; Korichneva, I.; Ma, J. Zinc Binding to MG53 Protein Facilitates Repair of Injury to Cell Membranes. J. Biol. Chem. 2015, 290, 13830–13839. [Google Scholar] [CrossRef]
- Hwang, M.; Ko, J.K.; Weisleder, N.; Takeshima, H.; Ma, J. Redox-dependent oligomerization through a leucine zipper motif is essential for MG53-mediated cell membrane repair. Am. J. Physiol. Cell Physiol. 2011, 301, C106–C114. [Google Scholar] [CrossRef]
- Park, E.Y.; Kwon, O.B.; Jeong, B.C.; Yi, J.S.; Lee, C.S.; Ko, Y.G.; Song, H.K. Crystal structure of PRY-SPRY domain of human TRIM72. Proteins Struct. Funct. Bioinform. 2010, 78, 790–795. [Google Scholar] [CrossRef]
- McNeil, P.L.; Khakee, R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992, 140, 1097–1109. [Google Scholar]
- Bansal, D.; Campbell, K.P. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004, 14, 206–213. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 1995, 131, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef] [PubMed]
- Alloush, J.; Weisleder, N. TRIM Proteins in Therapeutic Membrane Repair of Muscular Dystrophy. JAMA Neurol. 2013, 70, 928–931. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P. Membrane repair redux: Redox of MG53. Nat. Cell Biol. 2009, 11, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Seo, J.; Ko, Y.G.; Huh, Y.D.; Park, H. Lipid-binding properties of TRIM72. BMB Rep. 2012, 45, 26–31. [Google Scholar] [CrossRef]
- Zhu, H.; Lin, P.; De, G.; Choi, K.H.; Takeshima, H.; Weisleder, N.; Ma, J. Polymerase transcriptase release factor (PTRF) an-chors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 2011, 286, 12820–12824. [Google Scholar] [CrossRef]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 Regulates Membrane Budding and Exocytosis in Muscle Cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef]
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tu-bules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Middel, V.; Reischl, M.; Strähle, U.; Nienhaus, G.U. Distinct amino acid motifs carrying multiple positive charges regulate membrane targeting of dysferlin and MG53. PLoS ONE 2018, 13, e0202052. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata-Endo, H.; Kato, J.; Tonouchi, A.; Chung, Y.W.; Sun, J.; Stevens, L.A.; Zhu, J.; Aponte, A.M.; Springer, D.A.; San, H.; et al. Role of a TRIM72 ADP-ribosylation cycle in myocardial injury and membrane repair. JCI Insight 2018, 3, 97898. [Google Scholar] [CrossRef] [PubMed]
- Kohr, M.J.; Evangelista, A.M.; Ferlito, M.; Steenbergen, C.; Murphy, E. S-nitrosylation of TRIM72 at cysteine 144 is critical for protection against oxidation-induced protein degradation and cell death. J. Mol. Cell. Cardiol. 2014, 69, 67–74. [Google Scholar] [CrossRef]
- Fillmore, N.; Casin, K.M.; Sinha, P.; Sun, J.; Ma, H.; Boylston, J.; Noguchi, A.; Liu, C.; Wang, N.; Zhou, G.; et al. A knock-in mutation at cysteine 144 of TRIM72 is cardioprotective and reduces myocardial TRIM72 release. J. Mol. Cell. Cardiol. 2019, 136, 95–101. [Google Scholar] [CrossRef]
- Opie, L.H. Reperfusion injury and its pharmacologic modification. Circulation 1989, 80, 1049–1062. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Piper, H.M. Lethal reperfusion injury in acute myocardial infarction: Facts and unre-solved issues. Cardiovasc. Res. 2009, 83, 165–168. [Google Scholar] [CrossRef]
- Heusch, G. Postconditioning: Old wine in a new bottle? J. Am. Coll. Cardiol. 2004, 44, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Lemckert, F.A.; Bournazos, A.; Eckert, D.M.; Kenzler, M.; Hawkes, J.M.; Butler, T.L.; Ceely, B.; North, K.N.; Winlaw, D.S.; Egan, J.R.; et al. Lack of MG53 in human heart precludes utility as a biomarker of myocardial injury or endogenous cardio-protective factor. Cardiovasc. Res. 2016, 110, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.; Brown, R.H., Jr. Dysferlin in Membrane Trafficking and Patch Repair. Traffic 2007, 8, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Towler, M.C.; Kaufman, S.J.; Brodsky, F.M. Membrane Traffic in Skeletal Muscle. Traffic 2004, 5, 129–139. [Google Scholar] [CrossRef]
- Klinge, L.; Laval, S.; Keers, S.; Haldane, F.; Straub, V.; Barresi, R.; Bushby, K. From T-tubule to sarcolemma: Damage-induced dysferlin translocation in early myogenesis. FASEB J. 2007, 21, 1768–1776. [Google Scholar] [CrossRef]
- Sakamoto, A.; Ono, K.; Abe, M.; Jasmin, G.; Eki, T.; Murakami, Y.; Masaki, T.; Toyo-Oka, T.; Hanaoka, F. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: An animal model of disrupted dystrophin-associated glycoprotein complex. Proc. Natl. Acad. Sci. USA 1997, 94, 13873–13878. [Google Scholar] [CrossRef]
- Zhu, H.; Hou, J.; Roe, J.L.; Park, K.H.; Tan, T.; Zheng, Y.; Li, L.; Zhang, C.; Liu, J.; Liu, Z.; et al. Amelioration of ischemia-reperfusion-induced muscle injury by the recombinant human MG53 protein. Muscle Nerve 2015, 52, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, H.; Wu, D.; Hu, J.; Wu, W.; Zhang, Y.; Peng, X. A novel perspective for burn-induced myopathy: Mem-brane repair defect. Sci. Rep. 2016, 6, 31409. [Google Scholar] [CrossRef] [PubMed]
- Gushchina, L.V.; Bhattacharya, S.; McElhanon, K.E.; Choi, J.H.; Manring, H.; Beck, E.X.; Alloush, J.; Weisleder, N. Treatment with Recombinant Human MG53 Protein Increases Membrane Integrity in a Mouse Model of Limb Girdle Muscular Dystro-phy 2B. Mol. Ther. 2017, 25, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Nagre, N.; Herrera, J.; Pearson, A.C.; Pepper, I.; Morehouse, R.; Ji, H.L.; Jiang, D.; Hubmayr, R.D.; Zhao, X. TRIM72 promotes alveolar epithelial cell membrane repair and ameliorates lung fibrosis. Respir. Res. 2020, 21, 132. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, H.; Li, X.; Zhu, H.; Xu, Z.; Liu, L.; Ma, J.; Zhang, M. A Bioinspired Alginate-Gum Arabic Hydrogel with Micro-/Nanoscale Structures for Controlled Drug Release in Chronic Wound Healing. ACS Appl. Mater. Interfaces 2017, 9, 22160–22175. [Google Scholar] [CrossRef]
- Sermersheim, M.; Kenney, A.D.; Lin, P.H.; McMichael, T.M.; Cai, C.; Gumpper, K.; Adesanya, T.; Li, H.; Zhou, X.; Park, K.H.; et al. MG53 suppresses interferon-β and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat. Commun. 2020, 11, 3624. [Google Scholar] [CrossRef]
- Serracino-Inglott, F.; Habib, N.A.; Mathie, R.T. Hepatic ischemia-reperfusion injury. Am. J. Surg. 2001, 181, 160–166. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, M.; Xie, H.Y.; Zhou, L.; Meng, X.Q.; Shi, J.; Zheng, S. Role of Reactive Oxygen Species in Mediating Hepatic Ischemia-Reperfusion Injury and Its Therapeutic Applications in Liver Transplantation. Transplant. Proc. 2007, 39, 1332–1337. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.M.; Griffiths, M.; McAuley, D. Treatment of Acute Lung Injury: Current and Emerging Pharmacological Therapies. Semin. Respir. Crit. Care Med. 2013, 34, 487–498. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Kenney, A.D.; Li, Z.; Bian, Z.; Zhou, X.; Li, H.; Whitson, B.A.; Tan, T.; Cai, C.; Ma, J.; Yount, J.S. Recombinant MG53 Protein Protects Mice from Lethal Influenza Virus Infection. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Levraud, J.P.; Boudinot, P. Fish antiviral tripartite motif (TRIM) proteins. Fish Shellfish Immunol. 2019, 86, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef]
- Ohmine, S.; Sakuma, R.; Sakuma, T.; Thatava, T.; Takeuchi, H.; Ikeda, Y. The Antiviral Spectra of TRIM5α Orthologues and Human TRIM Family Proteins against Lentiviral Production. PLoS ONE 2011, 6, e16121. [Google Scholar] [CrossRef]
- Van Gent, M.; Sparrer, K.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by in-flammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Zhu, H.; Hsueh, W.; Whitson, B.A. Letter by Zhu et al Regarding Article, “Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis”. Circulation 2019, 140, e186–e187. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tissues | Phenotype of mg53−/− Mice | Therapeutic Application of rhMG53 Protein |
|---|---|---|
| SkeletalMuscle | muscle repair defect [3] reduced exercise capability [3,8] | muscle injury associated with exercise [3,9], muscular dystrophy [10] |
| Heart | defective cardiomyocyte repair [4,11] increased vulnerability to myocardial infarction | acute myocardial injury [12] chronic heart failure |
| Lung | defective alveolar structure and exacerbated lung injury under stress conditions [6] | acute lung injury, sepsis, chronic obstructive pulmonary disease [6,13,14] |
| Kidney | proximal tubular pathology [5] increased susceptibility to acute kidney injury | acute kidney injury [5] nephrotoxicity [15], chronic kidney disease |
| Cornea | reduced re-epithelialization and increased fibrosis following alkaline injury [7] | cornea injury and ulceration [7] prevention of cornea fibrosis |
| MG53 Species | Gene ID | Amino Acid (aa) | Identities (%) | Identities (%) | Protein Structure | ||
|---|---|---|---|---|---|---|---|
| RING | B-Box | SPRY | |||||
| Homo Sapiens | NP_001008275.2 | 477 | 100 | 100 | 100 | 100 |  |
| Pan Troglodytes | XP_001157628.2 | 477 | 100 | 100 | 100 | 100 |  |
| Macaca Mulatta (Isoform 2) | XP_001112866.1 | 477 | 98.95 | 100 | 100 | 97.93 |  |
| Canis Lupus Familiaris (Isoform X2) | XP_005621293.1 | 477 | 93.50 | 95.74 | 100 | 89.12 |  |
| Bos Taurus | XP_002698119.1 | 482 | 94.71 | 95.74 | 100 | 90.16 |  |
| Mus Musculus | NP_001073401.1 | 477 | 91.19 | 95.74 | 97.56 | 87.56 |  |
| Rattus Norvegicus | NP_001071143.1 | 477 | 90.78 | 93.62 | 97.56 | 86.53 |  |
| Xenopus Tropicalis | NP_001008188.1 | 477 | 59.14 | 53.19 | 72.50 | 62.63 |  |
 RING finger
RING finger  B-box zinc finger
B-box zinc finger  B-Box-type zinc finger
B-Box-type zinc finger  SPRY domain
SPRY domain  Poly(hydroxyalcanoate) granule associated protein (phasin)
Poly(hydroxyalcanoate) granule associated protein (phasin)  SPRY-associated domain (PRY).
SPRY-associated domain (PRY).Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Wang, L.; Yue, H.; Whitson, B.A.; Haggard, E.; Xu, X.; Ma, J. MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine. Cells 2021, 10, 122. https://doi.org/10.3390/cells10010122
Li Z, Wang L, Yue H, Whitson BA, Haggard E, Xu X, Ma J. MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine. Cells. 2021; 10(1):122. https://doi.org/10.3390/cells10010122
Chicago/Turabian StyleLi, Zhongguang, Liyang Wang, Huimin Yue, Bryan A. Whitson, Erin Haggard, Xuehong Xu, and Jianjie Ma. 2021. "MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine" Cells 10, no. 1: 122. https://doi.org/10.3390/cells10010122
APA StyleLi, Z., Wang, L., Yue, H., Whitson, B. A., Haggard, E., Xu, X., & Ma, J. (2021). MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine. Cells, 10(1), 122. https://doi.org/10.3390/cells10010122