Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
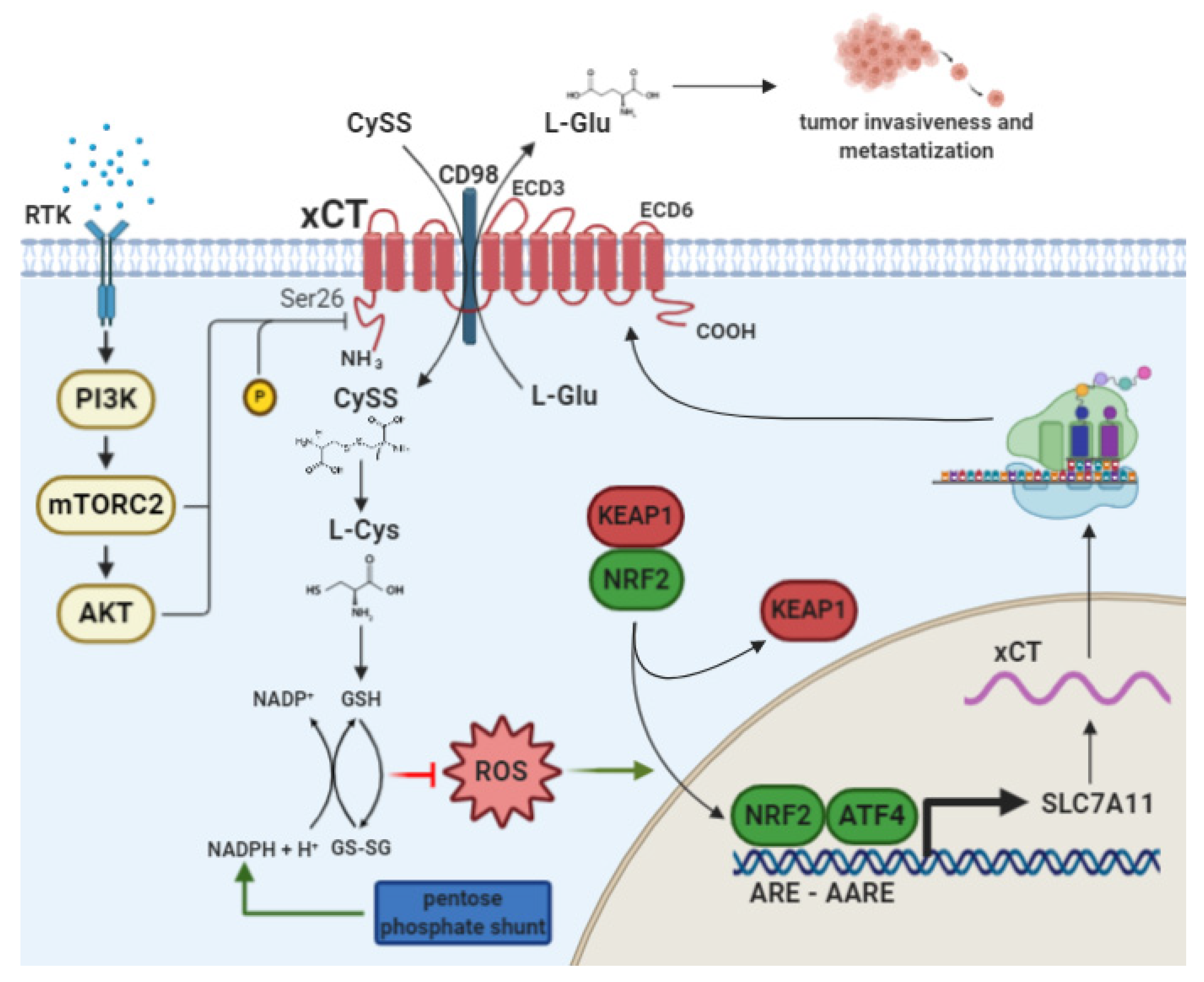
2. The Cystine/Glutamate Antiporter xCT
2.1. xCT Transcriptional Regulation by NRF2 and ATF4
2.2. xCT as a Player in Cancer Cell Metabolism and Tumor Progression
2.3. xCT as a Player in Chemoresistance
2.4. xCT as an Advocate for CSCs
2.5. xCT Targeting as an Anticancer Therapeutic Strategy
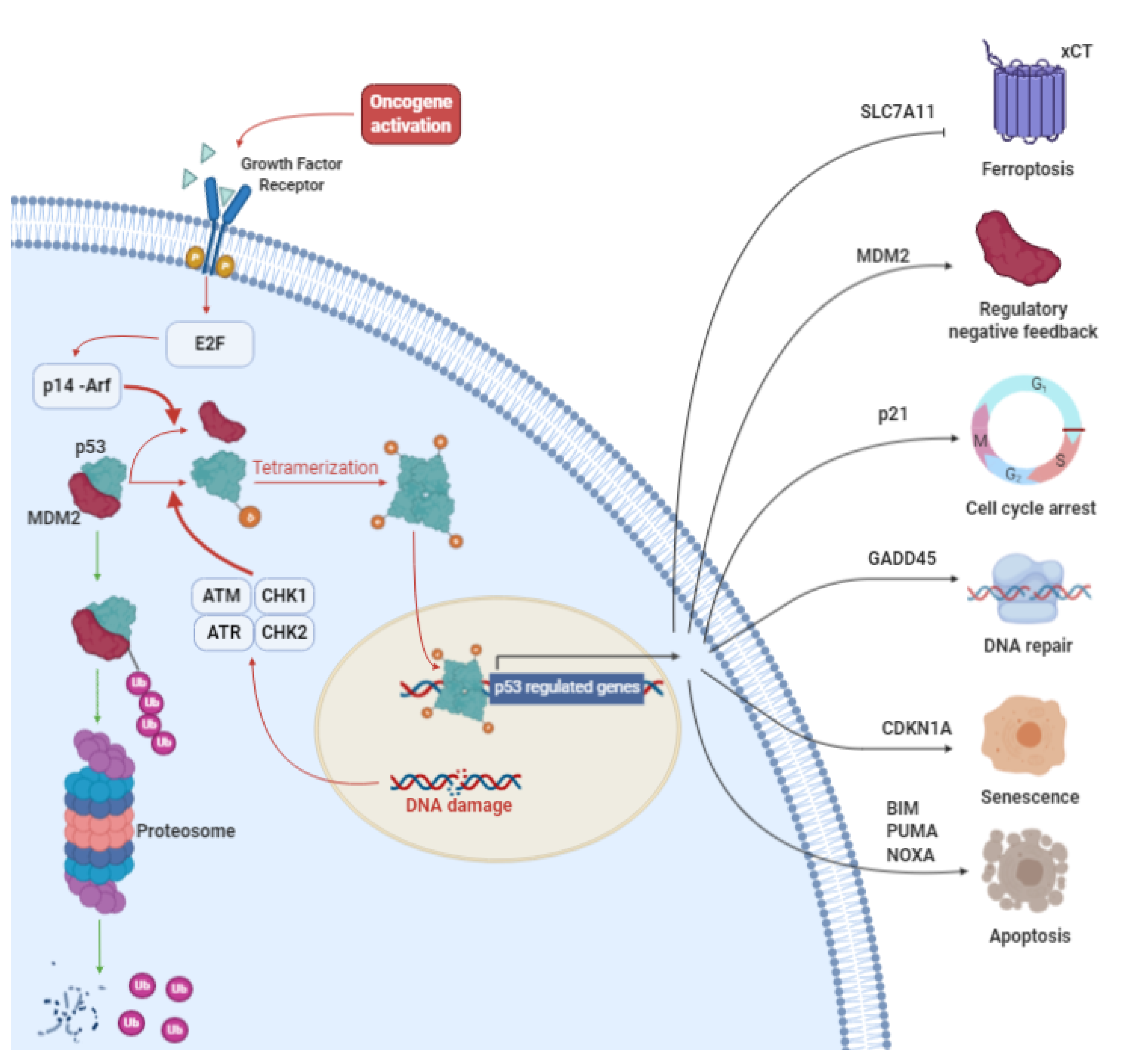
3. p53: More than a Genome Guardian
3.1. p53 in Cancer
3.2. Principles of p53 Targeted Therapy
3.3. PRIMA-1 and Its Analogues
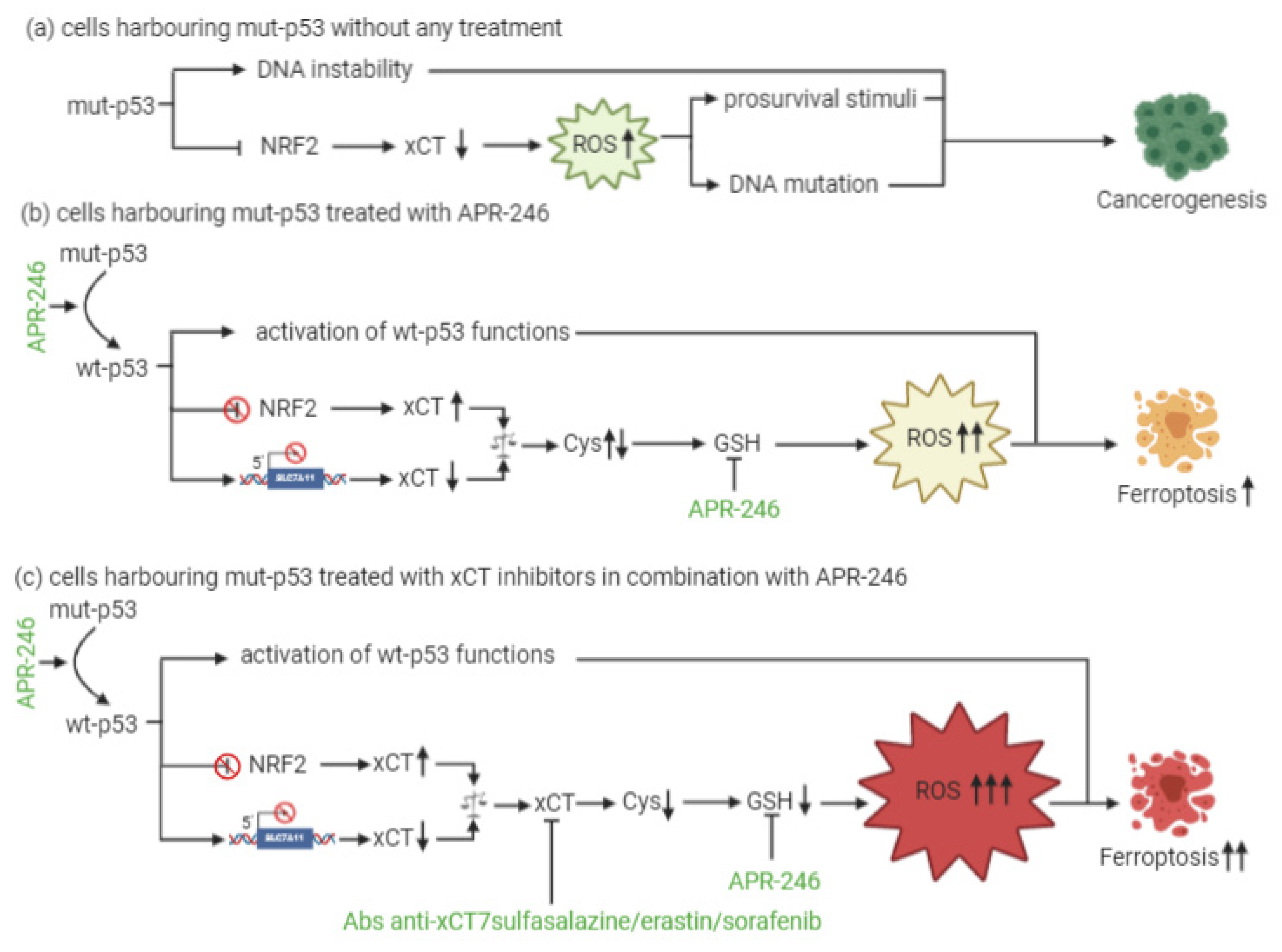
4. xCT and p53 Interplay in the Regulation of Ferroptosis
5. xCT Targeting and APR-246 Administration: A New Therapeutic Combinatorial Strategy
6. Additional Potential Targets for Combined Therapies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Li, Q.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, H.; Huang, C.; Lei, Y. Cancer drug resistance: Redox resetting renders a way. Oncotarget 2016, 7, 42740–42761. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Vander, H.M.G.; Berardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Ruiu, R.; Rolih, V.; Bolli, E.; Barutello, G.; Riccardo, F.; Quaglino, E.; Merighi, I.F.; Pericle, F.; Donofrio, G.; Cavallo, F.; et al. Fighting breast cancer stem cells through the immune-targeting of the xCT cystine–glutamate antiporter. Cancer Immunol. Immunother. 2019, 68, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Lien, E.C.; Ghisolfi, L.; Geck, R.C.; Asara, J.M.; Toker, A. Oncogenic PI3K promotes methionine dependency in breast cancer cells through the cystine-glutamate antiporter xCT. Sci. Signal. 2017, 10, 6604. [Google Scholar] [CrossRef]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione Biosynthesis. BBA-Gen. Subj. 2012, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Publ. Gr. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Sasaki, H.; Sato, H.; Kuriyama, M.K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Nomura, S.; Maebara, K.; Sato, K.; Tamba, M.; Bannai, S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem. Biophys. Res. Commun. 2004, 325, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Mimura, J.; Okada, T.; Sato, H.; Liu, T.; Maruyama, A.; Ohyama, C.; Itoh, K. Nrf2- and ATF4-Dependent Upregulation of xCT Modulates the Sensitivity of T24 Bladder Carcinoma Cells to Proteasome Inhibition. Mol. Cell. Biol. 2014, 34, 3421–3434. [Google Scholar] [CrossRef]
- Habib, E.; Linher, M.K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc- are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ben, S.I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Chen, D.; Fan, Z.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene 2017, 36, 5593–5608. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef]
- Dornier, E.; Rabas, N.; Mitchell, L.; Novo, D.; Dhayade, S.; Marco, S.; MacKay, G.; Sumpton, D.; Pallares, M.; Nixon, C.; et al. Glutaminolysis drives membrane trafficking to promote invasiveness of breast cancer cells. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Chen, R.; Song, Y.; Zhou, Z.; Tong, T.; Li, Y.; Fu, M.; Guo, X.; Dong, L.; He, X.; Qiao, H.; et al. Disruption of xCT inhibits cancer cell metastasis via the caveolin-1 /beta-catenin pathway. Oncogene 2009, 28, 599–609. [Google Scholar] [CrossRef]
- Polewski, M.D.; Reveron, T.R.F.; Cherryholmes, G.A.; Marinov, G.K.; Cassady, K.; Aboody, K.S. Increased expression of system xc- in glioblastoma confers an altered metabolic state and temozolomide resistance. Mol. Cancer Res. 2016, 14, 1229–1242. [Google Scholar] [CrossRef]
- Goji, T.; Takahara, K.; Negishi, M.; Katoh, H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 2017, 292, 19721–19732. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Liu, X.; Olszewski, K.; Zhang, Y.; Lim, E.W.; Shi, J.; Zhang, X.; Zhang, J.; Lee, H.; Koppula, P.; Lei, G.; et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020, 22, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. (Lond. Engl.) 2018, 38, 12. [Google Scholar] [CrossRef]
- Dai, Z.; Huang, Y.; Sadee, W.; Blower, P. Chemoinformatics Analysis Identifies Cytotoxic Compounds Susceptible to Chemoresistance Mediated by Glutathione and Cystine/Glutamate Transport System xc. J. Med. Chem. 2007, 10, 1896–1906. [Google Scholar] [CrossRef]
- Narang, V.S. Sulfasalazine-Induced Reduction of Glutathione Levels in Breast Cancer Cells: Enhancement of Growth-Inhibitory Activity of Doxorubicin. Chemotherapy 2007, 53, 210–217. [Google Scholar] [CrossRef]
- Wada, F.; Koga, H.; Akiba, J.; Niizeki, T.; Iwamoto, H.; Ikezono, Y.; Nakamura, T.; Abe, M.; Masuda, A.; Sakaue, T.; et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci. 2018, 109, 2801–2810. [Google Scholar] [CrossRef]
- Sehm, T.; Rauh, M.; Wiendieck, K.; Buchfelder, M.; Iiker, Y.; Savaskan, N.E. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 2016, 7, 74630. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Kuşoğlu, A.; Biray Avcı, Ç. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, E.; Conti, L.; Cavallo, F. Breast cancer stem cell antigens as targets for immunotherapy. Semin. Immunol. 2020, 47, 101386. [Google Scholar] [CrossRef]
- Ruiu, R.; Tarone, L.; Rolih, V.; Barutello, G.; Bolli, E.; Riccardo, F.; Cavallo, F.; Conti, L. Cancer Stem Cell Immunology and Immunotherapy: Harnessing the Immune System against Cancer’s Source, 1st ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 164, ISBN 9780128165751. [Google Scholar]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res. 2016, 76, 1–12. [Google Scholar] [CrossRef]
- Conti, L.; Lanzardo, S.; Arigoni, M.; Antonazzo, R.; Radaelli, E.; Cantarella, D.; Calogero, R.A.; Cavallo, F. The noninflammatory role of high mobility group box 1/toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. 2013, 27, 4731–4744. [Google Scholar] [CrossRef]
- Polewski, M.D.; Reveron, T.R.F.; Cherryholmes, G.A.; Marinov, G.K.; Aboody, K.S. SLC7A11 Overexpression in Glioblastoma Is Associated with Increased Cancer Stem Cell-Like Properties. Stem Cells Dev. 2017, 26, 1236–1246. [Google Scholar] [CrossRef]
- Horibe, S.; Kawauchi, S.; Tanahashi, T.; Sasaki, N.; Mizuno, S.; Rikitake, Y. CD44v-dependent upregulation of xCT is involved in the acquisition of cisplatin-resistance in human lung cancer A549cells. Biochem. Biophys. Res. Commun. 2018, 507, 426–432. [Google Scholar] [CrossRef]
- Gout, P.W.; Buckley, A.R.; Simms, C.R.; Bruchovsky, N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x-c cystine transporter: A new action for an old drug. Leukemia 2001, 15, 1633–1640. [Google Scholar] [CrossRef]
- Wahl, C.; Liptay, S.; Adler, G.; Schmid, R.M. Sulfasalazine: A potent and specific inhibitor of nuclear factor kappa B. J. Clin. Investig. 1998, 101, 1163–1174. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Holton, T.; Yuneva, M.; Louie, R.J.; Padró, M.; Daemen, A.; Hu, M.; Chan, D.A.; Ethier, S.P.; Veer, L.J.; et al. Glutamine Sensitivity Analysis Identifies the xCT Antiporter as a Common Triple-Negative Breast Tumor Therapeutic Target. Cancer Cell 2013, 24, 450–465. [Google Scholar] [CrossRef] [PubMed]
- FDA Sulfasalazine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/007073s128lbl.pdf (accessed on 24 December 2020).
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.; Welsch, M.; Skouta, R.; Lee, E.; Hayano, M.; Thomas, A.G.; Gleason, C.; Tatonetti, N.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 2014, 1–25. [Google Scholar] [CrossRef]
- Yagoda, N.; Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Larraufie, M.H.; Yang, W.S.; Jiang, E.; Thomas, A.G.; Slusher, B.S.; Stockwell, B.R. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorganic Med. Chem. Lett. 2015, 25, 4787–4792. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, H.; Daniels, J.D.; Zandkarimi, F.; Liu, H.; Brown, L.M.; Uchida, K.; Connor, O.A.; Stockwell, B.R. Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem. Biol. 2019, 26, 623–633. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar]
- FDA Sorafenib. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021923s020lbl.pdf (accessed on 23 December 2020).
- Yoshioka, R.; Fujieda, Y.; Suzuki, Y.; Kanno, O.; Nagahira, A.; Honda, T.; Murakawa, M.; Yukiura, H. Novel mouse model for evaluating in vivo efficacy of xCT inhibitor. J. Pharmacol. Sci. 2019, 140, 242–247. [Google Scholar] [CrossRef]
- Donofrio, G.; Tebaldi, G.; Lanzardo, S.; Ruiu, R.; Bolli, E.; Ballatore, A.; Rolih, V.; Macchi, F.; Conti, L.; Cavallo, F. Bovine herpesvirus 4-based vector delivering the full length xCT DNA efficiently protects mice from mammary cancer metastases by targeting cancer stem cells. Oncoimmunology 2018, 7, 1–16. [Google Scholar] [CrossRef]
- Quaglino, E.; Cavallo, F.; Conti, L. Cancer stem cell antigens as targets for new combined anti-cancer therapies. Int. J. Biochem. Cell Biol. 2020, 129, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bolli, E.; Rourke, J.P.; Conti, L.; Lanzardo, S.; Rolih, V.; Christen, J.M.; Barutello, G.; Forni, M.; Pericle, F.; Cavallo, F. A Virus-Like-Particle immunotherapy targeting Epitope-Specific anti-xCT expressed on cancer stem cell inhibits the progression of metastatic cancer in vivo. Oncoimmunology 2018, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Bolli, E.; Lorenzo, A.; Franceschi, V.; Macchi, F.; Riccardo, F.; Ruiu, R.; Russo, L.; Quaglino, E.; Donofrio, G.; et al. Immunotargeting of the xCT Cystine/Glutamate Antiporter Potentiates the Efficacy of HER2-Targeted Immunotherapies in Breast Cancer. Cancer Immunol. Res. 2020, 8, 1039–1053. [Google Scholar] [CrossRef]
- Habens, F.; Srinivasan, N.; Oakley, F.; Mann, D.A.; Ganesan, A.; Packham, G. Novel sulfasalazine analogues with enhanced NF-kB inhibitory and apoptosis promoting activity. Apoptosis 2005, 10, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Köstler, W.J.; Leitzel, K.; Ali, S.M.; Sperinde, J.; Weidler, J.; Paquet, A.; Sherwood, T.; Huang, W.; Bates, M.; et al. Quantitative HER2 protein levels predict outcome in fluorescence in situ hybridization tastatic breast cancer treated with trastuzumab. Cancer 2010, 116, 5168–5178. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef]
- Fazzari, J.; Balenko, M.D.; Zacal, N.; Singh, G. Identification of capsazepine as a novel inhibitor of system xc− and cancer-induced bone pain. J. Pain Res. 2017, 10, 915–925. [Google Scholar] [CrossRef]
- Rolih, V.; Caldeira, J.; Bolli, E.; Salameh, A.; Conti, L.; Barutello, G.; Riccardo, F.; Magri, J.; Lamolinara, A.; Parra, K.; et al. Development of a vlp-based vaccine displaying an XCT extracellular domain for the treatment of metastatic breast cancer. Cancers 2020, 12, 1492. [Google Scholar] [CrossRef]
- Pflaum, J.; Schlosser, S.; Müller, M. P53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 1–15. [Google Scholar] [CrossRef]
- Ghosh, A.; Stewart, D.; Matlashewski, G. Regulation of Human p53 Activity and Cell Localization by Alternative Splicing. Mol. Cell. Biol. 2004, 24, 7987–7997. [Google Scholar] [CrossRef]
- Müller, M.; Schleithoff, E.S.; Stremmel, W.; Melino, G.; Krammer, P.H.; Schilling, T. One, two, three-p53, p63, p73 and chemosensitivity. Drug Resist. Updat. 2006, 9, 288–306. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del, S.G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Reinisch, I.; Schupp, M.; Schulz, T.J.; Prokesch, A. P53 functions in adipose tissue metabolism and homeostasis. Int. J. Mol. Sci. 2018, 19, 2622. [Google Scholar] [CrossRef]
- Guo, A.; Salomoni, P.; Luo, J.; Shih, A.; Zhong, S.; Gu, W.; Pandolfi, P.P. The function of PML in p53-dependent apoptosis. Nat. Cell Biol. 2000, 2, 730–736. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef]
- Ryan, K.M. p53 and autophagy in cancer: Guardian of the genome meets guardian of the proteome. Eur. J. Cancer 2011, 47, 44–50. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Pivnyuk, A.D.; Zhivotovsky, B. P53-autophagy-metastasis link. Cancers 2018, 10, 148. [Google Scholar] [CrossRef]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon, C.C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta, G.D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Candi, E.; Agostini, M.; Melino, G.; Bernassola, F. How the TP53 Family Proteins TP63 and TP73 Contribute to Tumorigenesis: Regulators and Effectors. Hum. Mutat. 2014, 35, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Mavinahalli, J.N.; Madhumalar, A.; Beuerman, R.W.; Lane, D.P.; Verma, C. Differences in the transactivation domains of p53 family members: A computational study. BMC Genom. 2010, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sauer, M.; Bretz, A.C.; Beinoraviciute, K.R.; Beitzinger, M.; Burek, C.; Rosenwald, A.; Harms, G.S.; Stiewe, T. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res. 2008, 36, 1900–1912. [Google Scholar] [CrossRef] [PubMed]
- Reich, N.C.; Levine, A.J. Growth regulation of a cellular tumour antigen, p53, in nontransformed cells. Nature 1984, 308, 199–201. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef]
- Salvador, J.M.; Brown, C.J.D.; Fornace, A.J. Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 2013, 793, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Michael, D.; Oren, M. The p53 and Mdm2 families in cancer. Curr. Opin. Genet. Dev. 2002, 12, 53–59. [Google Scholar] [PubMed]
- Levine, A.J.; Hu, W.; Feng, Z. The P53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef]
- Christophorou, M.A.; Ringshausen, I.; Finch, A.J.; Swigart, L.B.; Evan, G.I. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006, 443, 214–217. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zleger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; Michael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Soussi, T.; Lozano, G. p53 mutation heterogeneity in cancer. Biochem. Biophys. Res. Commun. 2005, 331, 834–842. [Google Scholar]
- Muller, P.A.J.; Vousden, K.H. P53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar]
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar]
- Jackson, E.L.; Olive, K.P.; Tuveson, D.A.; Bronson, R.; Crowley, D.; Brown, M.; Jacks, T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005, 65, 10280–10288. [Google Scholar] [CrossRef] [PubMed]
- Bougeard, G.; Sesboüé, R.; Baert, D.S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugières, L.; Chompret, A.; Bressac, P.B.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538. [Google Scholar] [CrossRef]
- Milner, J.V.W. Addition of Fresh Medium Induces Cell Cycle and Conformation Changes in p53, a Tumour Suppressor Protein. Oncogene 1990, 5, 1683–1690. [Google Scholar] [PubMed]
- Sasaki, M.; Nie, L.; Maki, C.G. MDM2 binding induces a conformational change in p53 that is opposed by heat-shock protein 90 and precedes p53 proteasomal degradation. J. Biol. Chem. 2007, 282, 14626–14634. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yamada, S.; Ichwan, S.J.A.; Iseki, S.; Ohtani, K.; Otsu, M.; Ikeda, M.A. Inability of p53-reactivating compounds nutlin-3 and rita to overcome p53 resistance in tumor cells deficient in p53ser46 phosphorylation. Biochem. Biophys. Res. Commun. 2012, 417, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Mendrysa, S.M.; McElwee, M.K.; Michalowski, J.; O’Leary, K.A.; Young, K.M.; Perry, M.E. mdm2 Is Critical for Inhibition of p53 during Lymphopoiesis and the Response to Ionizing Irradiation. Mol. Cell. Biol. 2003, 23, 462–472. [Google Scholar] [CrossRef]
- Sriraman, A.; Radovanovic, M.; Wienken, M.; Najafova, Z.; Li, Y.; Dobbelstein, M. Cooperation of Nutlin-3a and a Wip1 inhibitor to induce p53 activity. Oncotarget 2016, 7, 31623–31638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Y.; Grinkevich, V.V.; Nikulenkov, F.; Bao, W.; Selivanova, G. Rescue of the apoptotic-inducing function of mutant p53 by small molecule RITA. Cell Cycle 2010, 9, 1847–1855. [Google Scholar] [CrossRef]
- Lehmann, S.; Bykov, V.J.N.; Ali, D.; Andreń, O.; Cherif, H.; Tidefelt, U.; Uggla, B.; Yachnin, J.; Juliusson, G.; Moshfegh, A.; et al. Targeting p53 in vivo: A first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 2012, 30, 3633–3639. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of mutant P53 and the cellular redox balance by APR-246 as a strategy for efficient cancer therapy. Front. Oncol. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef]
- Omar, S.I.; Tuszynski, J. The molecular mechanism of action of methylene quinuclidinone and its effects on the structure of p53 mutants. Oncotarget 2018, 9, 37137–37156. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Ottaggio, L.; Penna, I.; Foggetti, G.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 cytotoxicity correlates with nucleolar localization and degradation of mutant p53 in breast cancer cells. Biochem. Biophys. Res. Commun. 2010, 402, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Wenrui, D.; Li, G.; Xin, W.; Li, W.; Serge, P.; Nana, S.; Otterson, G.A.; Miguel, A.V.C. MicroRNA-34a is an important component of PRIMA-1-induced apoptotic network in human lung cancer cells. Int. J. Cancer 2010, 127, 313–320. [Google Scholar] [CrossRef]
- Yoshikawa, N.; Kajiyama, H.; Nakamura, K.; Utsumi, F.; Niimi, K.; Mitsui, H.; Sekiya, R.; Suzuki, S.; Shibata, K.; Callen, D.; et al. PRIMA-1 MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2543–2552. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190. [Google Scholar] [CrossRef]
- Teoh, P.J.; Bi, C.; Sintosebastian, C.; Tay, L.S.; Fonseca, R.; Chng, W.J. PRIMA-1 targets the vulnerability of multiple myeloma of deregulated protein homeostasis through the perturbation of ER stress via p73 demethylation. Oncotarget 2016, 7, 61806–61819. [Google Scholar] [CrossRef]
- Naoise, C.; Synnott, S.F.M.; Vladimir, J.N.; Bykov, J.; Crown, K.G.W.; Michael, J.D. The Mutant p53-Targeting Compound APR-246 Induces ROS-Modulating Genes in Breast Cancer Cells. Transl. Oncol. 2018, 11, 1343–1349. [Google Scholar]
- Peng, X.; Zhang, M.Q.Z.; Conserva, F.; Hosny, G.; Selivanova, G.; Bykov, V.J.N.; Arnér, E.S.J.; Wiman, K.G. APR-246/PRIMA-1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013, 4, 881. [Google Scholar] [CrossRef] [PubMed]
- Haffo, L.; Lu, J.; Bykov, V.J.N.; Martin, S.S.; Ren, X.; Coppo, L.; Wiman, K.G.; Holmgren, A. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhu, S.; Chen, P.; Hou, W.; Wen, Q.; Liu, J.; Xie, Y.; Liu, J.; Klionsky, D.J.; Kroemer, G.; et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc– Activity. Curr. Biol. 2018, 28, 2388–2399. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.; He, Q.; Zhao, S.; Zhang, G.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The regulation of ferroptosis by tumor suppressor p53 and its pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina, K.A.; Kung, C.P.; Murphy, M.E. The p53 tumor suppressor in the control of metabolism and ferroptosis. Front. Endocrinol. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda, W.K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef]
- Clemons, N.J.; Liu, D.S.; Duong, C.P.; Phillips, W.A. Inhibiting system xC− and glutathione biosynthesis–a potential Achilles’ heel in mutant-p53 cancers. Mol. Cell. Oncol. 2017, 4. [Google Scholar] [CrossRef]
- Sobhani, N.; Roviello, G.; Corona, S.P.; Scaltriti, M.; Ianza, A.; Bortul, M.; Zanconati, F.; Generali, D. The prognostic value of PI3K mutational status in breast cancer: A meta-analysis. J. Cell. Biochem. 2018, 119, 4287–4292. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Buontempo, F.; Cubrey, J.A. Drug discovery targeting the mTOR pathway. Clin. Sci. 2018, 132, 543–568. [Google Scholar] [CrossRef] [PubMed]
- Arqués, O.; Chicote, I.; Puig, I.; Tenbaum, S.P.; Argilés, G.; Dienstmann, R.; Fernández, N.; Caratù, G.; Matito, J.; Silberschmidt, D.; et al. Tankyrase Inhibition Blocks Wnt/b-Catenin Pathway and Reverts Resistance to PI3K and AKT Inhibitors in the Treatment of Colorectal Cancer. Clin. Cancer Res. 2016, 22, 644–656. [Google Scholar] [CrossRef]
- Tassone, B.; Saoncella, S.; Neri, F.; Ala, U.; Brusa, D.; Magnuson, M.A.; Provero, P.; Oliviero, S.; Riganti, C.; Calautti, E. Rictor/mTORC2 deficiency enhances keratinocyte stress tolerance via mitohormesis. Cell Death Differ. 2017, 24, 731–746. [Google Scholar] [CrossRef]
- Shetzer, Y.; Solomon, H.; Koifman, G.; Molchadsky, A.; Horesh, S.; Rotter, V. The paradigm of mutant p53-expressing cancer stem cells and drug resistance. Carcinogenesis 2014, 35, 1196–1208. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, L.; Gomez, C.R.; Wang, X.; Hayashi, R.; Appella, E.; Kopelovich, L.; Leo, A.B. Targeting cancer stem cells with p53 modulators. Oncotarget 2016, 7, 45079–45093. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Molecular Targets Other than xCT | Pre-Clinical Use In Vivo | Current Clinical Indications |
|---|---|---|---|
| Sulfasalazine (SAS) [40] | NF-κB [41] | Yes (but limited by low solubility and poor bioavailability) [42] | Second-line treatment of ulcerative colitis [43] |
| Erastin [44,45] | VDAC2, VDAC3 [46] | No (poor metabolic stability and low solubility) [47,48] | None |
| Piperazine Erastin (PE) [47] | Not investigated | Yes (Erastin analogue with increased water solubility and metabolic stability) [47] | None |
| Erastin Ketone Imidazole (IKE) [48] | Not investigated | Yes (Erastin analogue with increased potency, selectivity, water solubility and metabolic stability) [48,49] | None |
| Sorafenib [45] | Multiple kinases [50] | Yes [50] | Unresectable hepatocellular carcinoma; Advanced renal cell carcinoma; Locally recurrent or metastatic, progressive, differentiated thyroid carcinoma refractory to radioactive iodine treatment [51,52] |
| Anti-xCT vaccines [36,53,54,55,56] | Not investigated | Yes [36,53,54,55,56] | None |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magri, J.; Gasparetto, A.; Conti, L.; Calautti, E.; Cossu, C.; Ruiu, R.; Barutello, G.; Cavallo, F. Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells 2021, 10, 108. https://doi.org/10.3390/cells10010108
Magri J, Gasparetto A, Conti L, Calautti E, Cossu C, Ruiu R, Barutello G, Cavallo F. Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells. 2021; 10(1):108. https://doi.org/10.3390/cells10010108
Chicago/Turabian StyleMagri, Jolanda, Alessandro Gasparetto, Laura Conti, Enzo Calautti, Chiara Cossu, Roberto Ruiu, Giuseppina Barutello, and Federica Cavallo. 2021. "Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies" Cells 10, no. 1: 108. https://doi.org/10.3390/cells10010108
APA StyleMagri, J., Gasparetto, A., Conti, L., Calautti, E., Cossu, C., Ruiu, R., Barutello, G., & Cavallo, F. (2021). Tumor-Associated Antigen xCT and Mutant-p53 as Molecular Targets for New Combinatorial Antitumor Strategies. Cells, 10(1), 108. https://doi.org/10.3390/cells10010108