Targeting a Lipid Desaturation Enzyme, SCD1, Selectively Eliminates Colon Cancer Stem Cells through the Suppression of Wnt and NOTCH Signaling

 ,
,
Abstract
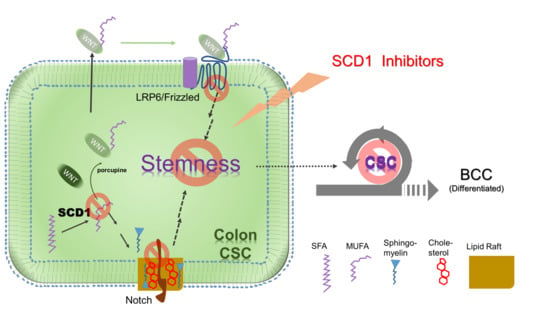
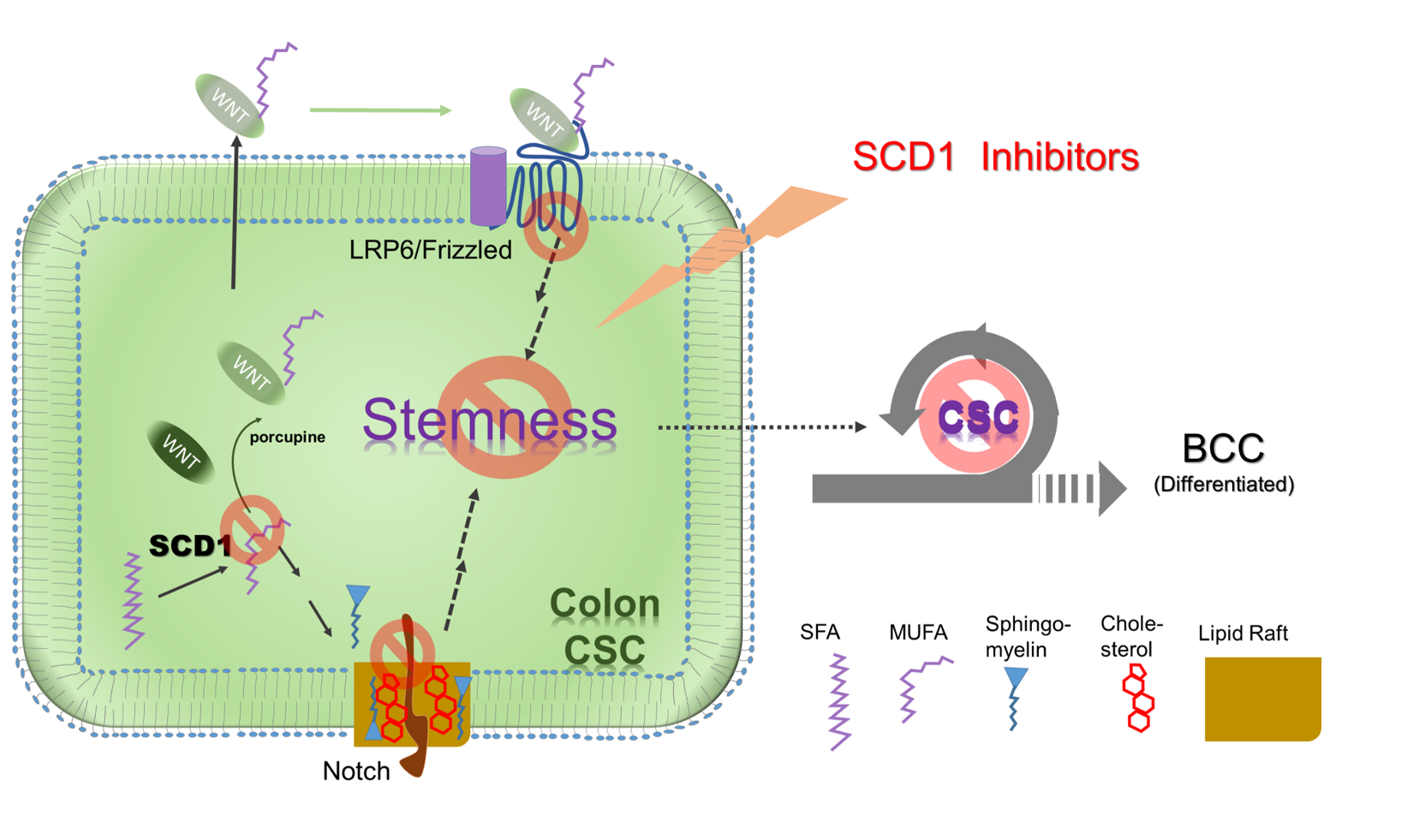{kind=link}
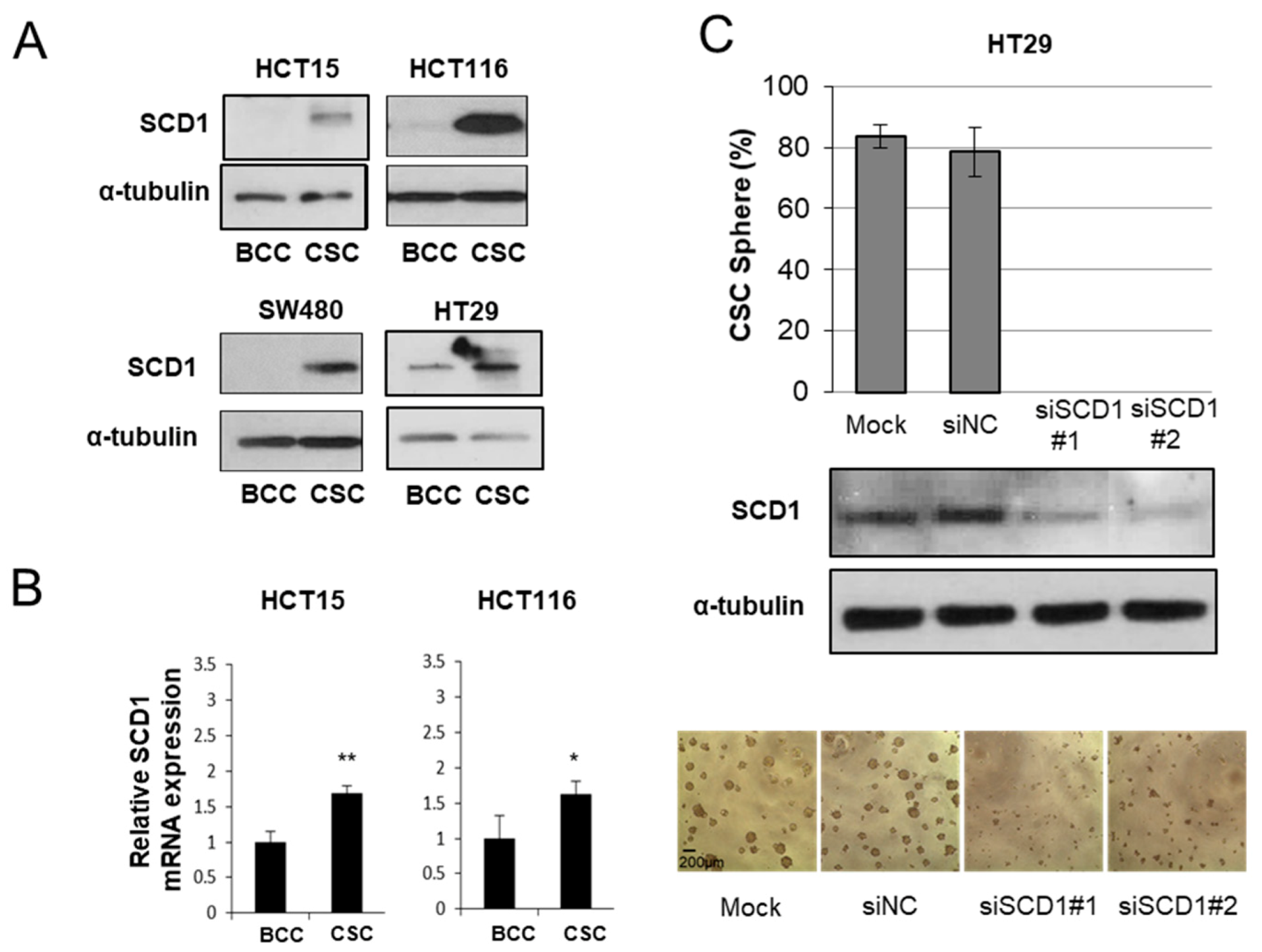{kind=link}
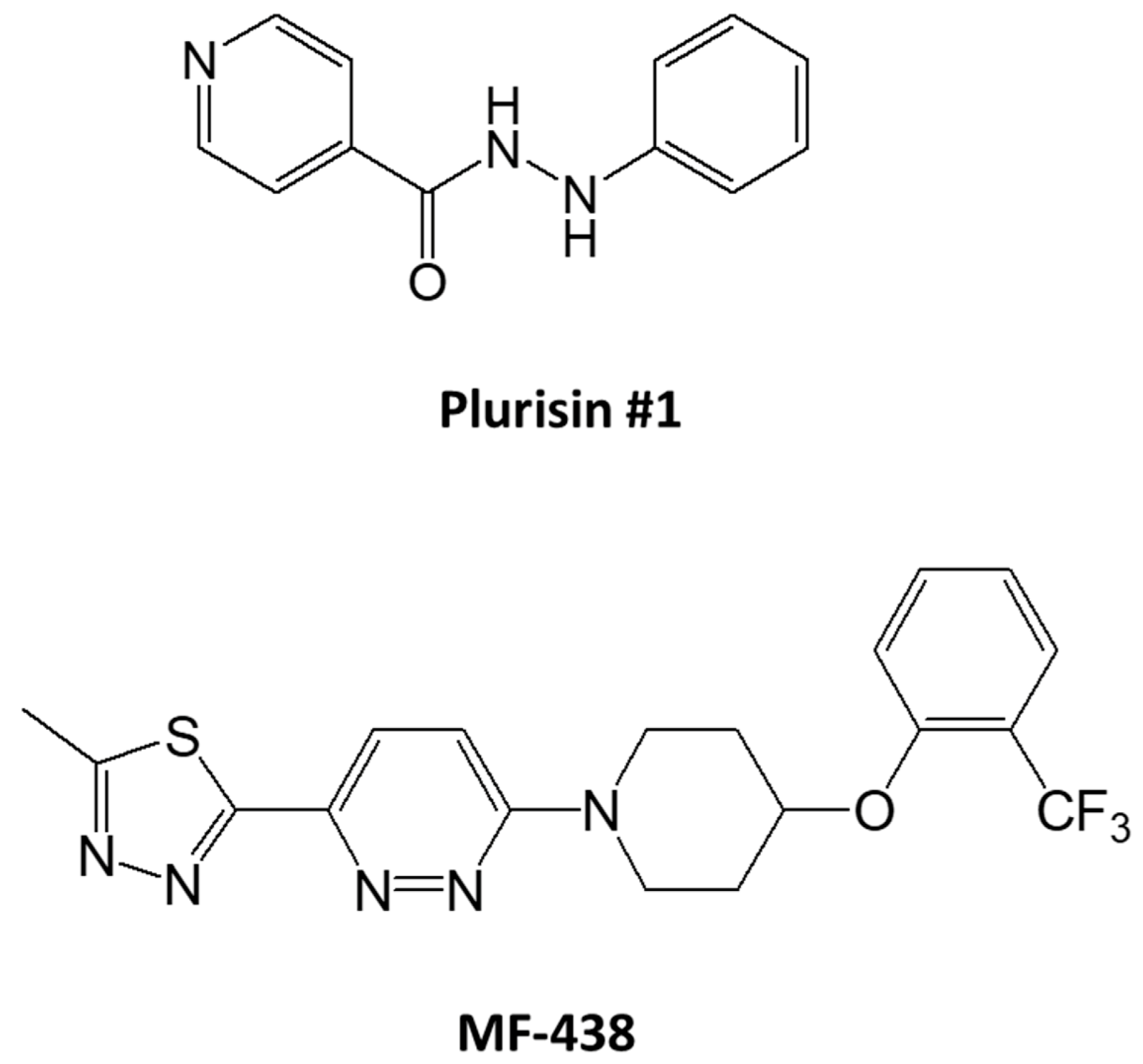{kind=link}
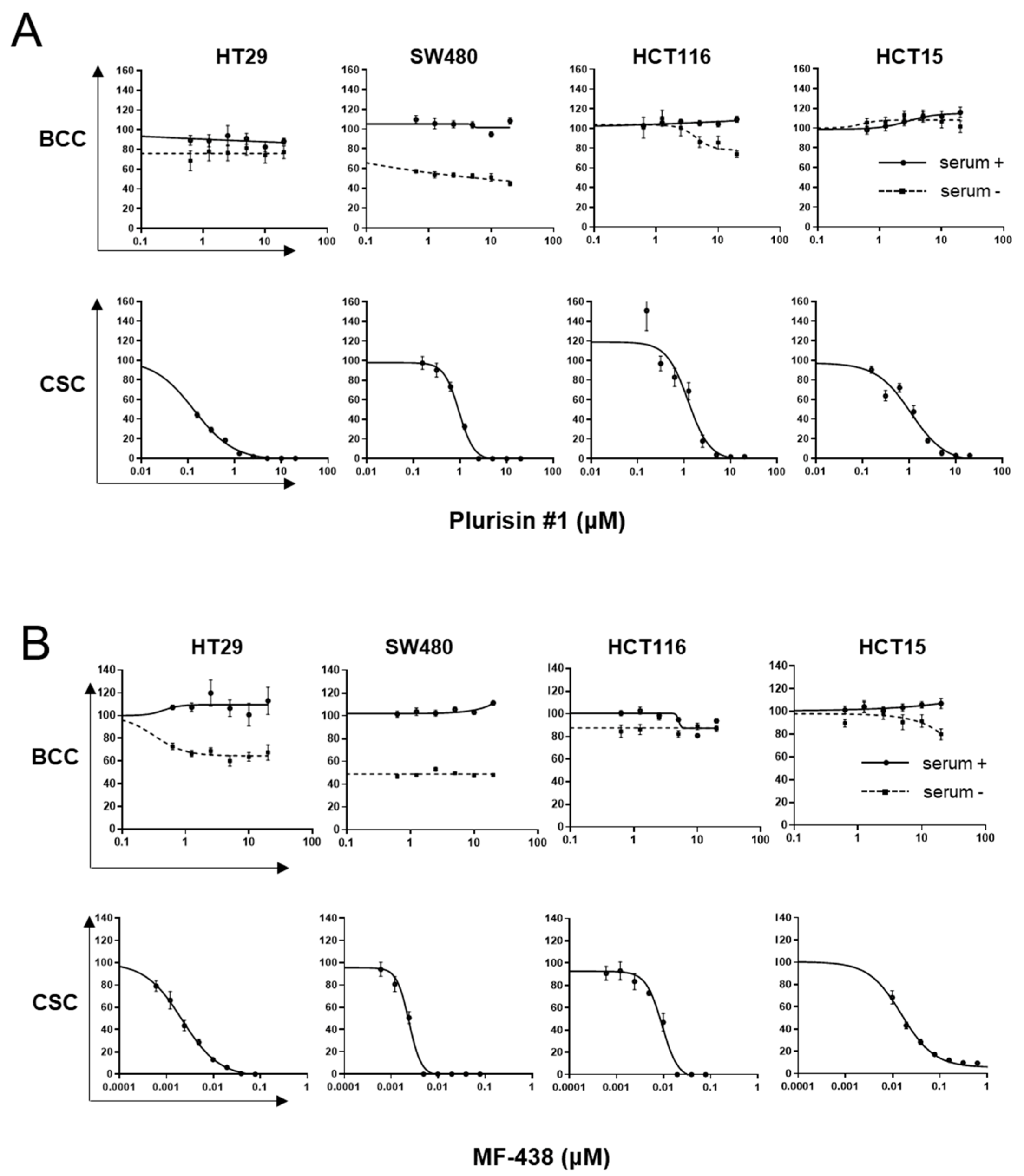{kind=link}
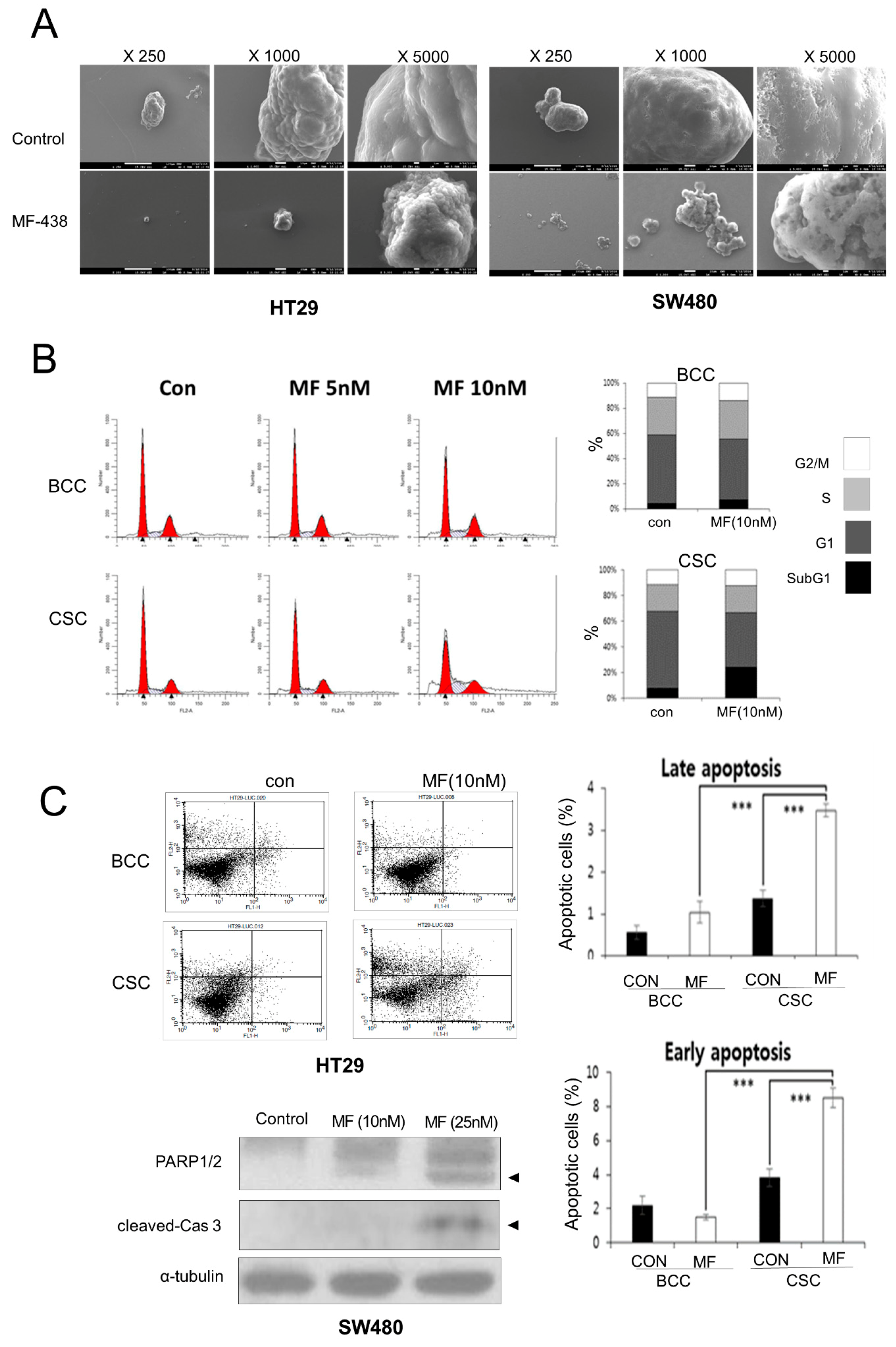{kind=link}
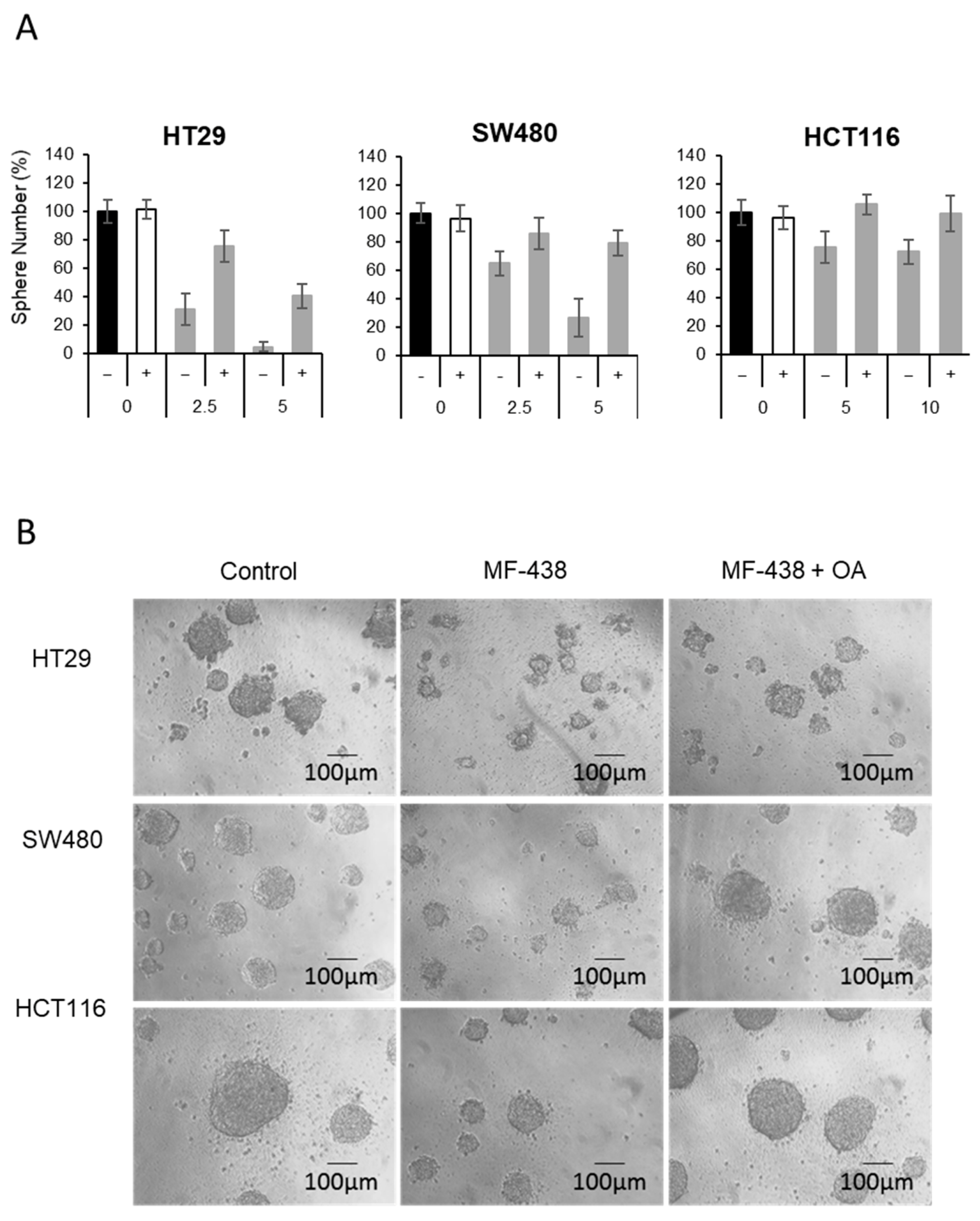{kind=link}
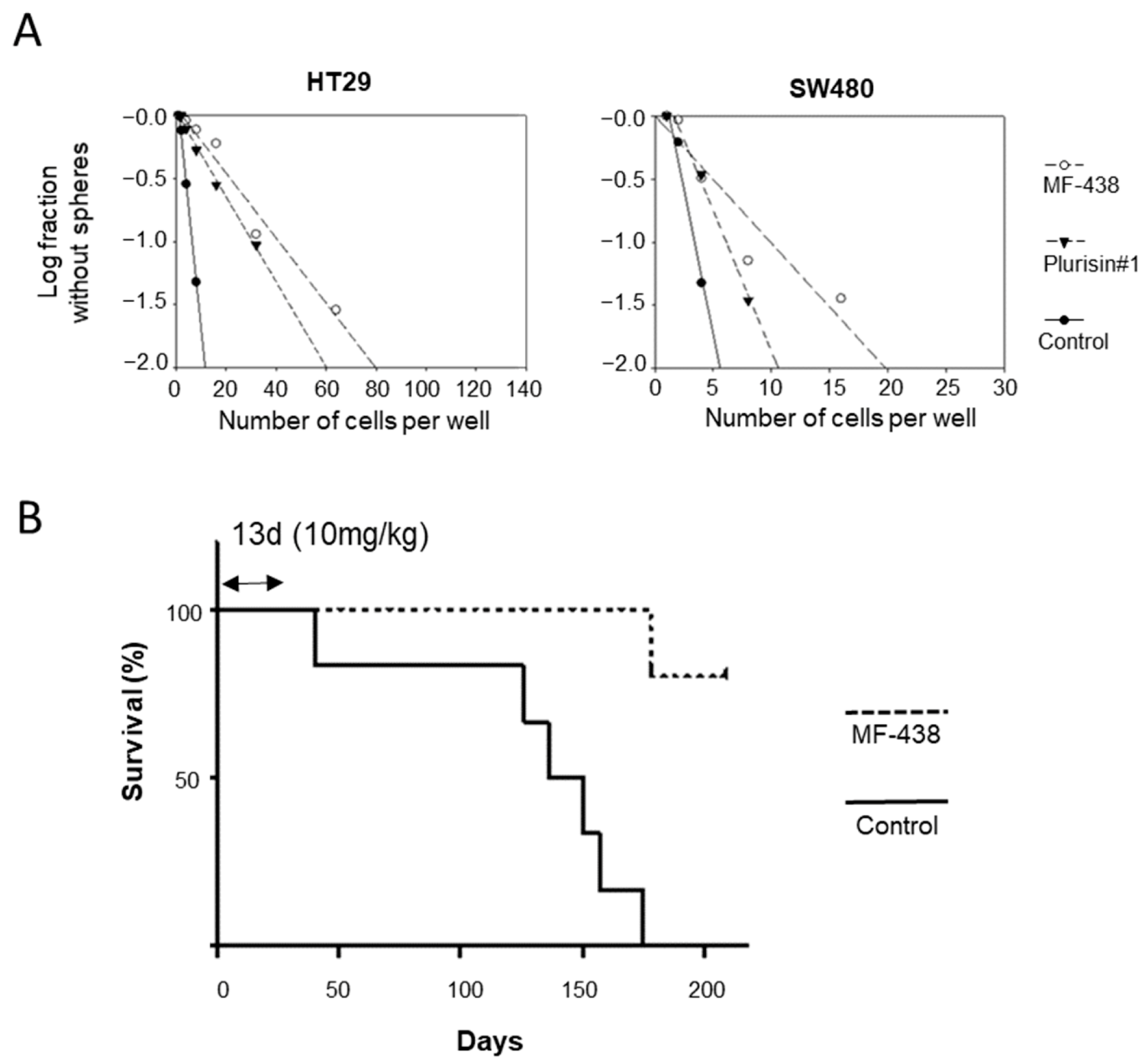{kind=link}
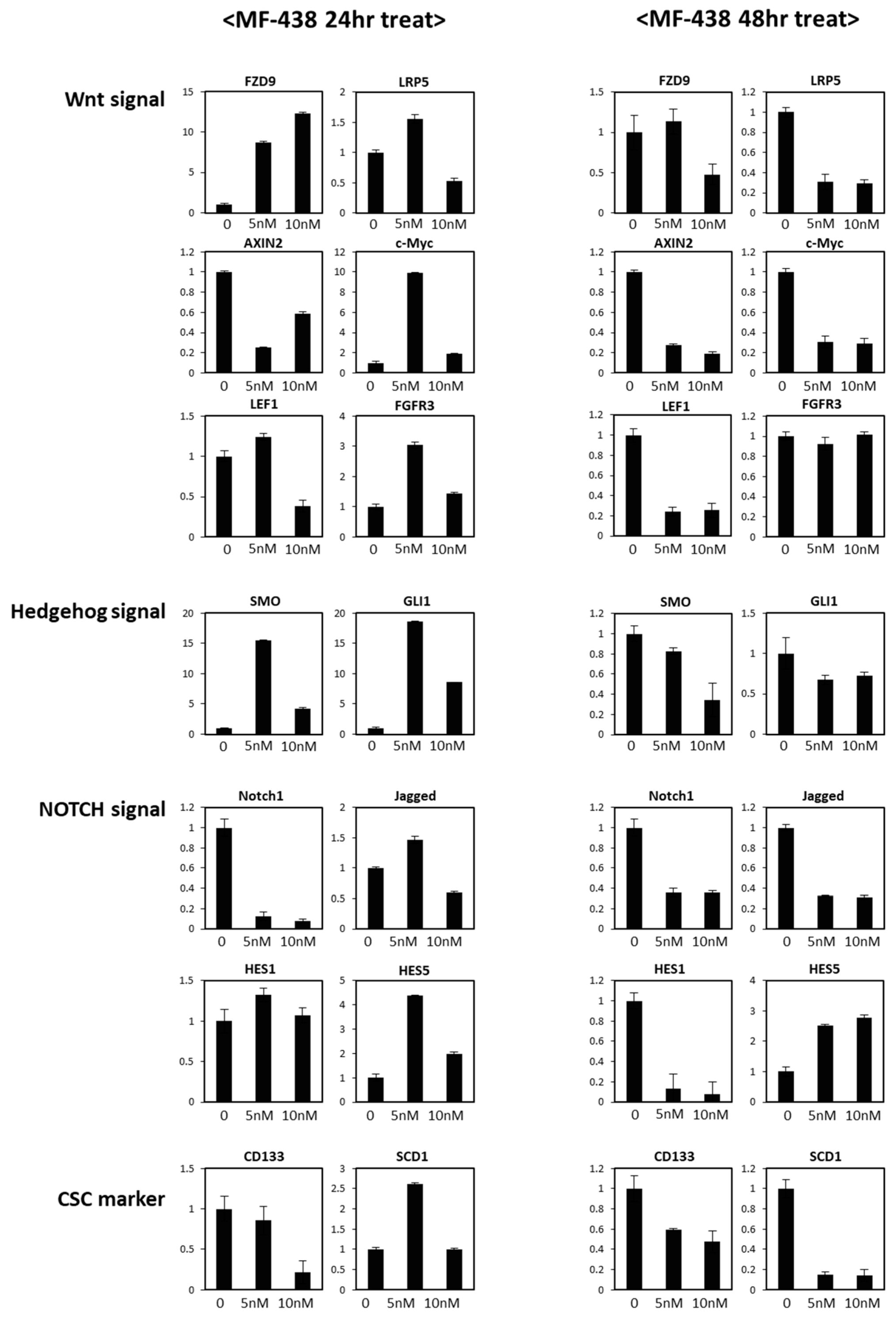{kind=link}
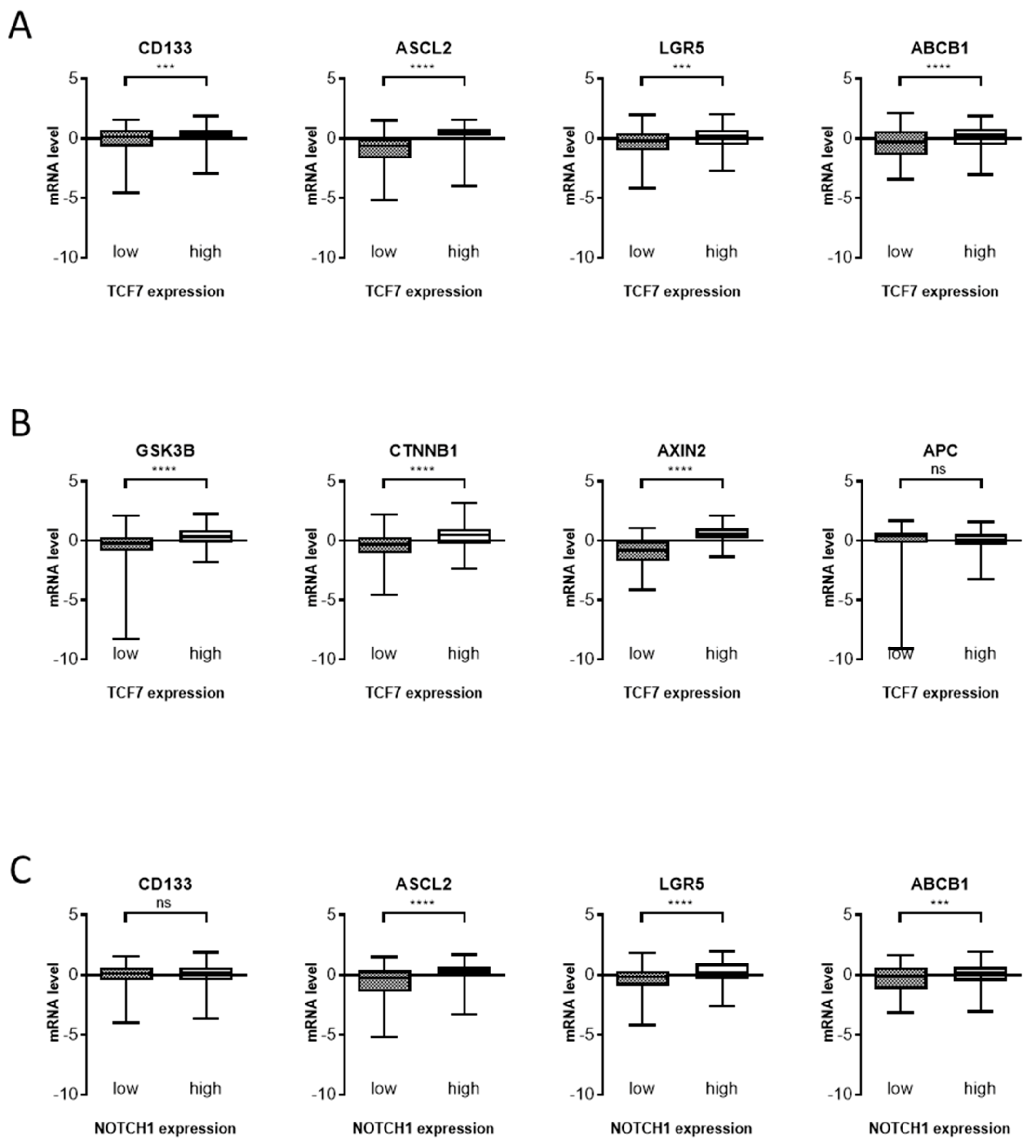{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Materials
2.2. Cell Proliferation Assay and CSC Sphere Proliferation Assay
2.3. Electron Microscopy, Cell Cycle Assay and Apoptosis Assay
2.4. Western Blotting
2.5. Limited Dilution Assay and Mouse Experiment
2.6. Quantitative RT-PCR
2.7. Bioinformatics Analysis of Clinical Data
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Peitzsch, C.; Tyutyunnykova, A.; Pantel, K.; Dubrovska, A. Cancer stem cells: The root of tumor recurrence and metastases. Semin. Cancer Biol. 2017, 44, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. N. Engl. J. Med. 2019, 380, 2237–2245. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.F. Clinical and Therapeutic Implications of Cancer Stem Cells. Reply. N. Engl. J. Med. 2019, 381, e19. [Google Scholar] [CrossRef]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, W.Y. Cancer stem cell targeting: Are we there yet? Arch. Pharm. Res. 2015, 38, 414–422. [Google Scholar] [CrossRef]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2019, 235, 790–803. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Tumor cell metabolism: The marriage of molecular genetics and proteomics with cellular intermediary metabolism; proceed with caution! Mol. Cancer 2006, 5, 59. [Google Scholar] [CrossRef]
- Choi, S.; Yoo, Y.J.; Kim, H.; Lee, H.; Chung, H.; Nam, M.H.; Moon, J.Y.; Lee, H.S.; Yoon, S.; Kim, W.Y. Clinical and biochemical relevance of monounsaturated fatty acid metabolism targeting strategy for cancer stem cell elimination in colon cancer. Biochem. Biophys. Res. Commun. 2019, 519, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Sacca, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef]
- Obniski, R.; Sieber, M.; Spradling, A.C. Dietary Lipids Modulate Notch Signaling and Influence Adult Intestinal Development and Metabolism in Drosophila. Dev. Cell 2018, 47, 98–111 e115. [Google Scholar] [CrossRef]
- Kim, W.Y. Therapeutic targeting of lipid synthesis metabolism for selective elimination of cancer stem cells. Arch. Pharm. Res. 2019, 42, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Lee, H.; Nam, M.H.; Jeong, E.; Kim, S.; Hong, Y.; Kim, N.; Yim, H.Y.; Yoo, Y.J.; Kim, J.S.; et al. Loss-of-function screens of druggable targetome against cancer stem-like cells. FASEB J. 2017, 31, 625–635. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314 e305. [Google Scholar] [CrossRef]
- Chen, L.; Ren, J.; Yang, L.; Li, Y.; Fu, J.; Li, Y.; Tian, Y.; Qiu, F.; Liu, Z.; Qiu, Y. Stearoyl-CoA desaturase-1 mediated cell apoptosis in colorectal cancer by promoting ceramide synthesis. Sci. Rep. 2016, 6, 19665. [Google Scholar] [CrossRef]
- Kim, H.; Yu, Y.; Choi, S.; Lee, H.; Yu, J.; Lee, J.H.; Kim, W.Y. Evodiamine Eliminates Colon Cancer Stem Cells via Suppressing Notch and Wnt Signaling. Molecules 2019, 24, 4520. [Google Scholar] [CrossRef]
- Jung, Y.; Park, H.; Zhao, H.Y.; Jeon, R.; Ryu, J.H.; Kim, W.Y. Systemic approaches identify a garlic-derived chemical, Z-ajoene, as a glioblastoma multiforme cancer stem cell-specific targeting agent. Mol. Cells 2014, 37, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Gan, Q.F.; Golan-Lev, T.; Arora, P.; Yanuka, O.; Oren, Y.S.; Leikin-Frenkel, A.; Graf, M.; Garippa, R.; Boehringer, M.; et al. Selective elimination of human pluripotent stem cells by an oleate synthesis inhibitor discovered in a high-throughput screen. Cell Stem Cell 2013, 12, 167–179. [Google Scholar] [CrossRef]
- Leger, S.; Black, W.C.; Deschenes, D.; Dolman, S.; Falgueyret, J.P.; Gagnon, M.; Guiral, S.; Huang, Z.; Guay, J.; Leblanc, Y.; et al. Synthesis and biological activity of a potent and orally bioavailable SCD inhibitor (MF-438). Bioorg. Med. Chem. Lett. 2010, 20, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Burattini, S.; Falcieri, E. Analysis of cell death by electron microscopy. Methods Mol. Biol. 2013, 1004, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef]
- Vinson, K.E.; George, D.C.; Fender, A.W.; Bertrand, F.E.; Sigounas, G. The Notch pathway in colorectal cancer. Int. J. Cancer 2016, 138, 1835–1842. [Google Scholar] [CrossRef]
- Blassberg, R.; Jacob, J. Lipid metabolism fattens up hedgehog signaling. BMC Biol. 2017, 15, 95. [Google Scholar] [CrossRef]
- Hakobyan, D.; Heuer, A. Key molecular requirements for raft formation in lipid/cholesterol membranes. PLoS ONE 2014, 9, e87369. [Google Scholar] [CrossRef] [PubMed]
- Levental, I.; Veatch, S. The Continuing Mystery of Lipid Rafts. J. Mol. Biol. 2016, 428, 4749–4764. [Google Scholar] [CrossRef] [PubMed]
- Silvius, J.R. Role of cholesterol in lipid raft formation: Lessons from lipid model systems. Biochim. Biophys. Acta 2003, 1610, 174–183. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Bringing order to chaos. J. Lipid Res. 2003, 44, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Sakane, H.; Yamamoto, H.; Kikuchi, A. LRP6 is internalized by Dkk1 to suppress its phosphorylation in the lipid raft and is recycled for reuse. J. Cell Sci. 2010, 123, 360–368. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Collu, G.M.; Hidalgo-Sastre, A.; Brennan, K. Wnt-Notch signalling crosstalk in development and disease. Cell Mol. Life Sci. 2014, 71, 3553–3567. [Google Scholar] [CrossRef]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.T.; ME, L.L. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef]
- Rios-Esteves, J.; Resh, M.D. Stearoyl CoA Desaturase Is Required to Produce Active, Lipid-Modified Wnt Proteins. Cell Rep. 2013, 4, 1072–1081. [Google Scholar] [CrossRef]
- Liu, J.; Pan, S.; Hsieh, M.H.; Ng, N.; Sun, F.; Wang, T.; Kasibhatla, S.; Schuller, A.G.; Li, A.G.; Cheng, D.; et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. USA 2013, 110, 20224–20229. [Google Scholar] [CrossRef]
- Mir, R.; Sharma, A.; Pradhan, S.J.; Galande, S. Regulation of Transcription Factor SP1 by the beta-Catenin Destruction Complex Modulates Wnt Response. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Srinivasan, T.; Tung, K.L.; Belmonte, J.M.; Wang, L.; Murthy, P.K.L.; Choi, J.; Rakhilin, N.; King, S.; Varanko, A.K.; et al. A Notch positive feedback in the intestinal stem cell niche is essential for stem cell self-renewal. Mol. Syst. Biol. 2017, 13, 927. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Shen, J. Presenilins are required for maintenance of neural stem cells in the developing brain. Mol. Neurodegener 2008, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Hayward, P.; Balayo, T.; Martinez Arias, A. Notch synergizes with axin to regulate the activity of armadillo in Drosophila. Dev. Dyn. 2006, 235, 2656–2666. [Google Scholar] [CrossRef]
- Kwon, C.; Cheng, P.; King, I.N.; Andersen, P.; Shenje, L.; Nigam, V.; Srivastava, D. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat. Cell Biol. 2011, 13, 1244–1251. [Google Scholar] [CrossRef]
- Ishiguro, H.; Okubo, T.; Kuwabara, Y.; Kimura, M.; Mitsui, A.; Sugito, N.; Ogawa, R.; Katada, T.; Tanaka, T.; Shiozaki, M.; et al. NOTCH1 activates the Wnt/beta-catenin signaling pathway in colon cancer. Oncotarget 2017, 8, 60378–60389. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Kim, H.; Choi, S.; Yu, J.; Lee, J.Y.; Lee, H.; Yoon, S.; Kim, W.-Y. Targeting a Lipid Desaturation Enzyme, SCD1, Selectively Eliminates Colon Cancer Stem Cells through the Suppression of Wnt and NOTCH Signaling. Cells 2021, 10, 106. https://doi.org/10.3390/cells10010106
Yu Y, Kim H, Choi S, Yu J, Lee JY, Lee H, Yoon S, Kim W-Y. Targeting a Lipid Desaturation Enzyme, SCD1, Selectively Eliminates Colon Cancer Stem Cells through the Suppression of Wnt and NOTCH Signaling. Cells. 2021; 10(1):106. https://doi.org/10.3390/cells10010106
Chicago/Turabian StyleYu, Yeongji, Hyejin Kim, SeokGyeong Choi, JinSuh Yu, Joo Yeon Lee, Hani Lee, Sukjoon Yoon, and Woo-Young Kim. 2021. "Targeting a Lipid Desaturation Enzyme, SCD1, Selectively Eliminates Colon Cancer Stem Cells through the Suppression of Wnt and NOTCH Signaling" Cells 10, no. 1: 106. https://doi.org/10.3390/cells10010106
APA StyleYu, Y., Kim, H., Choi, S., Yu, J., Lee, J. Y., Lee, H., Yoon, S., & Kim, W.-Y. (2021). Targeting a Lipid Desaturation Enzyme, SCD1, Selectively Eliminates Colon Cancer Stem Cells through the Suppression of Wnt and NOTCH Signaling. Cells, 10(1), 106. https://doi.org/10.3390/cells10010106