Genetic Identity Based on Whole-Genome SNP Array Data of Weedy Rice in Nagano, Japan


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Information for SNP Genotyping Array
2.3. DNA Isolation
2.4. Array Hybridization
2.5. SNP Genotype Calling
2.6. Population Structure and Genetic Diversity
2.7. Genome Divergence, Selection Sweeps, and Gene Flow Analysis
3. Results
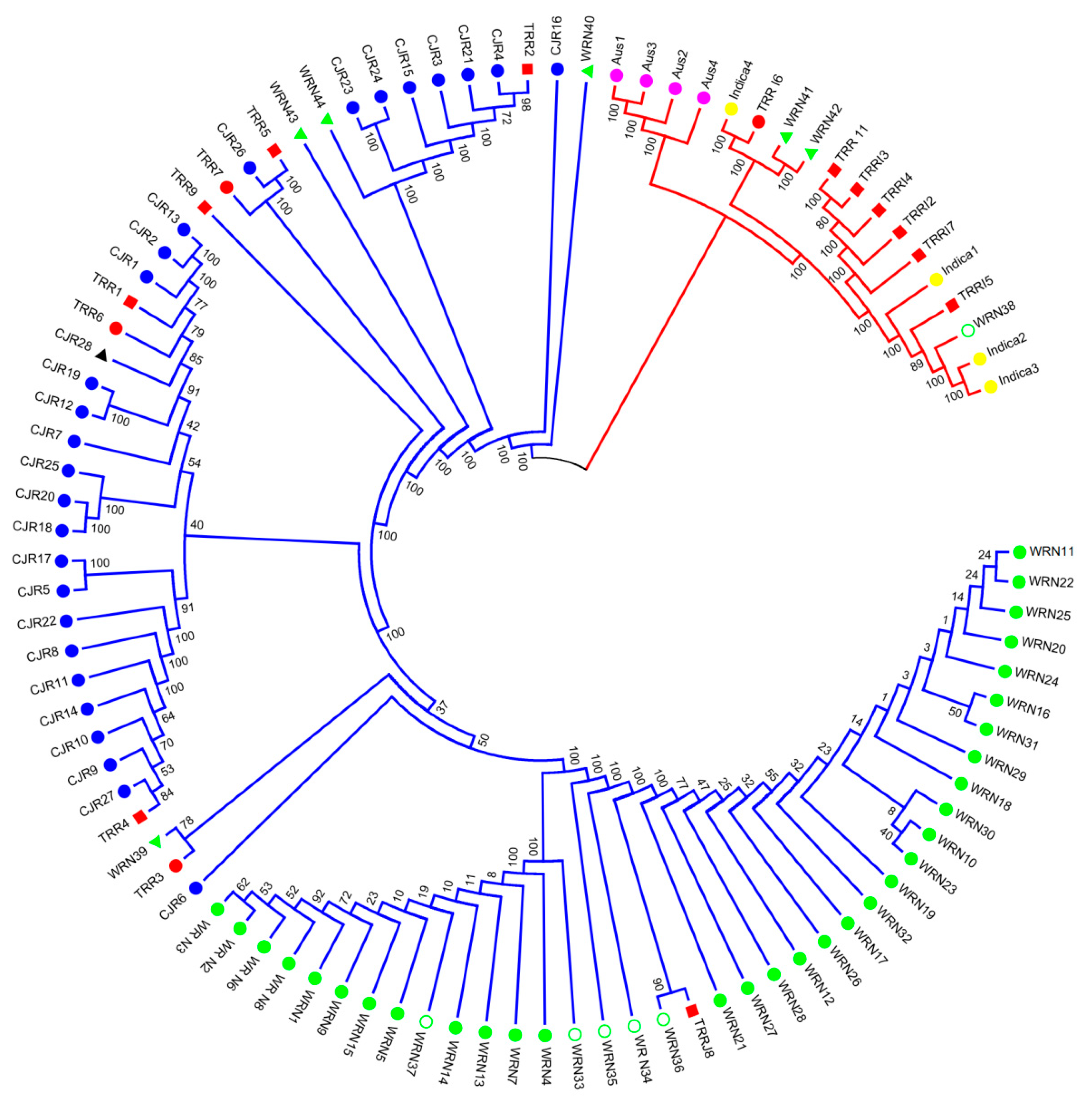
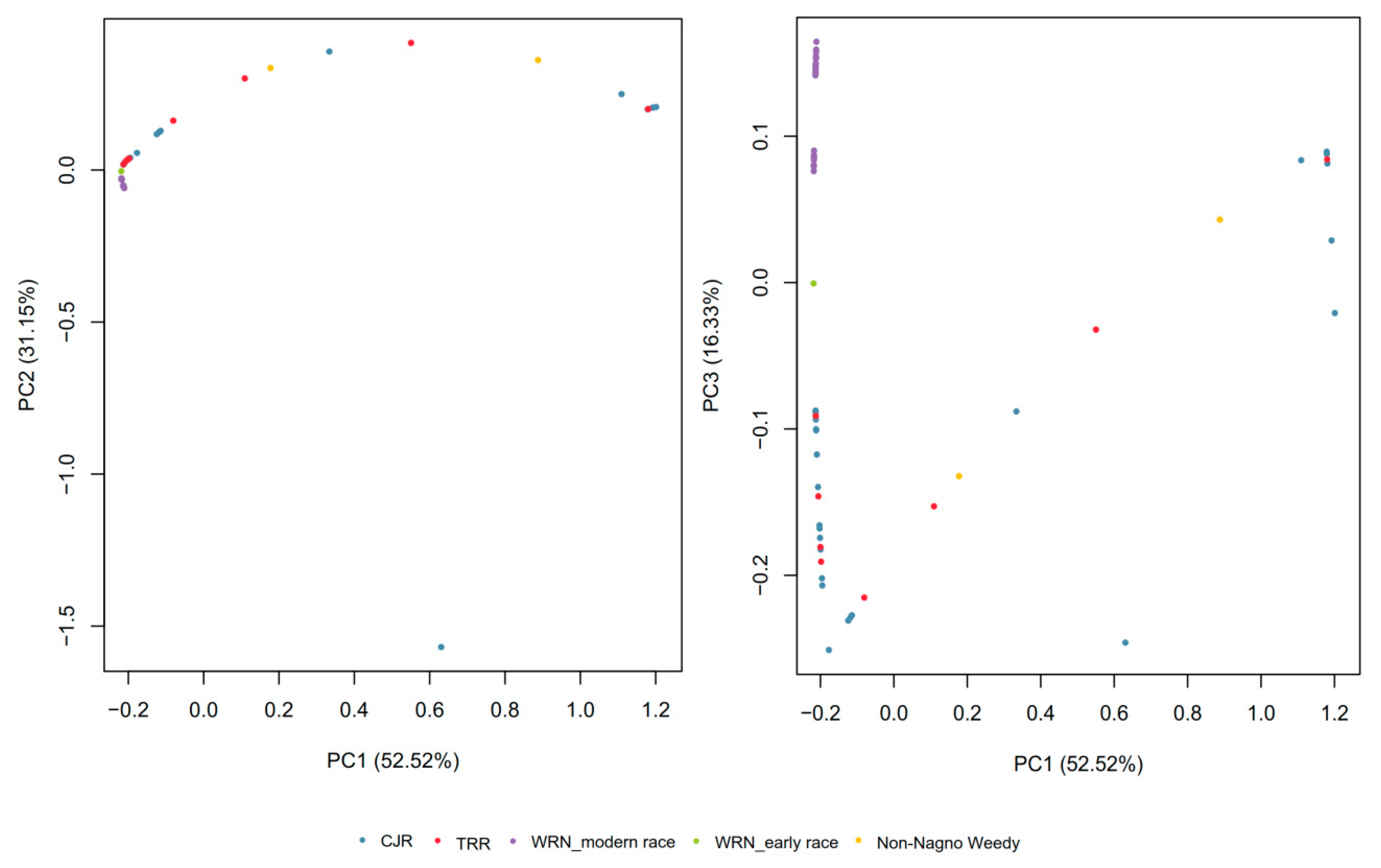
3.1. SNP Genotyping and Population Structure
3.2. Gene Diversity and Gene Flow
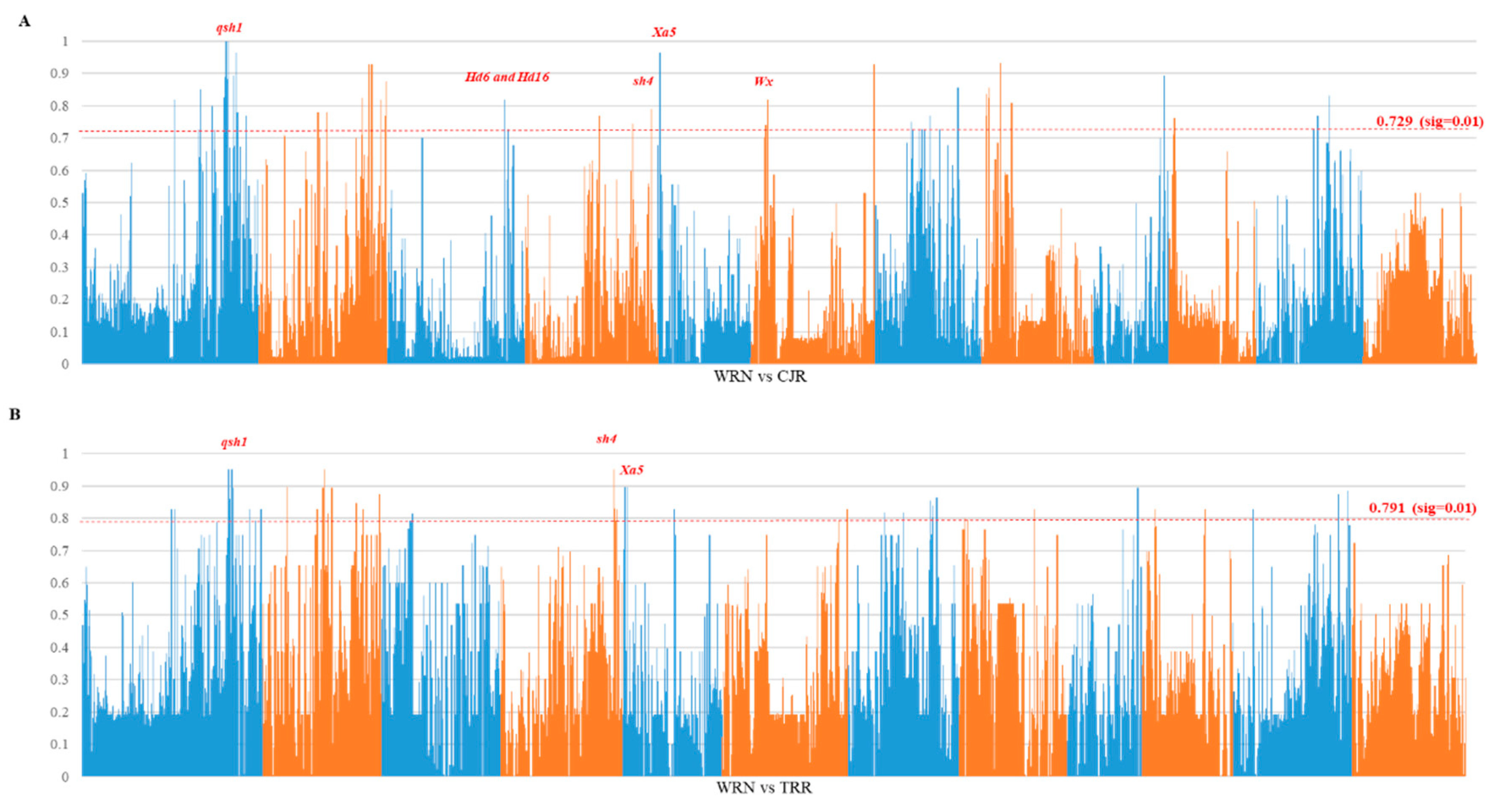
3.3. Genome Divergence and Selection Sweeps
4. Discussion
4.1. Origin of Weedy Rice in Nagano
4.2. Strategies for Weedy Rice Control
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, L.F.; Li, Y.L.; Jia, Y.L.; Caicedo, A.L.; Olsen, K.M. Signatures of adaptation in the weedy rice genome. Nat. Genet. 2017, 49, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhou, Y.J.; Mao, L.F.; Ye, C.; Wang, W.; Zhang, J.; Yu, Y.; Fu, F.; Wang, Y.; Qian, F.; et al. Genomic variation associated with local adaptation of weedy rice during de-domestication. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Miyajima, Y.; Takahashi, N. Red rice in Nagano. Agric. Tech. 1974, 29, 453–455. [Google Scholar]
- Tominaga, T.; Kurokawa, S. Research issues, challenges, and opportunities for weed management in Japan. Crop Prot. 2018. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xie, W.; Li, J.; Zhou, F.; Zhang, Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol. J. 2014, 12, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Ammiraju, J.S.; Luo, M.; Goicoechea, J.L.; Wang, W.; Kudrna, D.; Mueller, C.; Talag, J.; Kim, H.; Sisneros, N.B.; Blackmon, B.; et al. The Oryza bacterial artificial chromosome library resource: Construction and analysis of 12 deep-coverage large-insert BAC libraries that represent the 10 genome types of the genus Oryza. Genome Res. 2006, 16, 140–147. [Google Scholar] [CrossRef] [PubMed][Green Version]
- McNally, K.L.; Childs, K.L.; Bohnert, R.; Davidson, R.M.; Zhao, K.; Ulat, V.J.; Zeller, G.; Clark, R.M.; Hoen, D.R.; Bureau, T.E.; et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA 2009, 106, 12273–12278. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, A.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.F.; Crawford, G.W.; Jiang, L.P.; Chen, X.G. Rice domestication revealed by reduced shattering of archaeological rice from the Lower Yangtze valley. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Wu, Z.L.; Xing, Y.Y.; Zheng, F.G.; Guo, X.L.; Zhang, W.G.; Hong, M.M. Nucleotide sequence of rice waxy gene. Nucleic Acids Res. 1990, 18, 5898. [Google Scholar] [CrossRef] [PubMed]
- De Wet, J.M.J.; Harlan, J.R. Weeds and domesticates: Evolution in the man-made habitat. Econ. Bot. 1975, 29, 99–108. [Google Scholar] [CrossRef]
- Harlan, J.R. Crops and Man; American Society of Agronomy: Madison, WI, USA, 1992. [Google Scholar]
- Bres-Patry, C.; Lorieux, M.; Clément, G.; Bangratz, M.; Ghesquière, A. Heredity and genetic mapping of domestication-related traits in a temperate japonica weedy rice. Theor. Appl. Genet. 2001, 102, 118–126. [Google Scholar] [CrossRef]
- Lu, B.R.; Snow, A.A. Gene flow from genetically modified rice and its environmental consequences. BioScience 2005, 55, 669–678. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Heredia, S.M.; Leak-Garcia, J.A.; Heraty, J.M.; Burger, J.C.; Yao, L.; Nohzadeh-Malakshah, S.; Ridley, C.E. Crops gone wild: Evolution of weeds and invasives from domesticated ancestors. Evol. Appl. 2010, 3, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.N. Becoming weeds. Nat. Genet. 2017, 49, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Motohashi, R.; Tsuchimoto, S.; Fukuta, Y.; Ohtsubo, H.; Ohtsubo, E. Polyphyletic origin of cultivated rice: Based on the interspersion pattern of SINEs. Mol. Biol. Evol. 2003, 20, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Ebana, K.; Izawa, T. Inference of the japonica rice domestication process from the distribution of six functional nucleotide polymorphisms of domestication-related genes in various landraces and modern cultivars. Plant Cell Physiol. 2008, 49, 1283–1293. [Google Scholar] [CrossRef]
- Yu, G.; Bao, Y.; Shi, C.; Dong, C.; Ge, S. Genetic diversity and population differentiation of Liaoning weedy rice detected by RAPD and SSR markers. Biochem. Genet. 2005, 43, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, J.; Akasaka, M.; Takamtsu, M. Physiological and morphological characteristics of new type of weedy rice in Nagano. Japan. J. Crop Sci. 2013, 82 (Suppl. 1), 208–209. [Google Scholar]
- Ferrero, A. Weedy Rice, Biological Features and Control; FAO: Rome, Italy, 2003; Available online: http://www.fao.org/docrep/006/y5031e/y5031e00.htm#Contents (accessed on 16 July 2019).
- Delouche, J.C.; Labrada, R.; Rosell, C. Weedy Rices: Origin, Biology, Ecology and Control; FAO: Rome, Italy, 2007. [Google Scholar]
- Nagano prefecture. Nagano Prefecture Manual of Control of Weedy Rice in Nagano; Nagano prefecture: Nagano, Japan, 2012. [Google Scholar]
- Hosoi, J.; Ushiki, J.; Sakai, N.; Aoki, M.; Saito, K. Survival of seeds on the surface of paddy soil of weedy rice in Nagano. Jpn. J. Crop Sci. 2010, 79, 322–326. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop1 | Pop2 | Pop3 | 0pop | D | Z | BABA | ABBA | |
|---|---|---|---|---|---|---|---|---|
| Result | CJR | TRR | WRN | Outgroup | 0.0001 | 0.023 | 2845 | 2845 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, W.; Sun, J.; Hosoi, J.; Aoki, M.; Sakai, N.; Itani, T.; Xu, Z.; Tominaga, T. Genetic Identity Based on Whole-Genome SNP Array Data of Weedy Rice in Nagano, Japan. Agronomy 2019, 9, 472. https://doi.org/10.3390/agronomy9080472
Bi W, Sun J, Hosoi J, Aoki M, Sakai N, Itani T, Xu Z, Tominaga T. Genetic Identity Based on Whole-Genome SNP Array Data of Weedy Rice in Nagano, Japan. Agronomy. 2019; 9(8):472. https://doi.org/10.3390/agronomy9080472
Chicago/Turabian StyleBi, Wenjing, Jian Sun, Jun Hosoi, Masaharu Aoki, Nagao Sakai, Tomio Itani, Zhengjin Xu, and Tohru Tominaga. 2019. "Genetic Identity Based on Whole-Genome SNP Array Data of Weedy Rice in Nagano, Japan" Agronomy 9, no. 8: 472. https://doi.org/10.3390/agronomy9080472
APA StyleBi, W., Sun, J., Hosoi, J., Aoki, M., Sakai, N., Itani, T., Xu, Z., & Tominaga, T. (2019). Genetic Identity Based on Whole-Genome SNP Array Data of Weedy Rice in Nagano, Japan. Agronomy, 9(8), 472. https://doi.org/10.3390/agronomy9080472