Functional Annotation of Hypothetical Proteins Derived from Suppressive Subtraction Hybridization (SSH) Analysis Shows NPR1 (Non-Pathogenesis Related)-Like Activity


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and RNA Isolation
2.2. SSH Library Construction
2.3. DNA Sequencing and Initial Processing
2.4. Functional Annotation
2.4.1. Homology Search
2.4.2. Physicochemical Characterization
2.4.3. Subcellular Localization
2.4.4. Protein Classification and Function Prediction
2.4.5. Homology Modeling of miRNA and Target Proteins
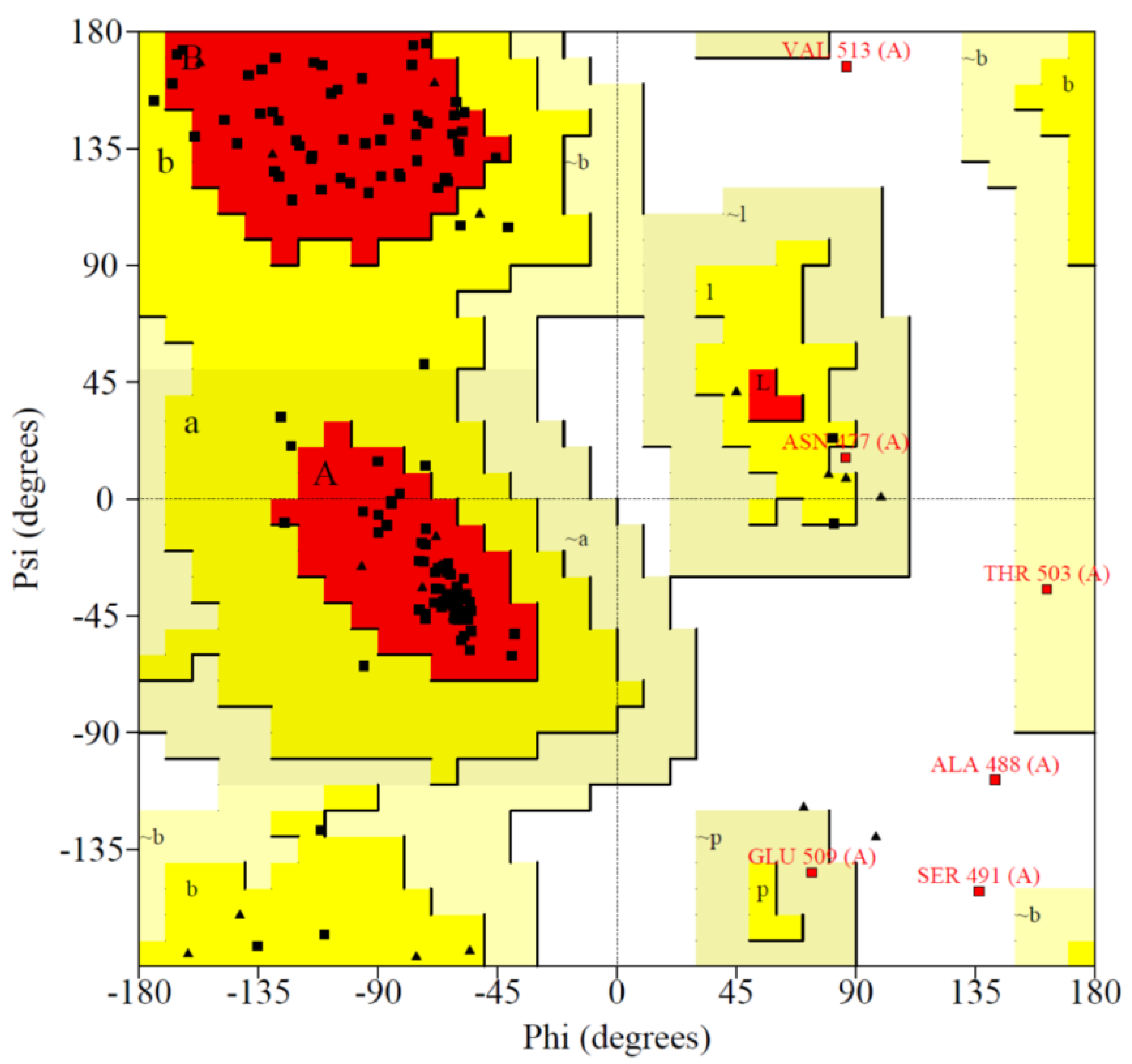
2.5. Model Validation
2.6. Molecular Dynamics Simulation
2.7. Analysis of Pockets and Clefts
2.8. Preparation of Phytoalexins
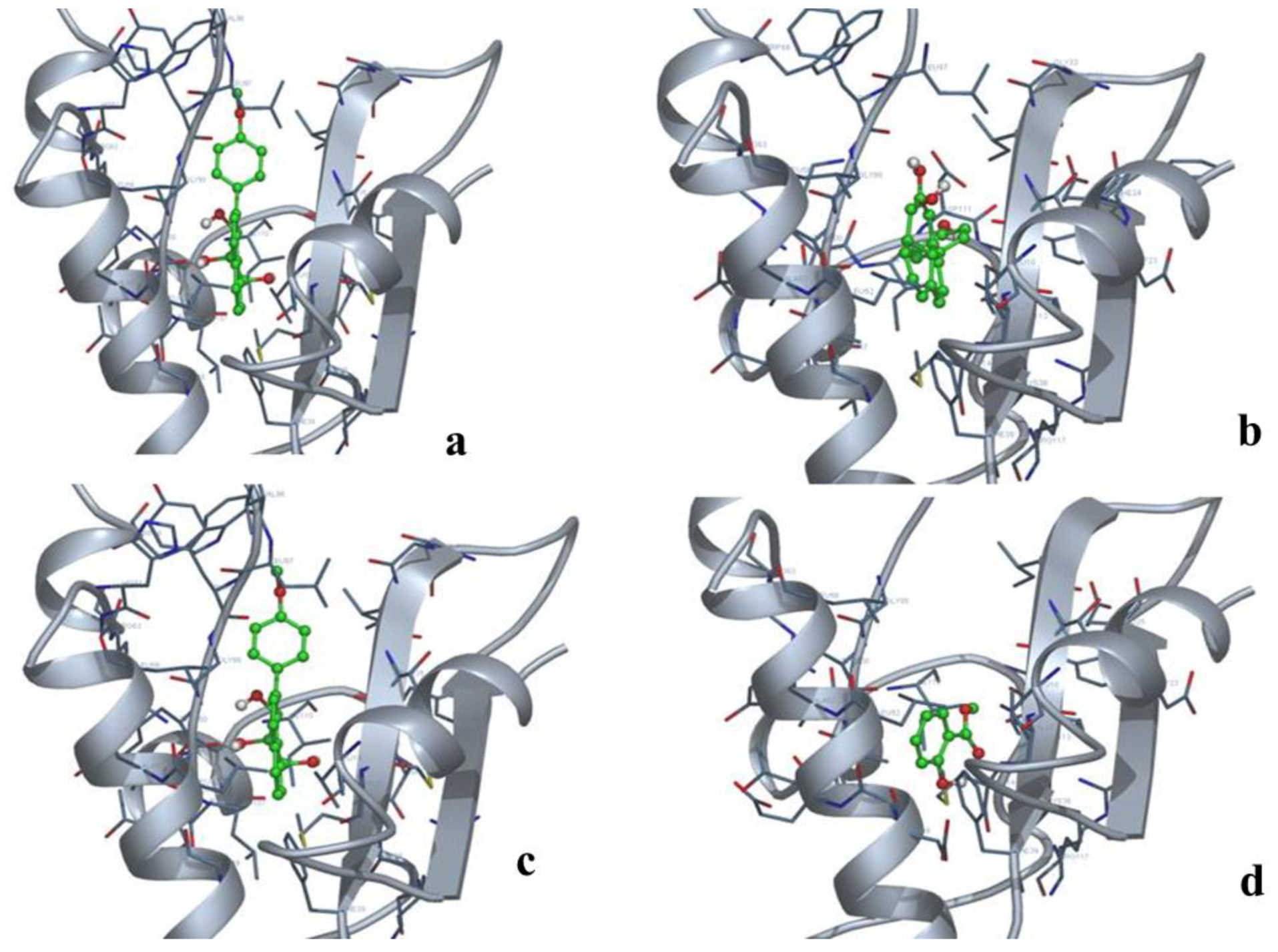
2.9. Docking and Analysis of the Interaction
2.10. Prediction of Rigid/Non-Rigid Nature of Interacting Residues
3. Results
3.1. SSH Analysis
3.2. Sequence Analysis
3.3. Homology Modeling and Sequential Docking
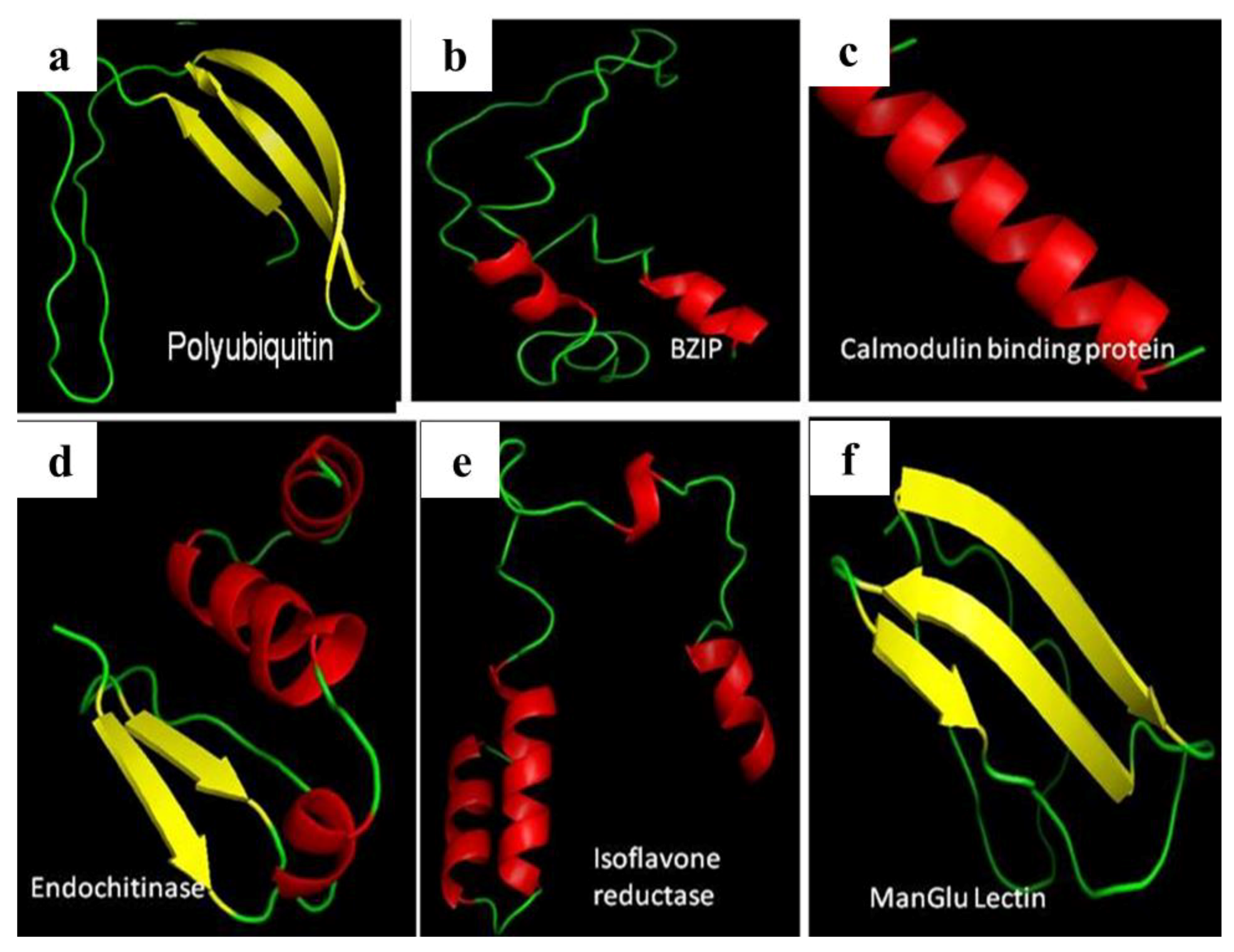
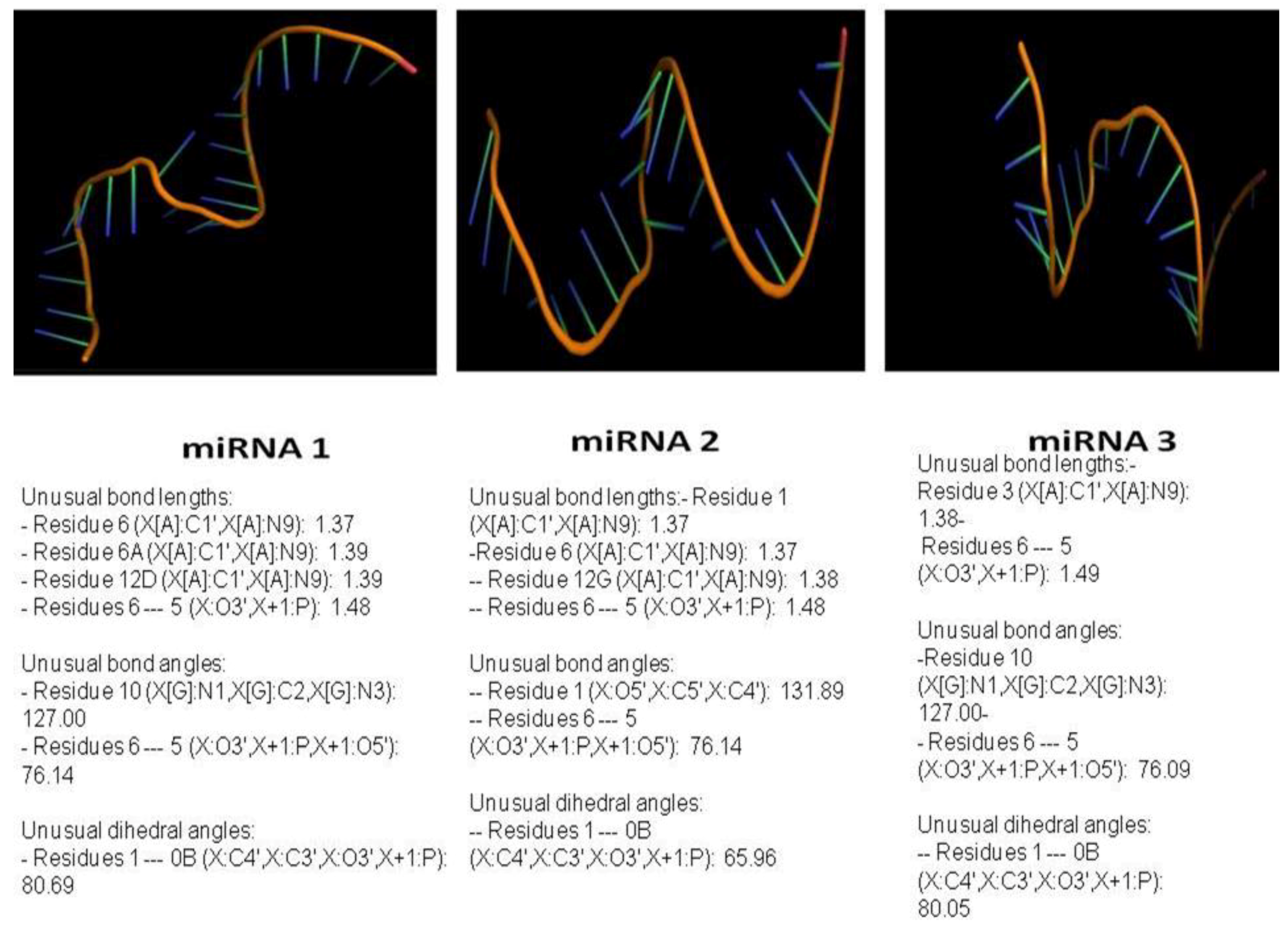
3.4. miRNA, Phytoalexin, and Polyubiquitin-Binding Capacity of Contig 21
3.5. Duality in Localization in Deciphering Contig 21 as NPR1 Homolog
3.6. Ubiquitination Activities of Contig 21
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, Y.H.; Chang, J.Y.; Liu, E.T.; Chao, C.P.; Huang, J.W.; Chang, P.F.L. Development of a molecular marker for specific detection of Fusarium oxysporum f. sp. cubense race 4. Eur. J. Plant Pathol. 2008, 123, 353–365. [Google Scholar] [CrossRef]
- Pegg, K.G.; Moore, N.Y.; Bentley, S. Fusarium wilt of banana in Australia: A review. Aust. J. Agric. Res. 1996, 47, 637–650. [Google Scholar] [CrossRef]
- Getha, K.; Vikineswary, S. Antagonistic effects of Streptomyces violaceusniger strain G10 on Fusarium oxysporum f.sp. cubense race 4: Indirect evidence for the role of antibiosis in the antagonistic process. J. Ind. Microbiol. Biotechnol. 2002, 28, 303–310. [Google Scholar] [PubMed]
- Monte, E. Understanding Trichoderma: Between biotechnology and microbial ecology. Int. Microbiol. 2001, 4, 1–4. [Google Scholar] [PubMed]
- Chandrasekaran, M.; Chun, S.C. Expression of PR-protein genes and induction of defense-related enzymes by Bacillus subtilis CBR05 in tomato (Solanum lycopersicum) plants challenged with Erwinia carotovora subsp. carotovora. Biosci. Biotechnol. Biochem. 2016, 80, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Harman, G.E.; Howell, C.R.; Viterbo, A. Trichoderma species—Opportunistic, virulent plant symbionts. Nat. Rev. Microbiol. 2004, 2, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Waghunde, R.R.; Shelake, R.M.; Sabalpara, A.N. Trichoderma: A significant fungus for agriculture and environment. Afr. J. Agric. Res. 2016, 11, 1952–1965. [Google Scholar]
- Mukherjee, P.K.; Buensanteai, N.; Moran-Diez, M.E.; Druzhinina, I.S.; Kenerley, C.M. Functional analysis of non-ribosomal peptide synthetases (NRPSs) in Trichoderma virens reveals a polyketide synthase (PKS)/NRPS hybrid enzyme involved in induced systemic resistance response in maize. Microbiology 2012, 158, 155–165. [Google Scholar] [CrossRef]
- Cotxarrera, L.; Trillas-Gay, M.I.; Steinberg, C.; Alabouvette, C. Use of sewage sludge compost and Trichoderma asperellum isolates to suppress Fusarium wilt of tomato. Soil Biol. Biochem. 2002, 34, 467–476. [Google Scholar] [CrossRef]
- Segarra, G.; Casanova, E.; Avile’s, M.; Trillas, M.I. Trichoderma asperellum strain T34 controls Fusarium wilt disease in tomato plants in soilless culture through competition for iron. Microb. Ecol. 2010, 59, 141–149. [Google Scholar] [CrossRef]
- Koonin, E.V.; Galperin, M.Y. Sequence-Evolution-Function: Computational Approaches in Comparative Genomics; Kluwer Academic Publishers: Philip Drive Norwell, MA, USA, 2003. [Google Scholar]
- Stephens, R.S.; Kalman, S.; Lammel, C.; Fan, J.; Marathe, R. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 1998, 282, 754–759. [Google Scholar] [CrossRef]
- Desler, C.; Suravajhala, P.; Sanderhoff, M.; Rasmussen, M.; Rasmussen, L.J. In silico screening for functional candidates amongst hypothetical proteins. BMC Bioinform. 2009, 10, 289. [Google Scholar] [CrossRef]
- Loewenstein, Y.; Raimondo, D.; Redfern, O.C.; Watson, J.; Frishman, D.; Linial, M.; Orengo, C.; Thornton, J.; Tramontano, A. Protein function annotation by homology-based inference. Genome Biol. 2009, 10, 207. [Google Scholar] [CrossRef]
- Mazandu, G.K.; Mulder, N.J. Function prediction and analysis of Mycobacterium tuberculosis hypothetical proteins. Int. J. Mol. Sci. 2012, 13, 7283–7302. [Google Scholar] [CrossRef]
- Shahbaaz, M.; Hassa, M.I.; Ahmad, F. Functional annotation of conserved hypothetical proteins from Haemophilus influenzae Rd KW20. PLoS ONE. 2013, 8, e84263. [Google Scholar] [CrossRef]
- Diatchenko, L.; Lau, Y.F.; Campbell, A.P.; Chenchik, A.; Moqadam, F.; Huang, B.; Lukyanov, S.; Lukyanov, K.; Gurskaya, N.; Sverdlov, E.D. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 6025–6030. [Google Scholar] [CrossRef]
- Fernandez, L.; Torregrosa, L.; Terrier, N.; Sreekantan, L.; Grimplet, J.; Davies, C.; Thomas, M.R.; Romieu, C.; Ageorges, A. Identification of genes associated with flesh morphogenesis during grapevine fruit development. Plant Mol. Biol. 2007, 63, 307–323. [Google Scholar] [CrossRef]
- Van Den Berg, N.; Berger, D.K.; Hein, I.; Birch, P.R.J.; Wingfield, M.J.; Viljoen, A. Tolerance in banana to Fusarium wilt is associated with early up-regulation of cell wall-strengthening genes in the roots. Mol. Plant Pathol. 2007, 8, 333–341. [Google Scholar] [CrossRef]
- Raman, T.; Gopalakrishnan, V.; Perumal, G. Identification of differentially expressed genes from Fusarium oxysporum f. sp cubense and Trichoderma asperellum (prr2) interaction in the susceptible banana cultivar Grand Naine. Turk. J. Bot. 2016, 40, 480–487. [Google Scholar] [CrossRef]
- Uma, S.; Sudhakar, B.; Thangavelu, R.; Backiyarani, S.; Saraswathi, M.S.; Saravankumar, A.S. Differential gene expression analysis for Sigatoka (Mycosphaerella eumusae) in resistant Musa acuminata cv. Manoranjitham (AAA). In Proceedings of the Plant & Animal Genomes XVIII Conference, San Diego, CA, USA, 9–13 January 2010. [Google Scholar]
- Portal, O.; Izquierdo, Y.; De Vleesschauwer, D.; Sanchez-Rodriguez, A.; Mendoza-Rodriguez, M. Acosta-Suarez. Analysis of expressed sequence tags derived from a compatible Mycosphaerella fijiensis-banana interaction. Plant Cell Rep. 2011, 30, 913–928. [Google Scholar] [CrossRef]
- Altschul, S.F.; Koonin, E.V. Iterated profile searches with PSI-BLAST—A tool for discovery in protein databases. Trends Biochem. Sci. 1998, 23, 444–447. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Tusnád, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. Signal-3L: A 3-layer approach for predicting signal peptides. Biochem. Biophys. Res. Commun. 2007, 363, 297–303. [Google Scholar] [CrossRef]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef]
- Timothy, L.; Bailey, E.C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA, 14–17 August 1994; AAAI Press: Menlo Park, CA, USA, 1994; pp. 28–36. [Google Scholar]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acid. Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Eswar, N.; Marti-Renom, M.A.; Webb, B.; Madhusudhan, M.S.; Eramian, D.; Shen, N.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling with MODELLER. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Volume 15, pp. 5.6.1–5.6.30. [Google Scholar]
- Bryson, K.; McGuffin, L.J.; Marsden, R.L.; Ward, J.J.; Sodhi, J.S.; Jones, D.T. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005, 33, W36–W38. [Google Scholar] [CrossRef]
- Marsden, R.L.; McGuffin, L.J.; Jones, D.T. Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci. 2002, 11, 2814–2824. [Google Scholar] [CrossRef]
- Maiti, R.; Gary, H.; Domselaar, V.; Zhang, H.; Wishart, D.S. SuperPose: A simple server for sophisticated structural superposition. Nucleic Acid. Res. 2004, 32, W590–W594. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comp. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Thompson, M.A. ArgusLab 4.0.1. Planaria Software; LLC: Seattle, WA, USA, 2004. [Google Scholar]
- Goswami, S.; Kumar, R.R.; Dubey, K.; Singh, J.P.; Tiwari, S.; Kumar, A.; Smita, S.; Mishra, D.C.; Kumar, S.; Grover, M.; et al. SSH Analysis of Endosperm Transcripts and Characterization of Heat Stress Regulated Expressed Sequence Tags in Bread Wheat. Front. Plant Sci. 2016, 7, 1230. [Google Scholar] [CrossRef]
- Badapanda, C. Suppression subtractive hybridization (SSH) combined with bioinformatics method: An integrated functional annotation approach for analysis of differentially expressed immune-genes in insects. Bioinformation 2013, 9, 216–221. [Google Scholar] [CrossRef]
- D’Hont, A.; Denoeud, F.; Aury, J.M.; Baurens, F.C.; Carreel, F.; Garsmeur, O.; Noel, B.; Bocs, S.; Droc, G.; Rouard, M.; et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 2012, 488, 213–217. [Google Scholar] [CrossRef]
- Sandhu, D.; Tasma, M.; Frasch, R.; Bhattacharyya, M.K. Systemic acquired resistance in soybean is regulated by two proteins, orthologous to Arabidopsis NPR1. BMC Plant Biol. 2009, 9, 105. [Google Scholar] [CrossRef]
- Després, C.; Chubak, C.; Rochon, A.; Clark, R.; Bethune, T.; Desveaux, D.; Fobert, P.R. The Arabidopsis NPR1 disease resistance protein is a novel cofactor that confers redox regulation of DNA binding activity to the basic domain/leucine zipper transcription factor TGA1. Plant Cell 2003, 15, 2181–2191. [Google Scholar] [CrossRef]
- Pumplin, N.; Voinnet, O. RNA silencing suppression by plant pathogens: Defence, counter-defence and counter-counter-defence. Nat. Rev. Microbiol. 2013, 11, 745–760. [Google Scholar] [CrossRef]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E. MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar] [CrossRef]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein–protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef]
- Mou, Z.; Fan, W.; Dong, X. Inducers of plant systemic acquired resistance regulate NPR1 function through redox changes. Cell 2003, 113, 935–944. [Google Scholar] [CrossRef]
- Pieterse, C.M.; van Loon, L.C. NPR1: The spider in the web of induced resistance signaling pathways. Curr. Opin. Plant Biol. 2004, 7, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Berrocal-Lobo, M.; Molina, A. Ethylene response factor 1 mediates Arabidopsis resistance to the soilborne fungus Fusarium oxysporum. Mol. Plant Microbe Interact. 2004, 17, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Shirasu, K. Ubiquitination in plant immunity. Curr. Opin. Plant Biol. 2010, 13, 402–408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Protein | BLASTX Results | E Value | Identity(%) |
|---|---|---|---|
| CONTIG5 | PREDICTED: uncharacterized protein LOC103975166 (Musa acuminata subsp. Malaccensis). ACCESSION XP_009388351 | 2 × 10−36 | 97% |
| CONTIG13 | hypothetical protein AUEXF2481DRAFT_37807 (Aureobasidium subglaciale EXF-2481). ACCESSION KEQ97279 | 9.5 | 36% |
| Contig 21 | PREDICTED: uncharacterized protein LOC103999244 (Musa acuminate subsp. Malaccensis). ACCESSION XP_009419208 | 7 × 10−58 | 100% |
| 12F | Unknown protein (Arabidopsis thaliana). ACCESSION AAK68824 | 2 × 10−30 | 66% |
| 49R | PREDICTED: uncharacterized protein LOC103975166 (Musa acuminate subsp. Malaccensis). ACCESSION XP_009388351 | 1 × 10−35 | 97% |
| 51R | PREDICTED: phytochrome E isoform X2 (Prunus mume). ACCESSION XP_008225375 | 8 × 10−29 | 71% |
| 68F | hypothetical protein HMPREF1063_05178, partial (Bacteroides dorei CL02T00C15). ACCESSION EIY16078 | 6 × 10−14 | 78% |
| 94F | PREDICTED: uncharacterized protein LOC103999244 (Musa acuminata subsp. Malaccensis). ACCESSION XP_009419208 | 4 × 10−38 | 100% |
| 964F | alpha-hemolysin (Burkholderia cenocepacia). ACCESSION ACJ54934 | 5 × 10−6 | 64% |
| 335R | PREDICTED: uncharacterized protein LOC103999244 (Musa acuminate subsp. Malaccensis). ACCESSION XP_009419208 | 7 × 10−41 | 100% |
| 542F | hypothetical protein ECP02989421_5339 (Escherichia coli P0298942.1). ACCESSION ENA26139 | 6 × 10−11 | 43% |
| 934F | hypothetical protein HMPREF1602_03442 (Escherichia coli 907889). ACCESSION ESD39518 | 2 × 10−7 | 35% |
| 959F | hypothetical protein, partial (Plasmodium chabaudi). ACCESSION XP_731877 | 6 × 10−5 | 53% |
| Name of the Docked Complex | Atomic Contact Energy |
|---|---|
| contig 21–calmodulin-binding protein | 288.75 |
| contig 21–manglu lectin | 302.46 |
| contig 21–bzip | 71.57 |
| contig 21–isoflavone reductase | 183.13 |
| contig 21–endochitinase | 227.07 |
| contig 21–polyubiquitin | 479.60 |
| contig 21–miRNA 1 | 804.86 |
| contig 21–miRNA 2 | 482.28 |
| contig 21–miRNA 3 | 494.75 |
| Name of the Protein | Binding Free Energy (kcal/mol) | Estimated Inhibition Constant, Ki (μM) | Electrostatic Energy (kcal/mol) | Total Intermolecular Energy (kcal/mol) | Frequency (%) | Interaction Surface |
|---|---|---|---|---|---|---|
| Contig 21–1 | −6.95 | 8.04 | −0.05 | −7.84 | 50 | 785.434 |
| Contig 21–2 | −7.58 | 2.77 | −0.04 | −8.14 | 50 | 679.576 |
| Contig 21–3 | −7.17 | 5.58 | −0.11 | −8.10 | 50 | 702.381 |
| Contig 21–4 | −7.29 | 4.53 | +0.04 | −7.49 | 50 | 664.13 |
| Residue Number | Name of the Residue | Individual B-Factor | Bi, N * |
|---|---|---|---|
| 34 | E | 0.227 | 0.0023 |
| 36 | D | 0.661 | 0.0354 |
| 37 | N | 0.808 | 0.0466 |
| 39 | G | 0.668 | 0.0359 |
| 52 | G | 0.58 | 0.0292 |
| 63 | T | 0.659 | 0.0352 |
| 66 | K | 0.737 | 0.0411 |
| 101 | R | 0.193 | −2.6675 |
| 102 | E | 0.084 | −0.0085 |
| 111 | S | 0.294 | 0.0074 |
| 113 | R | −0.076 | −0.0207 |
| 114 | F | −0.347 | −0.414 |
| Peptide | Position | Score |
|---|---|---|
| ANTQAGMKEAERLAD | 24 | 2.33 |
| DKNPTLVKTDEKTGE | 62 | 1.51 |
| TLVKTDEKTGEKKKRV | 66 | 1.01 |
| TDEKTGEKKRVLYGY | 70 | 1.02 |
| DEKTGEKKRVLYGYL | 71 | 0.96 |
| KIDFDMRKRAVIKSR | 93 | 1.09 |
| MRKRAVIKSRRELDL | 98 | 1.94 |
| ELDLSDCKTSFRFCH | 109 | 0.45 |
| TAGVLHVKGTCVSVL | 128 | 1.04 |
| GRTRVTIKERGLQRQ | 160 | 0.64 |
| LQRQRGGKPSYSGDG | 171 | 2.23 |
| FSGPSACKALNSEPL | 195 | 2.74 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandrasekaran, M.; Raman, C.; Karthikeyan, K.; Paramasivan, M. Functional Annotation of Hypothetical Proteins Derived from Suppressive Subtraction Hybridization (SSH) Analysis Shows NPR1 (Non-Pathogenesis Related)-Like Activity. Agronomy 2019, 9, 57. https://doi.org/10.3390/agronomy9020057
Chandrasekaran M, Raman C, Karthikeyan K, Paramasivan M. Functional Annotation of Hypothetical Proteins Derived from Suppressive Subtraction Hybridization (SSH) Analysis Shows NPR1 (Non-Pathogenesis Related)-Like Activity. Agronomy. 2019; 9(2):57. https://doi.org/10.3390/agronomy9020057
Chicago/Turabian StyleChandrasekaran, Murugesan, Chandrasekar Raman, Kandasamy Karthikeyan, and Manivannan Paramasivan. 2019. "Functional Annotation of Hypothetical Proteins Derived from Suppressive Subtraction Hybridization (SSH) Analysis Shows NPR1 (Non-Pathogenesis Related)-Like Activity" Agronomy 9, no. 2: 57. https://doi.org/10.3390/agronomy9020057
APA StyleChandrasekaran, M., Raman, C., Karthikeyan, K., & Paramasivan, M. (2019). Functional Annotation of Hypothetical Proteins Derived from Suppressive Subtraction Hybridization (SSH) Analysis Shows NPR1 (Non-Pathogenesis Related)-Like Activity. Agronomy, 9(2), 57. https://doi.org/10.3390/agronomy9020057