SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Maize with Network-Assisted Gene Prioritization


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Trial Conditions and Phenotyping
2.2. Population Structure, Linkage Disequilibrium (LD), and Haplotype Blocks
2.3. SNP- and Haplotype-Based GWAS
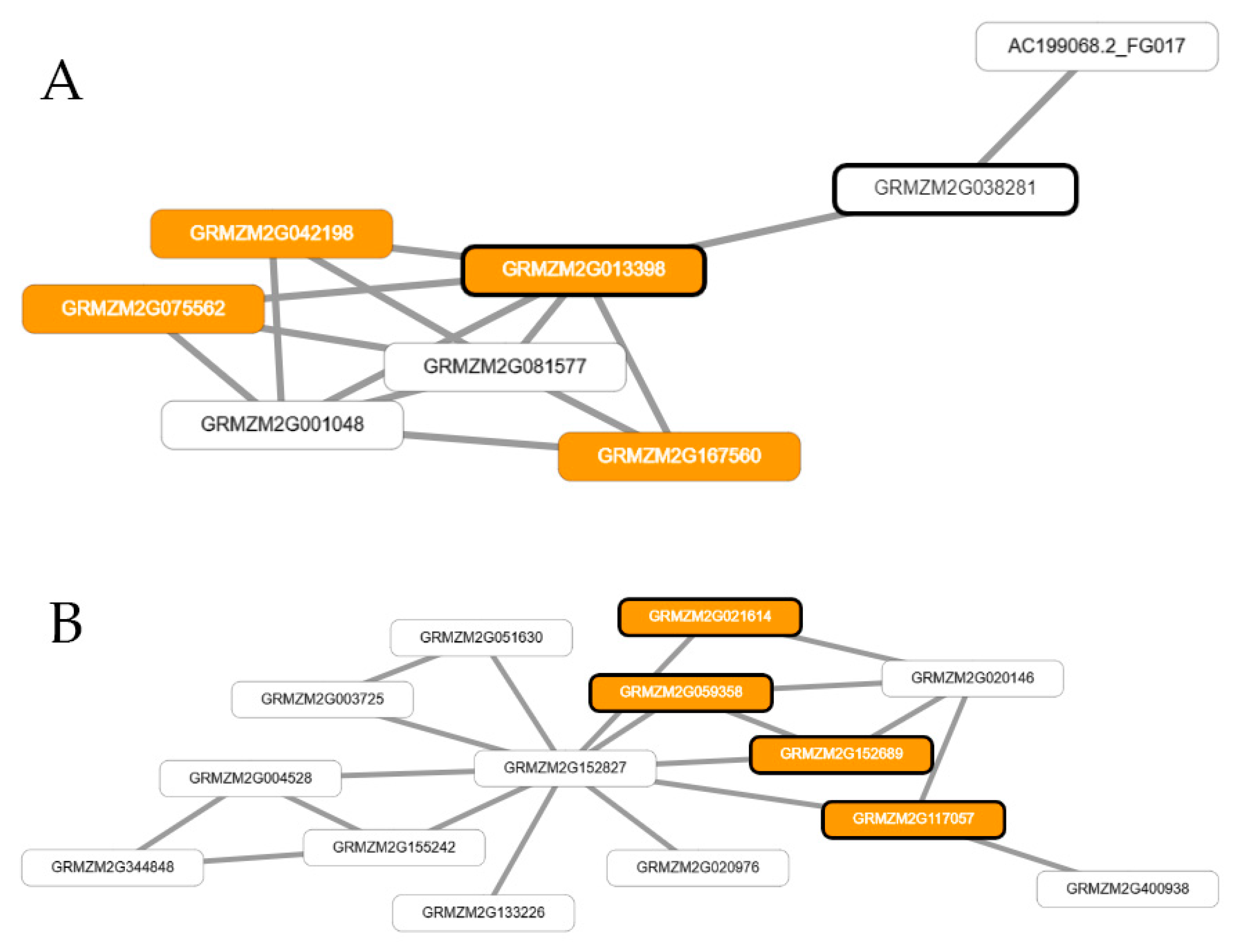
2.4. Prioritization of GWAS Candidate Genes and Inference of Co-Functional Networks for Flowering Traits in Maize
3. Results and Discussion
3.1. Genetic Structure
3.2. Linkage Disequilibrium
3.3. Haplotype Blocks
3.4. Genome-Wide Association Study and Network-Assisted Gene Prioritization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO. Save and Grow in Practice: Maize, Rice, Wheat—A Guide to Sustainable Production; Food and Agriculture Organization of the United Nations: Rome, Italy, 2016. [Google Scholar]
- Begcy, K.; Sandhu, J.; Walia, H. Transient heat stress during early seed development primes germination and seedling establishment in rice. Front. Plant. Sci. 2019, 9, 1768. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Rosentrater, K.A.; Bern, C. Effects of deterioration parameters on storage of maize: A review. J. Nat. Sci. Res. 2013, 3, 147. [Google Scholar]
- Ranum, P.; Pena-Rosas, J.P.; Garcia-Casal, M.N. Global maize production, utilization, and consumption. Ann. N. Y. Acad. Sci. 2014, 1312, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, C.; Mora, F.; Scapim, C.A.; Coan, M. Genome-wide haplotype-based association analysis of key traits of plant lodging and architecture of maize identifies major determinants for leaf angle: hapLA4. PLoS ONE 2019, 14, e0212925. [Google Scholar] [CrossRef]
- Coan, M.; Senhorinho, H.J.; Pinto, R.J.; Scapim, C.A.; Tessmann, D.J.; Williams, W.P.; Warburton, M.L. Genome-Wide Association Study of Resistance to Ear Rot by Fusarium verticillioides in a Tropical Field Maize and Popcorn Core Collection. Crop. Sci. 2018, 58, 564–578. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, H.; Wu, L.; Warburton, M.; Yan, J. Genome-Wide Association Studies in Maize: Praise and Stargaze. Mol. Plant. 2017, 10, 359–374. [Google Scholar] [CrossRef]
- Zhu, X.M.; Shao, X.Y.; Pei, Y.H.; Guo, X.M.; Li, J.; Song, X.Y.; Zhao, M.A. Genetic Diversity and Genome-Wide Association Study of Major Ear Quantitative Traits Using High-Density SNPs in Maize. Front. Plant. Sci. 2018, 9, 966. [Google Scholar] [CrossRef]
- Romay, M.C.; Millard, M.J.; Glaubitz, J.C.; Peiffer, J.A.; Swarts, K.L.; Casstevens, T.M.; Elshire, R.J.; Acharya, C.B.; Mitchell, S.E.; Flint-Garcia, S.A.; et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biol. 2013, 14, R55. [Google Scholar] [CrossRef]
- Liu, N.; Xue, Y.; Guo, Z.; Li, W.; Tang, J. Genome-Wide Association Study Identifies Candidate Genes for Starch Content Regulation in Maize Kernels. Front. Plant. Sci. 2016, 7, 1046. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Bradbury, P.; Liu, X.; Lu, F.; Romay, C.; Glaubitz, J.; Wu, X.; Peng, B.; Shi, Y.; et al. Identification of genetic variants associated with maize flowering time using an extremely large multi-genetic background population. Plant. J. 2016, 86, 391–402. [Google Scholar] [CrossRef]
- Lee, T.; Lee, S.; Yang, S.; Lee, I. MaizeNet: A co-functional network for network-assisted systems genetics in Zea mays. Plant. J. 2019, 99, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Ronald, P.C. A rapid DNA minipreparation method suitable for AFLP and other PCR applications. Plant. Mol. Biol. Rep. 1999, 17, 53–57. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Money, D.; Gardner, K.; Migicovsky, Z.; Schwaninger, H.; Zhong, G.Y.; Myles, S. LinkImpute: Fast and accurate genotype imputation for nonmodel organisms. G3 Genes Genom. Genet. 2015, 5, 2383–2390. [Google Scholar] [CrossRef]
- Yu, L.X.; Zheng, P.; Bhamidimarri, S.; Liu, X.P.; Main, D. The Impact of Genotyping-by-Sequencing Pipelines on SNP Discovery and Identification of Markers Associated with Verticillium Wilt Resistance in Autotetraploid Alfalfa (Medicago sativa L.). Front. Plant. Sci. 2017, 8, 89. [Google Scholar] [CrossRef]
- Gao, H.; Williamson, S.; Bustamante, C.D. A Markov chain Monte Carlo approach for joint inference of population structure and inbreeding rates from multilocus genotype data. Genetics 2007, 176, 1635–1651. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Breseghello, F.; Sorrells, M.E. Association Mapping of Kernel Size and Milling Quality in Wheat (Triticum aestivum L.) Cultivars. Genetics 2006, 172, 1165–1177. [Google Scholar] [CrossRef]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.D.M.J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2004, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Ballesta, P.; Maldonado, C.; Pérez-Rodríguez, P.; Mora, F. SNP and Haplotype-Based Genomic Selection of Quantitative Traits in Eucalyptus globulus. Plants 2019, 8, 331. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Endelman, J.B.; Jannink, J.L. Shrinkage estimation of the realized relationship matrix. G3 Genes Genom. Genet. 2012, 2, 1405–1413. [Google Scholar] [CrossRef]
- Contreras-Soto, R.I.; Mora, F.; de Oliveira, M.A.R.; Higashi, W.; Scapim, C.A.; Schuster, I. A genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis. PLoS ONE 2017, 12, e0171105. [Google Scholar] [CrossRef]
- Arriagada, O.; Amaral-Júnior, A.T.; Mora, F. Thirteen years under arid conditions: Exploring marker-trait associations in Eucalyptus cladocalyx for complex traits related to flowering, stem form and growth. Breed. Sci. 2018, 68, 367–374. [Google Scholar] [CrossRef]
- Hadfield, J. MCMCglmm Course Notes. 2012. Available online: http://cran.r-project.org/web/packages/MCMCglmm/vignettes/CourseNotes.pdf (accessed on 7 July 2019).
- Mora, F.; Zúñiga, P.E.; Figueroa, C.R. Genetic variation and trait correlations for fruit weight, firmness and color parameters in wild accessions of Fragaria chiloensis. Agronomy 2019, 9, 506. [Google Scholar] [CrossRef]
- Mora, F.; Ballesta, P.; Serra, N. Bayesian analysis of growth, stem straightness and branching quality in full-sib families of Eucalyptus globulus. Bragantia 2019, 78. [Google Scholar] [CrossRef]
- Hadfield, J.D. MCMC methods for multi-response generalized linear mixed models: The MCMCglmm R package. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Paes, G.P.; Viana, J.M.S.; Mundim, G.B. Linkage disequilibrium, SNP frequency change due to selection, and association mapping in popcorn chromosome regions containing QTLs for quality traits. Genet. Mol. Biol. 2016, 39, 97–110. [Google Scholar] [CrossRef]
- Yan, J.; Shah, T.; Warburton, M.L.; Buckler, E.S.; McMullen, M.D.; Crouch, J. Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 2009, 4, e8451. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, Y.; Liu, J.; Cao, M.; Wang, J.; Lan, H.; Xu, Y.; Lu, Y.; Pan, G.; Rong, T. The genetic architecture of flowering time and photoperiod sensitivity in maize as revealed by QTL review and meta-analysis. J. Integr. Plant. Biol. 2012, 54, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, B.; Liang, W.; Zheng, Y.; Qiu, F. Quantitative genetic analysis of flowering time, leaf number and photoperiod sensitivity in maize (Zea mays L.). J. Plant. Breed. Crop. Sci. 2011, 3, 168–184. [Google Scholar]
- Chardon, F.; Virlon, B.; Moreau, L.; Falque, M.; Joets, J.; Decousset, L.; Murigneux, A.; Charcosset, A. Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 2004, 168, 2169–2185. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zenda, T.; Wang, X.; Liu, G.; Jin, H.; Yang, Y.; Dong, A.; Duan, H. Comprehensive Meta-Analysis of Maize QTLs Associated with Grain Yield, Flowering Date and Plant Height Under Drought Conditions. J. Agric. Sci. 2019, 11, 1–19. [Google Scholar] [CrossRef]
- Coles, N.D.; McMullen, M.D.; Balint-Kurti, P.J.; Pratt, R.C.; Holland, J.B. Genetic control of photoperiod sensitivity in maize revealed by joint multiple population analysis. Genetics 2010, 184, 799–812. [Google Scholar] [CrossRef]
- Salvi, S.; Castelletti, S.; Tuberosa, R. An updated consensus map for flowering time QTLs in maize. Maydica 2009, 54, 501–512. [Google Scholar]
- Frey, F.P.; Presterl, T.; Lecoq, P.; Orlik, A.; Stich, B. First steps to understand heat tolerance of temperate maize at adult stage: Identification of QTL across multiple environments with connected segregating populations. Theor. Appl. Genet. 2016, 129, 945–961. [Google Scholar] [CrossRef]
- Buckler, E.S.; Holland, J.B.; Bradbury, P.J.; Acharya, C.B.; Brown, P.J.; Browne, C.; Ersoz, E.; Flint-Garcia, S.; Garcia, A.; Glaubitz, J.C.; et al. The genetic architecture of maize flowering time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef]
- Song, Y.H.; Shogo, I.; Takato, I. Flowering time regulation: Photoperiod-and temperature-sensing in leaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef]
- Pin, P.A.; Nilsson, O. The multifaceted roles of FLOWERING LOCUS T in plant development. Plant. Cell Environ. 2012, 35, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Y.; Yang, K.Z.; Wei, X.X.; Wang, X.Q. Revisiting the phosphatidylethanolamine-binding protein (PEBP) gene family reveals cryptic FLOWERING LOCUS T gene homologs in gymnosperms and sheds new light on functional evolution. New Phytol. 2016, 212, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Kawahigashi, H.; Ando, T.; Tonooka, T.; Handa, H. Molecular and functional characterization of PEBP genes in barley reveal the diversification of their roles in flowering. Plant. Physiol. 2009, 149, 1341–1353. [Google Scholar] [CrossRef]
- Wickland, D.P.; Hanzawa, Y. The FLOWERING LOCUS T/TERMINAL FLOWER 1 gene family: Functional evolution and molecular mechanisms. Mol. Plant 2015, 8, 983–997. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Chr | LD | N°SNP | Position (pb) First-Last SNP | Cambira | Sabaudia | ||||
|---|---|---|---|---|---|---|---|---|---|
| N°HB | SizeHB | Max(kb) | N°HB | SizeHB | Max(kb) | ||||
| 1 | 2.87 | 39,148 | 38,222–275,861,066 | 7180 | 32 | 498 | 7087 | 32 | 497 |
| 2 | 2.68 | 37,341 | 40,724–244,417,305 | 6874 | 35 | 500 | 6730 | 36 | 500 |
| 3 | 2.21 | 34,889 | 191,169–235,520,333 | 6370 | 35 | 487 | 6256 | 33 | 491 |
| 4 | 6.55 | 26,908 | 217,040–246,840,261 | 4790 | 42 | 500 | 4693 | 42 | 499 |
| 5 | 2.61 | 35,691 | 12,711–223,658,670 | 6584 | 33 | 493 | 6472 | 32 | 500 |
| 6 | 3.75 | 23,441 | 169,964–173,881,702 | 4368 | 53 | 466 | 4317 | 53 | 466 |
| 7 | 2.23 | 24,958 | 180,204–182,128,999 | 4683 | 28 | 500 | 4546 | 28 | 500 |
| 8 | 4.1 | 25,537 | 204,228–181,043,617 | 4703 | 50 | 498 | 4577 | 50 | 498 |
| 9 | 2.94 | 22,404 | 61,292–159,668,042 | 4091 | 35 | 500 | 3991 | 36 | 500 |
| 10 | 2.85 | 20,656 | 128,669–150,847,940 | 3760 | 52 | 498 | 3698 | 51 | 498 |
| Mean | 2.94 | 29,097 | - | 5340 | 40 | 494 | 5238 | 39 | 495 |
| Marker | Trait | Cambira | Sabaudia | ||||
|---|---|---|---|---|---|---|---|
| NM | Chr(NM) | PV% | NM | Chr(NM) | PV% | ||
| SNP | FF | 10 | 2(2), 3(1), 6(1), 7(4), 8(1), and 9(1) | 5.6–6.3 | 7 | 2(1), 3(1), 5(3), and 6(2) | 6.5–10.1 |
| MF | 5 | 2(2), 8(1), and 9(2) | 5.7–6.4 | 6 | 3(3), 5(1), and 6(2) | 6.5–8.5 | |
| ASI | 8 | 1(2), 3(1), 5(1), 6(1), 7(1), and 8(2) | 5.6–6.0 | 9 | 1(2), 3(2), 8(3), 9(1), and 10(1) | 6.5–9.9 | |
| Haplotype Blocks | FF | 11 | 1(1), 3(1), 5(1), 6(2), 7(1), 8(3), and 9(2) | 5.6–17.0 | 3 | 3(2) and 9(1) | 6.3–8.6 |
| MF | 7 | 2(1), 4(1), 7(1), 8(2), and 9(2) | 4.6–13.0 | 12 | 1(2), 4(4), 5(1), 7(3), 8(1), and 9(1) | 5.0–6.9 | |
| ASI | 4 | 1(1), 3(1), 8(1), and 10(1) | 5.6–7.8 | 7 | 2(2), 5(1), 7(2), 8(1), and 9(1) | 5.1–9.5 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, C.; Mora, F.; Bertagna, F.A.B.; Kuki, M.C.; Scapim, C.A. SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Maize with Network-Assisted Gene Prioritization. Agronomy 2019, 9, 725. https://doi.org/10.3390/agronomy9110725
Maldonado C, Mora F, Bertagna FAB, Kuki MC, Scapim CA. SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Maize with Network-Assisted Gene Prioritization. Agronomy. 2019; 9(11):725. https://doi.org/10.3390/agronomy9110725
Chicago/Turabian StyleMaldonado, Carlos, Freddy Mora, Filipe Augusto Bengosi Bertagna, Maurício Carlos Kuki, and Carlos Alberto Scapim. 2019. "SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Maize with Network-Assisted Gene Prioritization" Agronomy 9, no. 11: 725. https://doi.org/10.3390/agronomy9110725
APA StyleMaldonado, C., Mora, F., Bertagna, F. A. B., Kuki, M. C., & Scapim, C. A. (2019). SNP- and Haplotype-Based GWAS of Flowering-Related Traits in Maize with Network-Assisted Gene Prioritization. Agronomy, 9(11), 725. https://doi.org/10.3390/agronomy9110725